

Abstract
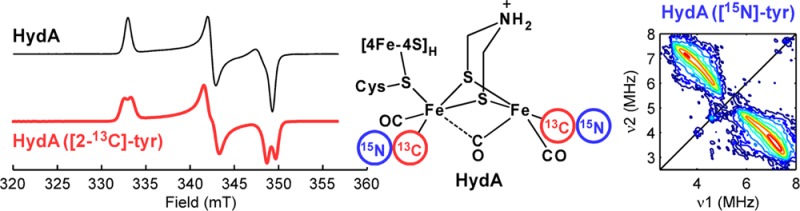
The two cyanide ligands in the assembled cluster of [FeFe] hydrogenase originate from exogenous l-tyrosine. Using selectively labeled tyrosine substrates, the cyanides were isotopically labeled via a recently developed in vitro maturation procedure allowing advanced electron paramagnetic resonance techniques to probe the electronic structure of the catalytic core of the enzyme. The ratio of the isotropic 13C hyperfine interactions for the two CN– ligands—a reporter of spin density on their respective coordinating iron ions—collapses from ≈5.8 for the Hox form of hydrogenase to <2 for the CO-inhibited form. Additionally, when the maturation was carried out using [15N]-tyrosine, no features previously ascribed to the nitrogen of the bridging dithiolate ligand were observed suggesting that this bridge is not sourced from tyrosine.
Hydrogenases catalyze the redox interconversion of protons and H2 and thus have received much focus as key elements in biological solar fuel production.1 The [FeFe] form of hydrogenase (HydA) is particularly active,1 and its catalytic H-cluster consists of a [4Fe-4S] cluster ([4Fe-4S]H) linked through a cysteine sulfur to a unique dinuclear iron cluster ([FeFe]H, Scheme 1).2 This subcluster possesses five inorganic ligands—two CN– and three CO—as well as a bridge recently assigned as dithiomethylamine (DTMA).3,4
Scheme 1.

Active HydA can be expressed in Escherichia coli only by also adding genes for three Fe-S containing maturase enzymes—HydE, HydF, and HydG—that are required for production of the [FeFe]H subcluster.5 Alternatively, synthetic dinuclear Fe clusters can be transferred to HydA apoprotein (containing only the [4Fe-4S]H subcluster) to produce active enzyme.4 We are utilizing a different technology: the HydE, HydF, and HydG maturases are added to a solution of apo-HydA for in vitro maturation and concurrent activation.6 This cell-free biosynthetic method allows for facile and precise isotope incorporation into the [FeFe]H subcluster.7
The Fe-bound CO and CN– ligands of the [FeFe]H subcluster are sourced from l-tyrosine (Tyr) and produced by HydG.8−10 In the present study, we use the cell-free biosynthetic method along with α-13C-Tyr ([2-13C]-Tyr) and [15N]-Tyr to specifically label the two CN– ligands with the magnetic nuclei 13C and 15N (I = 1/2).11,12 The hyperfine interaction (HFI) of these magnetic nuclei with the unpaired electrons distributed over the H-cluster serve as site-specific reporters of its electronic structure, important metrics for evaluating computational models of the H-cluster.
When poised in the active oxidation state known as Hox, the [4Fe-4S]H subcluster is diamagnetic with a formal charge of 2+,13 though the [4Fe-4S]H carries some unpaired density due to the exchange interaction with the [FeFe]H fragment. [FeFe]H itself is in a formally mixed-valence Fe(I,II) S = 1/2 state that is characterized by a rhombic electron paramagnetic resonance (EPR) spectrum (Figure 1A, top). While the overall oxidation state of the Hox form of the H-cluster is widely accepted, the distribution of the valences about the cluster is still debated. One formulation based on results from electronic structure calculations assigns a 1+ oxidation state to the Fe that is distal to the [4Fe-4S]H subcluster (Fed), leaving the proximal Fe ion (Fep) in the ferrous oxidation state.14 However, 57Fe electron nuclear double resonance (ENDOR) spectroscopic studies of HydA from Desulfovibrio desulfuricans (DdS) found that the spin density was shared more-or-less equally over both iron ions of [FeFe]H.15 Many computational models of the H-cluster have been judged based on the quality of the predicted magnetic parameters. Initially, only the 57Fe HFI were employed as a discriminating constraint.14,16 More recently, however, ligand HFI, from either the nearby, naturally abundant 14N nuclei or from 13C nuclei introduced by treatment of HydA with isotopically labeled 13CO gas, have been used to evaluate computer-generated structural models of the H-cluster.3,16,17 Unfortunately, in the case of the 14N hyperfine parameters, the assignment of the observed signals to specific nitrogen atoms is ambiguous owing to the high natural-abundance of 14N; and the 13CO-treatment aids only in characterizing the Hox-CO form. We therefore reasoned that studies of the electronic structure of Hox would be aided by selective incorporation of magnetic nuclei into the diatomic ligands of the [FeFe]H cluster.
Figure 1.
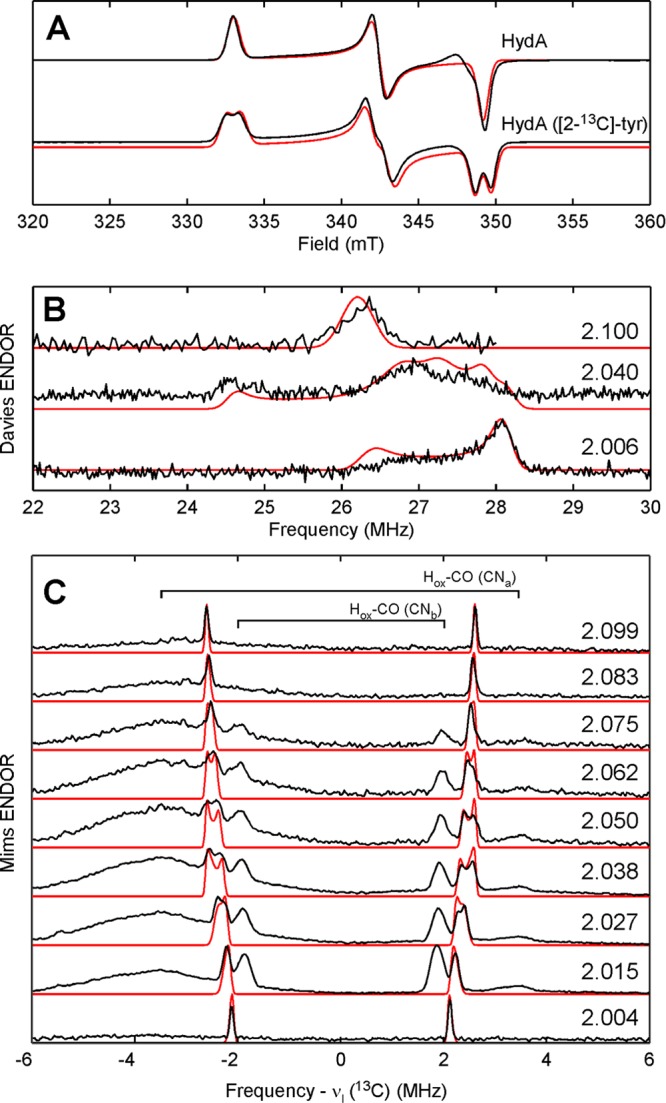
X-band (9.4 GHz) CW EPR spectra (A) of the Hox form of HydA matured using natural-abundance Tyr (top) or [2-13C]-Tyr (bottom). Davies ENDOR spectra (B) of HydA ([2-13C]-Tyr) collected at 1158, 1192, and 1212 mT (top to bottom). Corresponding g-values given in figure. Q-band (33.79 GHz) Mims ENDOR spectra (C) of HydA ([2-13C]-Tyr) collected at 1150, 1157, 1164, 1171, 1178, 1184, 1191, 1198, and 1205 mT (top to bottom). Corresponding g-values given in figure. Traces of experimental data are shown in black; simulations for the Hox form are presented in red.
The X-band continuous-wave (CW) EPR spectrum of in vitro matured HydA from Clostridium pasteurianum (CpI) poised in the Hox state is consistent with that published previously with g = 2.100, 2.040, 1.996 (Figure 1A). Using [2-13C]-Tyr in the maturation of HydA leads to a splitting of ≈1 mT centered at each g-value of this Hox signal (cf. top and bottom traces in Figure 1A).18 Q-band Davies ENDOR spectra acquired at field positions corresponding to each g-value (Figure 1B) confirm this strong 13C HFI by showing features at ≈27 MHz that have no counterpart in analogous spectra of HydA matured using natural-abundance tyrosine.19 The variation in shape and breadth of these features as a function of resonant field position results from orientation selection, i.e., at certain field positions, a discrete subset of molecular orientations of HydA are probed. Proper simulation of this behavior allows for the orientation of the corresponding 13C hyperfine tensor to be determined relative to the molecular g-tensor. These parameters are summarized in Table 1. The degree of 13C HFI anisotropy is consistent with that of other Fe-bound cyanides (cf. Table 1).
Table 1. 13C HFI and 15N HFI for CO and CN Bound to Fe-Centers.
| species | A13C (MHz) | [α, β, γ] (deg)a | assignment | reference |
|---|---|---|---|---|
| CpI Hox ([2-13C]-Tyr) | [30.9, 23.3, 30.2] | [60, 120, 170] | CNd | this work |
| [5.22, 5.24, 4.16] | [30, 90, 0] | CNp | this work | |
| CpI Hox-CO ([2-13C]-Tyr) | [7.0, 7.0, 7.2] | [0, 0, 0] | CNa | this work |
| [3.75, 3.75, 3.90] | [0, 0, 0] | CNb | this work | |
| DdS Hox-13CO | [15.6, 16.6, 19.2] | COext | (17) | |
| [8.5, 9.8, 3.9] | CObridge | (17) | ||
| [3.2, 3.7, 4.4] | COd | (17) | ||
| Mb-13CN | [−23.0, −27.6, −28.7] | Fe(III)-CN | (21) | |
| Pf Fd-13CN | [−4.5, −4.5, +0.1] | [4Fe-4S]+-CN | (22) |
| species | A15N (MHz) | [α, β, γ] (deg) | assignment | reference |
|---|---|---|---|---|
| CpI Hox ([15N]-Tyr) | [0.8, 6.3, −1.2] | [45, −20, 0] | CNd | this work |
| DdS Hox | [2.1, 5.3, −0.6]b | [41, 24, 0] | CNd | (3) |
| [1.4, 2.7, 2.0]b | [40, 25, 0] | DTMA | (3) | |
| [−3.4, 2.0, −1.0]b | [0, 4, 20] | Lys | (3) | |
| DdS Hox-CO | [0.56, −0.28, 0.79]b | [0, −10, 0] | (17) | |
| Mb-C15N | [n.d., n.d., 5.25] | Fe(III)-CN | (23) | |
| Pf Fd-C15N | [+1.8, +1.0, −2.4] | [4Fe-4S]+-CN | (22) |
Euler angles are relative to g-frame defined by g1 < g2 < g3. For Hox, this corresponds to gz < gy < gx as we assign the local z-axis of Fed to the Fe-CObridge bonding vector.
Determined by scaling the experimentally determined 14N HFI by the ratio of the 15N/14N Larmor frequencies (1.4028).
Abbreviations: Mb = myoglobin; Pf Fd = [4Fe-4S] ferredoxin from Pyrococcus furiosus; n.d. = not determined.
Orientation-selected Mims ENDOR spectra (Figure 1C) reveal three distinct classes of more weakly coupled 13C nuclei (Aiso = 3.80, 4.87, and ≈7.0 MHz). These features are centered about the 13C Larmor frequency and split by the magnitude of the HFI. Analogous data sets collected for CO-treated samples (Figures S3 and S4) possess similar features at ±1.8 and ±3.6 MHz, confirming that they arise from the two cyanide ligands in the Hox-CO form of hydrogenase (labeled as CNa and CNb since we cannot distinguish between the Fep-bound and Fed-bound cyanides at this time). Note the absence of contributions from Hox-CO to the ENDOR spectra acquired at the extreme field positions (g = 2.099 and 2.004) of Hox (Figure 1C). This results from the relative narrowness of the Hox-CO signal. This narrowness is also why we see strong contributions from Hox-CO even though the contamination is relatively small. The remaining features centered at ±2.2 MHz in Figure 1C are thus ascribed to the other CN– ligand in Hox.
Based on the crystallographic results,2 Fed possesses a square pyramidal local geometry whose z-axis points along the bond between the Fed ion and the bridging CO. For the six-coordinate Fep, the identity of the local z-axis is less obvious, but computational results suggest that it is aligned along the Fep-CObridge bond.14 As the two terminal CN– ligands appear to be bound in the same position relative to the local z-axis of their respective Fe ions, the ratio of the isotropic 13C HFI should serve as a reporter of the relative spin density on each iron. Again, based on earlier computational results, we assign the larger 13C HFI as arising from the distal Fe-bound cyanide of Hox. For the proximal Fe-bound cyanide, we measure Aiso = 4.87 MHz. This ratio of ≈5.8 correlates approximately with the Fed:Fep ratio of computed Mulliken spin populations.14,16 For Hox–CO, the Aiso(13CNa):Aiso(13CNb) ratio drops to <2 (see magnetic parameters listed in Table 1) indicating a much more even distribution of spin density over the two Fe ions than what was observed for Hox that is again consistent with computational results.14,16 Interestingly, the 13C HFI tensors for the two CN– ligands in the Hox–CO form lack significant anisotropy compared to other Fe-bound cyanides (cf. Table 1)
X- and Q-band HYSCORE spectra for natural-abundance Hox (Figure 2, top) are essentially identical to those obtained earlier by Silakov et al.3 When the in vitro maturation of HydA is performed with 15N-labeled tyrosine ([15N]-Tyr), the nitrogens of the cyanide ligands become selectively isotopically labeled.9 The corresponding HYSCORE data are strikingly different from those of natural-abundance Hox (cf. top and bottom plots in Figure 2) signaling that the majority of features arise from tyrosine-derived nitrogens. The correlation ridges in the Q-band spectrum of Hox ([15N]-Tyr) are well-simulated with the hyperfine parameters A(15N) = [0.8, 6.3, −1.2] MHz (Figure S5). Given the rather large magnitude of Aiso(15N), this nitrogen is likely that in the Fed-bound cyanide. We observe no 15N-derived features that we could assign to cyanides in the Hox–CO form.
Figure 2.

X-band (left) and Q-band (right) HYSCORE spectra of the Hox form of HydA matured using natural-abundance ([14N]-Tyr, top) or with [15N]-Tyr (bottom).
The biosynthetic origin of the putative DTMA bridge is presently unknown. One proposal suggests that HydG can assemble this bridging ligand from two molecules of tyrosine.20 Analysis of 14N HYSCORE spectra of DdS HydA poised in the Hox state led to the assignment of a set of correlation ridges to the DTMA amino nitrogen (A(14N) = [1.0, 1.9, 1.4] MHz).3 By scaling this reported 14N HFI by the ratio of the 15N/14N Larmor frequencies, we can simulate the X-band HYSCORE spectrum as if the DTMA had been 15N-labeled (see Figures S6 and S7). The predicted correlation ridges corresponding to the 15N-DTMA nitrogen are not found in the experimental HYSCORE spectrum of Hox ([15N]-Tyr) suggesting either that tyrosine is not the source of the DTMA nitrogen or that the previously reported 14N HFI parameters for DdS HydA are not appropriate for CpI Hox.
Using isotopically labeled tyrosine substrates in conjunction with the in vitro biosynthetic route to generate the H-cluster gives us the flexibility to site-specifically label the cyanide ligands with 13C and 15N. The signals we observe from 15N are unambiguously attributed to the nitrogen of an Fe-bound cyanide. Further, comparison of the two cyanide 13C couplings is consistent with just one of the Fe ions (Fed) of [FeFe]H carrying the majority of unpaired electron spin in the Hox state. As such, the relatively large rhombicity of the Hox EPR signal can be understood as arising from the asymmetry in the equatorial ligand set for the low-spin 3d7 Fed spin center. Thus, the difference in g-shifts for gy and gx (0.0367 vs 0.0947) is attributed to the difference in the energies of the Fed-3dxz → Fed-3dz2 and the Fed-3dyz → Fed-3dz2 transitions, respectively.24 If we orient the g-tensor for Hox as follows: gz is oriented along of z-axis of Fed, and gx and gy are made to bisect the Fed-S and Fed-S bonding vectors and the Fed-COd and Fed-CNd bonding vectors, respectively; then the unique axis of the 13C hyperfine tensor for CNd is found to point approximately along the Fed-CNd bond, as expected (Figure S8).25 This finding supports our electronic structure description of Hox; namely, that the unpaired electron largely resides in a molecular orbital of 3dz2 character centered on the Fed ion.
Based on the similar magnitudes of the 13CN HFI, the electron spin becomes distributed more evenly over both iron ions after inhibition with free CO. This more delocalized spin topology leads to a collapse of the g-matrix rhombicity. Analogously, the rather narrow EPR signal for the formally mixed-valence Cu(I,II) CuA cluster in nitrous oxide reductase is understood as a weighted sum of the hypothetical mononuclear g-matrices of each Cu site.26 In the case of Hox-CO, we do not know the values for the intrinsic g-matrix for the two Fe ions. However, we can use the Hoxg-values as a first estimate. Upon forming Hox-CO, delocalization of the unpaired electron spin cancels out some of the anisotropy from each site-specific g-matrix, leading to the axial (g = 2.072, 2.006, 2.006), molecular g-matrix. The nearly isotropic HFI tensors for the two CN– ligands in Hox-CO result from this same mechanism of anisotropy cancellation. These findings are in agreement with earlier computational models14,16 that indicate a dramatic delocalization of unpaired spin density in going from the Hox form to Hox-CO
Acknowledgments
This work was funded by National Institutes of Health (GM104543 to R.D.B.) and the Division of Material Sciences and Engineering (J.R.S. award no. DE-FG02-09ER46632) of the Office of Basic Energy Sciences of the U.S. Department of Energy. D.L.M.S. acknowledges support from the National Institutes of Health (F32GM111025 from the NIGMS).
Supporting Information Available
Details of experimental procedures and data analysis methods. Supplemental EPR spectra and corresponding simulations. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Vincent K. A.; Parkin A.; Armstrong F. A. Chem. Rev. 2007, 107, 4366. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Lanzilotta W. N.; Lemon B. J.; Seefeldt L. C. Science 1998, 282, 1853. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Wenk B.; Reijerse E.; Lubitz W. Phys. Chem. Chem. Phys. 2009, 11, 6592. [DOI] [PubMed] [Google Scholar]
- Berggren G.; Adamska A.; Lambertz C.; Simmons T. R.; Esselborn J.; Atta M.; Gambarelli S.; Mouesca J. M.; Reijerse E.; Lubitz W.; Happe T.; Artero V.; Fontecave M. Nature 2013, 499, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard E. M.; Mus F.; Betz J. N.; Byer A. S.; Duffus B. R.; Peters J. W.; Broderick J. B. Biochemistry 2014, 53, 4090. [DOI] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Shiigi S. A.; Swartz J. R. Methods Mol. Biol. (N. Y., NY, U. S.) 2014, 1122, 49. [DOI] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Myers W. K.; Suess D. L. M.; Stich T. A.; Pelmenschikov V.; Shiigi S. A.; Cramer S. P.; Swartz J. R.; Britt R. D.; George S. J. Science 2014, 343, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson K. D.; Duffus B. R.; Beard T. E.; Peters J. W.; Broderick J. B. Eur. J. Inorg. Chem. 2011, 935. [Google Scholar]
- Kuchenreuther J. M.; George S. J.; Grady-Smith C. S.; Cramer S. P.; Swartz J. R. PLoS One 2011, 6, e20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Myers W. K.; Stich T. A.; George S. J.; NejatyJahromy Y.; Swartz J. R.; Britt R. D. Science 2013, 342, 472. [DOI] [PubMed] [Google Scholar]
- Driesener R. C.; Challand M. R.; McGlynn S. E.; Shepard E. M.; Boyd E. S.; Broderick J. B.; Peters J. W.; Roach P. L. Angew. Chem. 2010, 49, 1687. [DOI] [PubMed] [Google Scholar]
- Shepard E. M.; Duffus B. R.; George S. J.; McGlynn S. E.; Challand M. R.; Swanson K. D.; Roach P. L.; Cramer S. P.; Peters J. W.; Broderick J. B. J. Am. Chem. Soc. 2010, 132, 9247. [DOI] [PubMed] [Google Scholar]
- Popescu C. V.; Munck E. J. Am. Chem. Soc. 1999, 121, 7877. [Google Scholar]
- Fiedler A. T.; Brunold T. C. Inorg. Chem. 2005, 44, 9322. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Reijerse E. J.; Albracht S. P. J.; Hatchikian E. C.; Lubitz W. J. Am. Chem. Soc. 2007, 129, 11447. [DOI] [PubMed] [Google Scholar]
- Greco C.; Silakov A.; Bruschi M.; Ryde U.; De Gioia L.; Lubitz W. Eur. J. Inorg. Chem. 2011, 1043. [Google Scholar]
- Silakov A.; Wenk B.; Reijerse E.; Albracht S. P. J.; Lubitz W. J. Biol. Inorg. Chem. 2009, 14, 301. [DOI] [PubMed] [Google Scholar]
- A modest (20% of overall spectral intensity) of the axial signal (g = 2.072, 2.006, 2.006) arising from Hox-CO was removed by subtraction. Hox-CO contamination is common and can be seen by other methods such as infrared absorption spectroscopy.9
- This ENDOR transition at 27 MHz is approximately equal to twice the 13C Larmor frequency at this field; therefore the ENDOR transtion in other spin manifold is expected at <1 MHz though it is not evident in our ENDOR data. However, both 13C spin-flip transitions are observed in the Q-band HYSCORE spectrum (Figure S2).
- Pilet E.; Nicolet Y.; Mathevon C.; Douki T.; Fontecilla-Camps J. C.; Fontecave M. FEBS Lett. 2009, 583, 506. [DOI] [PubMed] [Google Scholar]
- Van Doorslaer S.; Trandafir F.; Harmer J. R.; Moens L.; Dewilde S. Biophys. Chem. 2014, 190–191, 8. [DOI] [PubMed] [Google Scholar]
- Telser J.; Smith E. T.; Adams M. W. W.; Conover R. C.; Johnson M. K.; Hoffman B. M. J. Am. Chem. Soc. 1995, 117, 5133. [Google Scholar]
- Mulks C. F.; Scholes C. P.; Dickinson L. C.; Lapidot A. J. Am. Chem. Soc. 1979, 101, 1645. [Google Scholar]
- Weil J. A.; Bolton J. R.. Electron paramagnetic resonance: elementary theory and practical applications; John Wiley & Sons: Hoboken, NJ, 2007. [Google Scholar]
- Telser J.; Smith E. T.; Adams M. W. W.; Conover R. C.; Johnson M. K.; Hoffman B. M. J. Am. Chem. Soc. 1995, 117, 5133. [Google Scholar]
- Neese F.; Zumft W. G.; Antholine W. E.; Kroneck P. M. H. J. Am. Chem. Soc. 1996, 118, 8692. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.