

Abstract
Background
Cancer cells show enhanced glycolysis and inhibition of oxidative phosphorylation, even in the presence of sufficient oxygen (aerobic glycolysis). Glycolysis is much less efficient for energy production than oxidative phosphorylation, and the reason why cancer cells selectively use glycolysis remains unclear.
Methods
Biospecimens were collected from 45 hepatocellular carcinoma patients. Protein samples were prepared through subcellular localization or whole-cell lysis. Protein synthesis was measured by SDS-PAGE and immunoblotting. mRNA transcription was measured using quantitative RT-PCR. Statistical correlation among immunoblotting data and clinicolaboratory factors were analyzed using SPSS.
Results
Enzymes for oxidative phosphorylation (SDHA and SDHB) were frequently decreased (56 and 48 % of patients, respectively) in hepatocellular carcinomas. The lowered amount of the SDH protein complex was rarely accompanied by stabilization of HIF1α and subsequent activation of the hypoxia response. On the other hand, protein synthesis of G6PD and TKT, enzymes critical for pentose phosphate pathway (PPP), was increased (in 45 and 55 % of patients, respectively), while that of ALDOA, an enzyme for mainstream glycolysis, was eliminated (in 55 % of patients). Alteration of protein synthesis was correlated with gene expression for G6PD and TKT, but not for TKTL1, ALDOA, SDHA or SDHB. Augmented transcription and synthesis of PPP enzymes were accompanied by nuclear accumulation of NRF2.
Conclusion
Hepatocellular carcinomas divert glucose metabolism to the anabolic shunt by activating transcription factor NRF2.
Electronic supplementary material
The online version of this article (doi:10.1007/s00535-013-0895-x) contains supplementary material, which is available to authorized users.
Keywords: Aerobic glycolysis, Succinate dehydrogenase, HIF-1α, Pentose phosphate pathway, NRF2
Introduction
Human cells maintain their homeostasis through energy production via glucose catabolism. In the presence of oxygen, most differentiated cells primarily catabolize glucose to carbon dioxide by oxidation of glycolytic pyruvate in the mitochondrial tricarboxylic acid (TCA) cycle (oxidative phosphorylation). On the other hand, cancer cells ‘ferment’ glucose into lactate (glycolysis) even when access to sufficient oxygen is available [1]. This phenomenon is known as aerobic glycolysis or the Warburg effect [1]. In terms of ATP production, oxidative phosphorylation is far more efficient than glycolysis, and it has long remained unclear why cancer cells selectively use aerobic glycolysis.
It is becoming more evident that actively proliferating cells consume glucose not only for ATP production, but also for the synthesis of nucleotides or fatty acids [2–4]. Cancer cells have a strong demand for replicating all of their components, and meet those needs by exploiting a specific branch of glycolysis [5]. The pentose phosphate pathway (PPP) is a glycolysis shunt that produces the ribose and nicotinamide adenine dinucleotide phosphate required for the synthesis of macromolecules [5, 6].
Hepatocellular carcinoma (HCC) is a typical hypervascular tumor and appears to have good access to oxygen [7]. However, the significance of aerobic glycolysis in HCC remains unclear. Fludeoxyglucose-positron emission tomography (FDG-PET) is a diagnostic imaging that exploits the excess glucose uptake by carcinoma cells over normal (differentiated) cells. While some studies have reported that FDG-PET is inferior to X-ray computed tomography in terms of accuracy [8], others have indicated that FDG-PET is indeed useful for prognostication [9]. Molecular analyses of HCC glycolysis have been performed using both microarray [10] and immunohistochemistry [11, 12] techniques. Accumulating evidence suggests a possible causal role of aerobic glycolysis in HCC malignancy.
In the present study, we investigated the enzymes involved in glucose catabolism in HCC specimens. The synthesis of each enzyme protein was frequently altered, and such alteration was mostly consistent with the Warburg effect, that is, SDHA and SDHB were reduced and synthesis of PPP enzymes was augmented. Unexpectedly, ALDOA, an enzyme involved in mainstream glycolysis, was eliminated. The decrease of mitochondrial enzymes was not accompanied by HIF-1α activation, but augmented synthesis of PPP enzymes and loss of ALDOA were accompanied by nuclear accumulation of NRF2. Our findings confirm that HCC utilizes ‘metabolic reprogramming’ as a fundamental strategy for tumor growth by activating oncogenic signaling.
Methods
This study had the approval of the Institutional Review Board (provided ID number: 24089) on the basis of the Ethical Guidelines for Clinical Research of the Ministry of Health, Labour and Welfare in Japan (http://www.mhlw.go.jp/seisakunitsuite/bunya/hokabunya/kenkyujigyou/i-kenkyu/index.html).
Moreover, this study is registered with the UMIN Clinical Trials Registry (provided ID number: UMIN000011465).
Patients and tumor specimens
Biospecimens were collected from patients newly diagnosed as having HCC that were undergoing surgical resection at Dokkyo Medical University Hospital. None of the patients had received prior treatment for their disease, including chemotherapy or radiotherapy. All cases were collected regardless of histologic grade, but those showing predominance of necrotic and inflammatory cells were excluded. Each tumor specimen had accompanying non-tumor tissue, which was cirrhosis in most cases. After pathological diagnosis, specimens from 45 patients were used for the present analysis, which was conducted under strict adherence to ethical guidelines.
Preparation of protein samples
Protein samples were prepared either through subcellular fractionation or whole-cell lysis. To separate subcellular localized proteins, we used an NE-PER Nuclear Protein Extraction Kit (Thermo Scientific, Rockford, IL). Tissue specimens were weighed and minced with surgical scissors, and the minced tissue was immersed in hypotonic solution and homogenized in a Dounce homogenizer. After cell swelling/bursting and brief centrifugation, cytoplasmic (mitochondrial as well) and nuclear fractions were separated. Nuclear proteins were solubilized in RIPA buffer (25 mM Tris/HCl pH 7.5, 150 mM NaCl, 1 % NP-40, 1 % sodium deoxycholate), and genomic DNA was fragmented by sonication (Bioruptor, Diagenode, Liege, Belgium). Proteins were denatured in lithium dodecyl sulfate (NuPAGE Sample Buffer, Life Technologies, Carlsbad, CA, USA) and reduced in 50 mM dithiothreitol. Starting with various specimen weights, and adding various amounts of reagents, the protein concentrations were adjusted to around 10 mg/ml (cytoplasm/mitochondria) or 5 mg/ml (nucleus). Successful fractionation was confirmed by immunoblotting for GAPDH (cytoplasm) or Histone H3 (nucleus). Alternatively, minced specimens were directly disrupted by sonication (Bioruptor) in denaturation buffer (7 M urea, 2 M thiourea, 2 % Triton X-100) and insoluble debris was removed by centrifugation.
SDS PAGE and immunoblotting
Proteins were size-fractionated by electrophoresis in 8 or 12 % sodium dodecyl sulfate polyacrylamide gel, then wet-transferred to a PVDF membrane (Immobilon-P, Merck Millipore, Billerica, MA, USA). After blocking with 2 % skim milk or 5 % bovine serum albumin TBST (Tris/HCl pH 7.4, 150 mM NaCl, 0.1 % Tween20), the membranes were incubated with each respective primary antibody at 4 °C overnight. Non-specific binding was washed away with TBST and the protein-antibody complex was visualized using horseradish peroxidase-conjugated secondary antibody (#7074 or 7076, Cell Signaling Technology, Beverly, MA, USA) and the luminescence reaction (ECL, ECL prime or ECL select, GE Healthcare, Little Chalfont, United Kingdom). Images were taken with a CCD camera and stored in the machine (LAS500, GE Healthcare).
Primary antibodies
For the information about primary antibodies used in Western blotting, see the Supplemental Table 1.
Reverse transcription-polymerase chain reaction
Total RNA was purified from biospecimens using the guanidine isothiocyanate method (Isogen Plus, Takara Bio Inc., Shiga, Japan). mRNA was subsequently reverse-transcribed to cDNA (Prime Script-RT, Takara Bio Inc.) and the amount of each mRNA was measured with a real-time PCR machine (ABI PRISM7000 realtime PCR system, Applied Biosystems, Foster City, CA). The TaqMan probes used are as follows: Eukaryotic 18S rRNA (#4333760F, Life Technologies), ALDOA (#Hs00605108_g1, Applied Biosystems), G6PD (#Hs00166169_m1, Applied Biosystems), TKT (#Hs01115545_m1, Applied Biosystems), SDHB (#Hs01042482-m1, Applied Biosystems) and TKTL1 (#Hs00202061_m1, Applied Biosystems). For SDHA amplification and detection, we utilized web-based qPCR design software (Universal Probe Assay Design Center, Roche Applied Science, Penzberg, Germany, https://qpcr.probefinder.com/organism.jsp). Universal probe #70 was used for SDHA specific detection and the primer sequences of upper and lower primers are as follows: Forward 5′-TCCACTACATGACGGAGCAG-3′ and Reverse 5′-CCATCTTCAGTTCTGCTAAACG-3′. Size of SDHA product was 82 bp.
Data quantification and statistical analysis
Data from Western blotting was quantified with IQTL software (version8.1, GE healthcare). We calculated the relative abundance of protein as the ratio of tumor to non-tumor band intensity but the data of extremely low amounts were defined as ‘under detection’. Some molecules were synthesized exclusively in tumor tissue (e.g., PKM2 or G6PD) and the data were presented as ‘−’, ‘+’, ‘++’ or ‘+++’. The calculated ratio was subsequently normalized using GAPDH as a standard (ratio of target protein)/(ratio of GAPDH). Although we initially tested the feasibility of using GAPDH or β-actin as a standard molecule, GAPDH found to be superior (see Supplemental Table 2 for the standard deviation of each ratio). For nuclear proteins, Histone H3 served as a standard. Data from real-time RT-PCR was quantified using the 2−ΔΔCt method [13]. Dependencies between groups were analyzed using the chi-squared test, dividing each variable into two groups (e.g., PKM2 synthesized or not). Statistical analyses were performed using the SPSS statistical software package, version 20.0 (SPSS Inc., Chicago, IL, USA. Differences at P < 0.05 were considered to be statistically significant.
Results
Inhibition of oxidative phosphorylation in HCC
First, we compared the synthesis of subunits of succinate dehydrogenase (SDH) complex that are functional in the TCA cycle. In comparison with their non-tumor tissue counterparts, synthesis of SDHA and SDHB proteins was reduced in the HCC tumor specimens (Fig. 1). Twenty-two (among 39 examined, 56 %) and 19 (among 40, 48 %) patients exhibited more than 40 % decrease for SDHA and SDHB, respectively. These data suggested that SDH activity was repressed and that the TCA cycle was arrested. To examine whether the decrease of SDH proteins was due to non-specific protein degradation, we checked isocitrate dehydrogenase (IDH), another enzyme involved in oxidative phosphorylation. IDH1 and IDH2 are localized to the cytoplasm and mitochondria, respectively, and we compared the synthesis of each protein between HCC and each respective non-tumor counterpart tissue sample. Although some patients exhibited decreased synthesis of IDH1 (11/37 = 30 %) and IDH2 (12/38 = 32 %), the frequency was not as remarkable as that observed for SDHA and SDHB.
Fig. 1.

Frequent reduction of SDHA and SDHB synthesis in HCCs. Fifty μg of protein of whole-tissue lysate were loaded in each well, and Western blotting for TCA cycle enzymes was performed. Tumor/non-tumor paired samples were placed side by side. The band intensity was quantified with densitometry and the ratio between tumor/non-tumor were presented. GAPDH serves as an internal control. Eight representative specimens are shown
Pentose phosphate pathway is activated but mainstream glycolysis is inhibited in HCC
Next we examined the synthesis of the enzyme proteins glucose-6-phosphate dehydrogenase (G6PD) and transketolase (TKT) in HCC. G6PD and TKT are critical enzymes in the glycolysis shunt, the pentose phosphate pathway. We also examined the synthesis of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and aldolase (ALDOA), which are enzymes involved in mainstream glycolysis and therefore we expected them to serve as internal controls. Remarkably, synthesis of both G6PD and TKT was increased in HCCs relative to the non-tumor counterpart tissues (Fig. 2). Thirteen (among 29 examined, 45 %) and 21 (among 38, 55 %) patients exhibited this phenomenon for G6PD and TKT, respectively. On the other hand, synthesis of ALDOA was nearly eliminated (Fig. 2). The ratio of patients who had lost ALDOA relative to the total number of patients examined was 55 % (16/29). In contrast, synthesis of GAPDH was almost identical in tumor and non-tumor tissues (Fig. 2) in all of the patients examined (tumor/non-tumor: mean = 0.96, standard deviation = 0.27, n = 41).
Fig. 2.
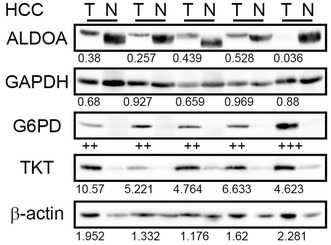
Frequently increased synthesis of G6PD and TKT, and loss of ALDOA synthesis, in HCCs. Fractionated cytoplasmic/mitochondrial proteins were loaded in each well and Western blotting for glycolysis/PPP enzymes was performed. Tumor/non-tumor paired samples were placed side by side. The band intensity was quantified with densitometry and the ratio between tumor/non-tumor was presented. β-actin serves as a loading control. Five representative specimens are shown
The hypoxia-inducible factor HIF-1α is not activated in HCCs
Next, we examined the synthesis and accumulation of HIF-1α protein in nuclear extracts from HCCs. However, we failed to detect the HIF-1α protein in most of the HCC patients (2/29 positive, data not shown). As a positive control, HIF-1α protein was easily detected in the lysate of Huh7 cells that had been incubated in a hypoxic chamber (O2 1 %, 4 h, data not shown). Surgically resected specimens were exposed to ambient oxygen before preservation in a freezer, and the proteins were subsequently purified. To exclude the possibility that HIF-1α protein would have been degraded during sample preparation, we checked the events downstream of HIF-1α activation. When we compared the protein synthesis of cytochrome oxidase IV (COX4-1), a negative target of HIF-1α, 18 % of the patients (7/39) displayed a reduced level of COX4-1 protein (Fig. 3). We also examined the synthesis of PKM2 and LDHA, positive targets of HIF-1α. However, synthesis of the PKM2 protein was observed in 15 % (6/41) of the HCC patients (Fig. 3) and for LDHA, only 8 % of the patients (3/39) displayed augmented protein synthesis (Fig. 3).
Fig. 3.

Occasional degradation of COX4-1, synthesis of PKM2 and augmented LDHA synthesis in HCCs. Fifty μg of protein of whole-tissue lysate were loaded in each well, and Western blotting for HIF-1 targets/modulators was performed. Tumor/non-tumor paired samples were placed side by side. The band intensity was quantified with densitometry and the ratio between tumor/non-tumor were presented. GAPDH serves as an internal control. Eight representative specimens are shown
Frequent nuclear accumulation of NRF2 protein in HCCs
Next, we examined the nuclear localization of a transcription factor NRF2. In contrast to HIF-1α, which was hardly detectable by the same method, NRF2 protein was detected in tumor tissue and occasionally in non-tumor tissue (Fig. 4). We used two different monoclonal antibodies recognizing distinct epitopes, but the data mostly overlapped (Fig. 4). The ratio of patients who possessed nuclear NRF2 relative to the total examined was 22/30 (73 % for NRF2 C-terminal) and 22/30 (77 % for phosphorylated S40 of NRF2). However, synthesis of KEAP1, an E3 uniquitin ligase against NRF2, varied (increase: 7/24, decrease: 9/24, unchanged: 8/24, data not shown).
Fig. 4.
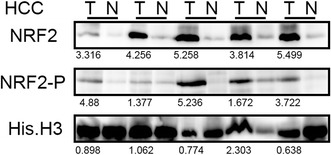
Frequent accumulation of NRF2 proteins in HCC nuclei. Fractionated nuclear proteins were loaded in each well, and Western blotting for NRF2 was performed. Tumor/non-tumor paired samples were placed side by side. The band intensity was quantified with densitometry and the ratio between tumor/non-tumor was presented. Histone H3 serves as an internal control. Five representative specimens are shown
Augmented synthesis of PPP proteins were correlated with mRNA induction
Next, we measured mRNA transcription of genes that encode G6PD, TKT, ALDOA, SDHA or SDHB protein. As expected, gene transcription of G6PD was increased and correlated well with protein synthesis (Supplementary Fig. 1). The TKT protein is encoded by three distinct genes, TKT, TKTL1 or TKTL2. Although TKTL1 mRNA is known to be induced in some tumors (for review, see Riganti et al. [14]), only trace amounts of the TKTL1 mRNA were expressed, and we determined that the data were out of quantification range (data not shown). However, the TKT gene was abundantly expressed and further increased in tumor tissue (Supplementary Fig. 2), indicating that G6PD and TKT could be upregulated by a transcription factor, NRF2. On the other hand, transcription of the ALDOA gene was poorly correlated with protein synthesis (Supplementary Fig. 3). Fifty-eight percent of patients (7/12) displayed increased ALDOA expression, although most of these patients displayed loss of ALDOA protein (Supplementary Fig. 3). Thus, transcription of the ALDOA gene and the synthesis of ALDOA protein displayed an inverse relationship. We also examined the SDHA and SDHB gene transcription. However, neither SDHA (Supplementary Fig. 4) nor SDHB (Supplementary Fig. 5) had any apparent relationship between mRNA transcription and protein synthesis. Fifty percent (6/12) and 67 % (8/12) of patients displayed an inverse relationship with SDHA and SDHB, respectively.
SDH reduction did not correlate with hypoxia response, while increased synthesis of PPP enzymes correlated with activation of NRF2
Finally, statistical analysis was performed to examine the dependencies among HIF-1α activation, and alteration of LDHA, PKM2, SDHA, SDHB and COX4-1 (Table 1). HIF-1a activation was significantly correlated with PKM2 synthesis (P = 0.02). Synthesis of PKM2 was inversely correlated with decreased synthesis of SDHB (P = 0.033*). Decreased COX4-1 synthesis was significantly correlated with decreased synthesis of SDHA and SDHB (P = 0.009 and P = 0.001, respectively), and decreased SDHA synthesis was significantly correlated with decreased SDHB synthesis (P < 0.001).
Table 1.
Dependencies between HIF-1α activation and alteration of glycolytic enzyme
| HIF-1α | LDHA | PKM2 | SDHA | SDHB | COX4-1 | |
|---|---|---|---|---|---|---|
| HIF1α | ||||||
| LDHA | 0.773 | |||||
| PKM2 | 0.02 | 0.290 | ||||
| SDHA | 0.747 | 0.920 | 0.067 | |||
| SDHB | 0.727 | 0.234 | 0.033* | <0.001 | ||
| COX4-1 | 0.197 | 0.971 | 0.944 | 0.009 | 0.001 |
Asterisk denotes inverse correlation
Bold number denotes statistically significant
Chi-square test, P < 0.05
HIF1α Hypoxia-induced factor 1α, LDHA Lactate dehydrogenase, PKM2 Pyruvate kinase M2, SDH Succinate dehydrogenase
We next analyzed the dependencies between NRF2 activation (nuclear NRF2) and other protein alterations (Fig. 5). NRF2 activation was significantly correlated with G6PD synthesis (P = 0.044, Table 2) and with TKT (P = 0.04, Table 2), but not with other variables (data not shown).
Fig. 5.
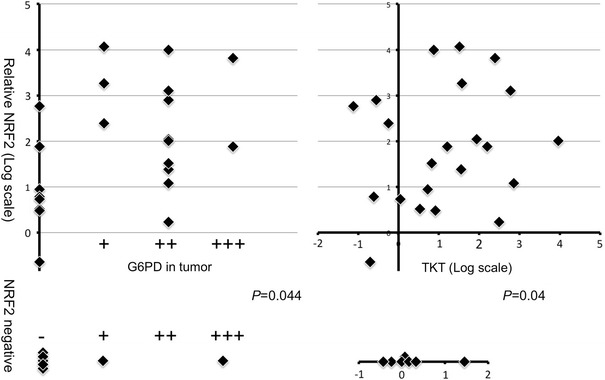
Relationships between NRF2 activation and G6PD or TKT increase. Upper Scatter plot of relative abundance of protein shows nuclear NRF2 (y-axis) and glycolysis enzymes (x-axis). Quantitative data are presented in log-scale (base = 2). Lower For patients who displayed undetectable NRF2 protein, protein levels of enzymes are shown as one-variable plots. P value is calculated with the chi-square test
Table 2.
Dependencies between activation of NRF2 and pentose phosphate pathway enzymes
| Variable | Activated NRF2C-terminal (n = 21) | Not activated NRF2C-terminal (n = 8) | P |
|---|---|---|---|
| G6PD | |||
| Activated | 14 | 2 | |
| Not activated | 7 | 6 | 0.044 |
| TKT | |||
| Activated | 14 | 2 | |
| Not activated | 7 | 6 | 0.04 |
Chi-squared test
G6PD Glucose-6-phosphate dehydrogenase, TKT transketalase
Statistical analysis was also performed to compare the Western blotting data with various clinicolaboratory factors, such as tumor differentiation, number of tumors, tumor size, UICC TNM staging (7th edition), tumor markers (AFP, PIVKA-II), AST, ALT, Alb, CRP, PLT, and hepatitis viral infection (all the information are presented in Supplementary Table 2). Increased expression of G6PD was significantly correlated with moderate or poor differentiation (P = 0.007). Augmented NRF2 activation was significantly correlated with invasive growth (P = 0.041). PIVKA-2 was significantly correlated with NRF2 activation and an increase of TKT (P = 0.004, P = 0.014). AFP was significantly correlated with an increase of G6PD and a decrease of IDH1 (P = 0.018, P = 0.015). ICGR15 was significantly correlated with IDH1 decrease (P = 0.008). Hepatitis virus C infection was significantly correlated with ALDOA decrease (P = 0.036, patients who had HCV vs. those who had HBV).
Discussion
In this study, we collected specimens of HCC from Japanese patients and examined the synthesis of enzymes involved in glucose metabolism. In comparison to other solid tumors, the tissue composition of HCC is relatively uniform in both the tumor and non-tumor components. Data obtained from subcellular fractionation and Western blotting could reflect the metabolic status of HCC in situ. Our present findings are mostly consistent with Warburg’s original report, although we discovered some unexpected phenomena as well.
Overall, the liver is a hypervascular organ, and thus HCC is likely to have a good oxygen supply during tumor formation. Nevertheless, HCC intensively induces angiogenesis and grows into an apparently vascular-rich tumor. One possible explanation is that HIF-1 is activated under normoxic conditions and induces vascular endothelial growth factor. We observed that synthesis of SDHA and SDHB was frequently reduced (22/39 and 19/40, respectively) in HCCs (Fig. 1). SDHB is a tumor suppressor of familial paraganglioma or pheochromocytoma [15, 16], and loss of its function leads to succinate accumulation [17]. Succinate is a substrate of SDH and its accumulation results in inhibition of prolyl hydroxylase and subsequent stabilization of HIF-1α [17]. Activation of HIF-1α, in turn, induces LONP1, a protease that degrades mitochondrial protein cytochrome oxidase IV (COX4-1) and inhibits oxidative phosphorylation [18]. Moreover, HIF-1α activation enhances LDHA gene expression [3], and the cooperative activation of HIF-1α and PKM2 is the driving force of aerobic glycolysis [19]. Thus, SDH function and HIF-1 are closely related to each other, and we had expected that SDHA/SDHB reduction would be accompanied by HIF-1 activation. However, we concluded that this was not the case, based on certain items of evidence. First, HIF-1α protein was detected in only 7 % of HCC patients (2/29). Second, augmented synthesis of a transcription target (LDHA) was also a rare event in HCCs (8 % = 3/39). Third, although activation of PKM2 was occasional (15 % = 6/41), it was inversely correlated with SDHA/SDHB reduction (Table 1). Indeed, statistical analysis indicated that alteration of COX4-1 was correlated with SDHA/SDHB decrease (Table 1), and this specific variable could partly explain the decrease of SDH (mitochondrial remodeling). Other variables (HIF-1α, PKM2 and LDHA), however, were not positively correlated with SDHA/SDHB decrease. It is generally accepted that hypoxia-inducible factors play crucial roles in hepatocellular carcinogenesis [20]. Previous studies used immunohistochemistry to detect HIF-1α in HCC specimens [21–23]. We cannot exclude the possibility that HIF-1α was activated below the detection range of our present method, or that other HIF proteins were activated instead [24]. However, we speculate that activation of HIF-1α is not a predominant factor of HCCs in Japanese patients.
The accumulation of NRF2 in HCC nuclei (73 % = 22/30 for NRF2 C-terminal and 77 % = 23/30 for phosphorylated S40 of NRF2) was another major discovery. Recently, comprehensive exome sequencing of HCC has been carried out, and frequently mutated genes have been reported [25]. Among four newly identified genes, NFE2L2 encodes NRF2 protein [25]. NRF2 was originally identified as a transcription factor that responds to various stress signals, such as oxidative stress [26]. Upon activation by such stresses, NRF2 induces a series of detoxifying enzymes and antioxidant genes [26]. Under normal conditions, E3 ubiquitin ligase KEAP1 maintains NRF2 at a very low level through the ubiquitin–proteasome degradation system [27, 28]. In addition to the well-known target genes of NRF2, Motohashi and colleagues have identified PPP enzymes (G6PD, PGD, TKT and TALDO1) as novel targets of NRF2 [29]. Our findings are consistent with their report: NRF2 translocation to nuclei involved an increase of G6PD and TKT (Figs. 2, 4). Interestingly, their study verified that oncogenic PI3 kinase signaling augments Nrf2 translocation to murine hepatocyte nuclei [29]. We also observed occasional NRF2 accumulation in nuclei of non-tumor cirrhotic tissue, but the amount was mostly higher in tumor tissue (77 % = 17/22 for NRF2 C-terminal or 70 % = 16/23 for phosphorylated S40 of NRF2). These data suggest that the oncogenic signal is needed to augment NRF2 translocation in HCCs.
Constitutive stabilization/activation of NRF2 protein is a prognostic indicator in some cancers [30–32]. Several different mechanisms can explain NRF2 activation: (1) point mutation in the KEAP1 or NFE2L2 gene [30, 31, 33, 34]; (2) synthesis of an inhibitor of KEAP1-NRF2 interaction [35–37]; (3) promoter methylation and silencing of the KEAP1 gene [38, 39]; (4) succination of KEAP1 and subsequent dissociation of KEAP1-NRF2 interaction [40]. Two studies have reported that the rate of point mutation in the NFE2L2 gene is 6.4 % [25] and in the KEAP1 gene is 8.9 % [41]. Although we did not carry out mutational screening in this series of Japanese HCC cases, the modest frequency of point mutation is unlikely to explain the high incidence of nuclear translocation of NRF2 (Supplementary Table 2). Alternatively, chronic inflammation results in deregulation of autophagy, leading to accumulation of an inhibitor of KEAP1-NRF2 interaction [36, 42]. The possibility that increased succinate caused by loss of SDHA/SDHB might result in KEAP1 succination is also intriguing [40].
Unexpectedly, we observed that ALDOA protein was frequently eliminated in HCCs (Fig. 2). Three isozymes are encoded by three different genes: ALDOA, ALDOB and ALDOC. It has been reported that the expression of ALDOB mRNA is reduced in HCCs [43], whereas that of ALDOA mRNA is increased [44], thus fitting the conventional concept of enhanced glycolysis. Indeed, as for mRNA expression, we confirmed that this was the case in Japanese HCC specimens (Supplementary Fig. 3). Nevertheless, the synthesis of ALDOA protein did not correlate with mRNA expression (Supplementary Fig. 3). Of note, the band of ALDOA was migrated more slowly in tumor specimens compared to non-tumor counterparts (Fig. 2). It is possible that ALDOA protein is constantly degraded due to post-translational modification, such as ubiquitination. Aldolase catalyses a reversible aldol reaction utilizing not only fructose 1,6-bisphosphate, but also sedoheptulose 1,7-bisphosphate as substrates. The latter is located in the non-oxidative arm of PPP and this specific branch is known to be regulated by oncogenic signaling [45]. Future studies will need to clarify whether ALDOA is simply a bystander or an active driver of carcinogenesis.
There are a couple of differences between the classical definition of the Warburg effect and our present observations (Supplementary Fig. 6). First, increased expression of LDHA (an enzyme for lactate production) was a rare event (8 % = 3/39). Second, synthesis of PKM2 (a driver of aerobic glycolysis) was occasional (15 % = 6/41), but inversely correlated with a reduction of SDH proteins. Third, ALDOA (an enzyme for mainstream glycolysis) was frequently eliminated (55 % = 16/29). Currently, we are extending our analysis to other gastrointestinal tumors such as colorectal carcinomas (CRCs). In contrast to HCCs, CRCs frequently synthesized PKM2 protein and increased ALDOA protein (Supplementary Fig. 6, N. Shibuya, K. Inoue and K. Kubota, manuscript in preparation). Increased PPP enzymes, however, were observed in CRCs as well (Supplementary Fig. 6). Data indicate that the Warburg effect, at least in HCC, does not fit the stereotypic form originally proposed. If HCC does not augment mainstream glycolysis, the net glusose uptake of tumor cells could be similar to those of surrounding hepatocytes. Although further evidence is needed, this could be the reason why the usefulness of FDG-PET is relatively low compared to other tumors (difficult to distinguish the change of FDG uptake).
Lastly, but not least, metabolic reprogramming seems to be a fundamental survival strategy of cancer cells [46]. In the near future, a more comprehensive understanding of aerobic glycolysis may lead to the design of novel therapeutic strategies for HCC.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
SDHB and ALDOA reduction was originally discovered through an attempt to characterize advanced glycation end products (AGEs) in HCCs. Therefore, we sincerely thank Prof. M. Takeuchi for use of the affinity-purified AGE antibody. We are also grateful to Dr. K. Akimoto and Ms. S. Satoh for technical assistance. This study was funded by a research grant, Society for Biomarker, Japan and a project research grant (#2012-02-2), Dokkyo Medical University.
Conflict of interest
The authors declare that they have no conflict of interest.
Abbreviations
- HCC
Hepatocellular carcinoma
- PPP
Pentose phosphate pathway
- TCA
Tricarboxylic acid cycle
- SDH
Succinate dehydrogenase
- FDG-PET
Fludeoxyglucose-positron emission tomography
- IDH
Isocitrate dehydrogenase
- G6PD
Glucose-6-phosphate dehydrogenase
- TKT
Transketolase
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- CRC
Colorectal Carcinoma
References
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Weinhouse S. The warburg hypothesis fifty years later. Zeitschrift Für Krebsforschung Und Klinische Onkologie. 1976;87:115–126. doi: 10.1007/BF00284370. [DOI] [PubMed] [Google Scholar]
- 3.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 4.Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009;324:1029–33. [DOI] [PMC free article] [PubMed]
- 6.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 7.Semela D, Dufour J. Angiogenesis and hepatocellular carcinoma. J Hepatol. 2004;41:864–880. doi: 10.1016/j.jhep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Khan MA, Combs CS, Brunt EM, Lowe VJ, Wolverson MK, Solomon H, et al. Positron emission tomography scanning in the evaluation of hepatocellular carcinoma. J Hepatol. 2000;32:792–797. doi: 10.1016/S0168-8278(00)80248-2. [DOI] [PubMed] [Google Scholar]
- 9.Shiomi S, Nishiguchi S, Ishizu H, Iwata Y, Sasaki N, Tamori A, et al. Usefulness of positron emission tomography with fluorine-18-fluorodeoxyglucose for predicting outcome in patients with hepatocellular carcinoma. Am J Gastroenterol. 2001;96:1877–1880. doi: 10.1111/j.1572-0241.2001.03888.x. [DOI] [PubMed] [Google Scholar]
- 10.Hamaguchi T, Iizuka N, Tsunedomi R, Hamamoto Y, Miyamoto T, Iida M, et al. Glycolysis module activated by hypoxia-inducible factor 1α is related to the aggressive phenotype of hepatocellular carcinoma. Int J Oncol. 2008;33:725. [PubMed] [Google Scholar]
- 11.Daskalow K, Pfander D, Weichert W, Rohwer N, Thelen A, Neuhaus P, et al. Distinct temporospatial expression patterns of glycolysis-related proteins in human hepatocellular carcinoma. Histochem Cell Biol. 2009;132:21–31. doi: 10.1007/s00418-009-0590-4. [DOI] [PubMed] [Google Scholar]
- 12.Kitamura K, Hatano E, Higashi T, Narita M, Seo S, Nakamoto Y, et al. Proliferative activity in hepatocellular carcinoma is closely correlated with glucose metabolism but not angiogenesis. J Hepatol. 2011;55:846–857. doi: 10.1016/j.jhep.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 13.Arocho A, Chen B, Ladanyi M, Pan Q. Validation of the 2-DeltaDeltaCt calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn Mol Pathol. 2006;15:56–61. doi: 10.1097/00019606-200603000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radical Biol Med. 2012;53:421–436. doi: 10.1016/j.freeradbiomed.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- 16.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda R, Zhang H, Kim J, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 19.Bayley J, Devilee P. The warburg effect in 2012. Curr Opin Oncol. 2012;24:62–67. doi: 10.1097/CCO.0b013e32834deb9e. [DOI] [PubMed] [Google Scholar]
- 20.Nath B, Szabo G. Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology. 2012;55:622–633. doi: 10.1002/hep.25497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, et al. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–421. doi: 10.1016/S0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang G, Yang L, Lu W. Expression of hypoxia-inducible factor 1α and vascular endothelial growth factor in hepatocellular carcinoma: impact on neovascularization and survival. World J Gastroenterol. 2005;11:1705–1708. doi: 10.3748/wjg.v11.i11.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie H, Song J, Liu K, Ji H, Shen H, Hu S, et al. The expression of hypoxia-inducible factor-1α in hepatitis B virus-related hepatocellular carcinoma: correlation with patients’ prognosis and hepatitis B virus X protein. Dig Dis Sci. 2008;53:3225–3233. doi: 10.1007/s10620-008-0296-9. [DOI] [PubMed] [Google Scholar]
- 24.Bangoura G, Liu Z, Qian Q, Jiang C, Yang G, Jing S. Prognostic significance of HIF-2alpha/EPAS1 expression in hepatocellular carcinoma. World J Gastroenterol. 2007;13:3176. doi: 10.3748/wjg.v13.i23.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 27.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi A, Kang M, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 30.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1–NRF2 interaction in non-small-cell lung cancer. PLoS Medicine. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135: 1358–1368. e4. [DOI] [PubMed]
- 32.Jiang T, Chen N, Zhao F, Wang X, Kong B, Zheng W, et al. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010;70:5486–5496. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 34.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci. 2008;105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen W, Sun Z, Wang X, Jiang T, Huang Z, Fang D, et al. Direct interaction between Nrf2 and p21 Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 37.Lau A, Wang X, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang R, An J, Ji F, Jiao H, Sun H, Zhou D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun. 2008;373:151–154. doi: 10.1016/j.bbrc.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, et al. Loss of kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010;9:336–346. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoo NJ, Kim HR, Kim YR, An CH, Lee SH. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology. 2012;60:943–952. doi: 10.1111/j.1365-2559.2012.04178.x. [DOI] [PubMed] [Google Scholar]
- 42.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinoshita M, Miyata M. Underexpression of mRNA in human hepatocellular carcinoma focusing on eight loci. Hepatology. 2002;36:433–438. doi: 10.1053/jhep.2002.34851. [DOI] [PubMed] [Google Scholar]
- 44.Castaldo G, Calcagno G, Sibillo R, Cuomo R, Nardone G, Castellano L, et al. Quantitative analysis of aldolase A mRNA in liver discriminates between hepatocellular carcinoma and cirrhosis. Clin Chem. 2000;46:901–906. [PubMed] [Google Scholar]
- 45.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.