

Abstract
Olefin metathesis has emerged as a promising strategy for modulating the stability and activity of biologically relevant compounds; however, the ability to control olefin geometry in the product remains a challenge. Recent advances in the design of cyclometalated ruthenium catalysts has led to new strategies for achieving such control with high fidelity and Z selectivity, but the scope and limitations of these catalysts on substrates bearing multiple functionalities, including peptides, remained unexplored. Herein, we report an assessment of various factors that contribute to both productive and nonproductive Z-selective metathesis on peptides. The influence of sterics, side-chain identity, and preorganization through peptide secondary structure are explored by homodimerization, cross metathesis, and ring-closing metathesis. Our results indicate that the amino acid side chain and identity of the olefin profoundly influence the activity of cyclometalated ruthenium catalysts in Z-selective metathesis. The criteria set forth for achieving high conversion and Z selectivity are highlighted by cross metathesis and ring-closing metathesis on diverse peptide substrates. The principles outlined in this report are important not only for expanding the scope of Z-selective olefin metathesis to peptides but also for applying stereoselective olefin metathesis in general synthetic endeavors.
Introduction
Olefin metathesis is a highly versatile tool for the generation of carbon–carbon bonds, and a variety of applications have evolved around its implementation.1−5 The broad utility of olefin metathesis is a consequence of the exceptional selectivity, activity, and functional-group compatibility of select metathesis catalysts, highlighted by carbon–carbon bond formation on a variety of complex substrates including small molecules,6−11 natural products,12,13 organic and inorganic materials,14−18 and even proteins.19,20 The use of metathesis in biological applications is an emerging field of research, in part, because of advances in the genetic21−23 and chemical24−26 incorporation of alkene-containing amino acids into peptides and proteins. This has enabled the installation of a variety of carbon–carbon bonds with high fidelity for applications in peptide stapling,27−34 as surrogates of hydrogen bonding,35−37 and as methods for modifying peptides and proteins used to mimic physiologically relevant post-translational modifications.20,38 The application of metathesis for stabilizing peptide secondary structure and for performing selective protein modifications has implications for imparting greater metabolic stability, cellular permeability, and higher binding affinity toward biological targets.30,39−41 Indeed, this strategy has led to the development of “stapled” peptides used as inhibitors of HIV fusion42 and assembly,43−46 as modulators of signaling pathways involved in cancer,47−50 and as selective activators of enzymes involved in diabetes.51,52
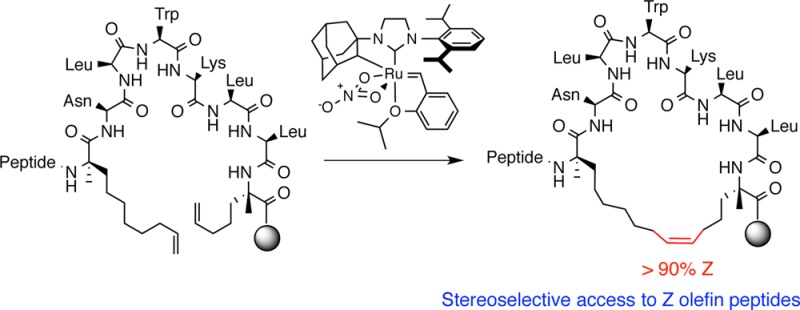
Despite the tremendous success of metathesis in peptide and peptidomimetic research, the ability to control olefin geometry in the product has been met with limited success.53−55 Most metathesis catalysts exhibit minimal kinetic selectivity, and thus, the product distribution reflects the thermodynamic stability of each olefin isomer.56 In many cases, a mixture of E and Z isomers is formed that is often inseparable. This imposes challenges for examining the influence of alkene geometry on the stability and activity of diverse compounds. In pursuit of catalysts with greater control over olefin products, we uncovered a series of cyclometalated ruthenium catalysts that could achieve high conversions with exquisite Z selectivity (Figure 1).57−62 The origin of Z selectivity for cyclometalated ruthenium catalysts involves approach of the olefin from a side-bound position (i.e., cis to the NHC ligand and trans to the chelating adamantyl) and is favored through a combination of steric and electronic effects imposed by the NHC ligand.63 Although catalysts 1 and 2 demonstrate excellent selectivity in olefin metathesis, their activity on complex substrates, including peptides, remained unexplored. To this end, we sought to initiate a comprehensive evaluation of Z-selective metathesis on peptides using newly developed cyclometalated ruthenium catalysts. Through the combined efforts of homodimerization, cross metathesis and ring-closing metathesis, we have developed guidelines for assessing the influence of amino acids and peptides on catalyst activity and selectivity. These principles were applied for carrying out Z-selective metathesis on challenging substrates including peptides that comprise parallel β-sheets and on stapling of α-helical peptides.
Figure 1.

Z-selective cyclometalated ruthenium catalysts.
Results and Discussion
Assessing the Amino Acid Substrate Scope by Z-Selective Homodimerization
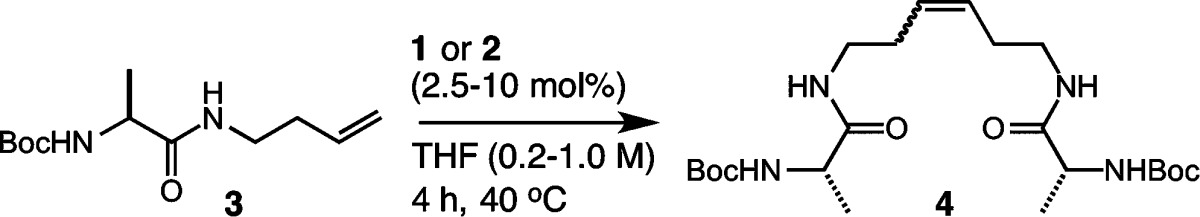
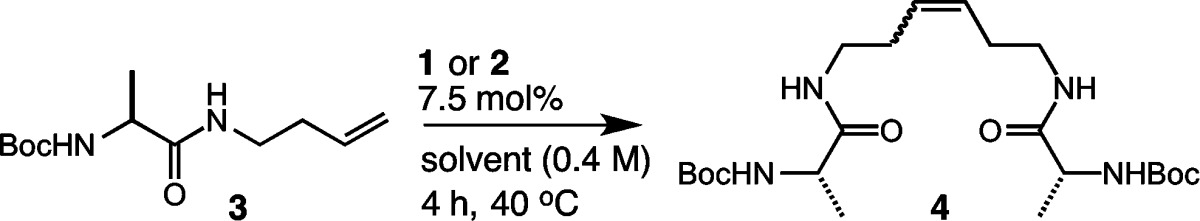
Our goal for expanding the utility of Z-selective ruthenium catalysts was to examine the influence of amino acids on the selectivity and activity of catalysts 1 and 2. Catalysts bearing N-adamantyl substituents and bidentate nitrato ligands were found to be critical for achieving high Z selectivity,59 and we set out to determine whether such catalysts could be applied to substrates bearing multiple functionalities and with varying steric profiles. To benchmark the reactivities of catalysts 1 and 2, we chose to investigate the homodimerization of protected amino acids modified with homoallyl functionality (Table 1).64 Our initial studies focused on the use of alanine 3, as we anticipated that amino acids bearing unhindered and aliphatic side chains would provide an ideal platform for comparative studies. We began with a catalyst loading of 2.5 mol % in tetrahydrofuran (THF) at 40 °C, and this afforded the homodimerization product 4 in 53% yield after 4 h using catalyst 1 and 58% yield in the presence of catalyst 2 (entry 1). Gratifyingly, the Z selectivity remained high (∼90%) throughout the course of the reaction.65 An increase in catalyst loading was then examined at varying concentrations. Catalyst loadings of 5 mol % afforded product 4 in 65% yield with 92% Z selectivity in the presence of catalyst 2 (entry 2). Increasing the concentration led to modest improvements in yields while maintaining high Z selectivity (entries 3 and 4). Ultimately, we found that using a catalyst loading of 7.5 mol % afforded product 4 in 76% yield with 94% Z selectivity (entry 6), and these conditions proved to be optimal among the various reaction conditions explored. We next examined the solvent dependence of the activities of catalysts 1 and 2 in homodimerization, as this is an important consideration in view of the solubility profiles of many peptidic substrates (Table 2). Several polar and nonpolar solvents were investigated reflecting those most often used in peptide synthesis. Coordinating solvents [e.g., acetonitrile (MeCN)] were less active in promoting Z-selective metathesis as compared to noncoordinating solvents [e.g., dicholorethane (DCE)] (entries 2 and 3). Polar solvents including dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and N-methylpyrrolidone (NMP) were tolerated by catalysts 1 and 2, affording product 4 in yields ranging from 55% to 67% and 90% Z selectivity (entries 4–6). Protic solvents including methanol (MeOH), ethanol (EtOH), and aqueous tert-butanol (t-BuOH) mixtures yielded highly enriched Z-olefin products (entries 7–9).66 Other polar solvents including nitromethane (MeNO2) resulted in low conversions (entry 10), presumably by activating decomposition pathways of the cyclometalated ruthenium catalysts.67 It is worth noting that, across the variety of reaction conditions explored, the Z selectivity remained consistently high.
Table 1. Optimization of Homodimerization for Homoallyl-Modified Alanine 3.
| yielda (%) |
Z selectivityb (%) |
|||||
|---|---|---|---|---|---|---|
| entry | catalyst (mol %) |
concentration (M) |
1 | 2 | 1 | 2 |
| 1 | 2.5 | 0.4 | 53 | 58 | 89 | 93 |
| 2 | 5.0 | 0.2 | 61 | 63 | 86 | 92 |
| 3 | 5.0 | 0.4 | 62 | 63 | 91 | 90 |
| 4 | 5.0 | 1.0 | 60 | 72 | 91 | 91 |
| 5 | 7.5 | 0.2 | 68 | 71 | 90 | 94 |
| 6 | 7.5 | 0.4 | 74 | 76 | 91 | 94 |
| 7 | 7.5 | 1.0 | 72 | 72 | 90 | 91 |
| 8 | 10 | 0.4 | 72 | 70 | 89 | 93 |
| 9 | 10 | 1.0 | 71 | 72 | 90 | 92 |
Isolated yields.
Determined by 1H or 13C NMR spectroscopy.
Table 2. Solvent Effects on Homodimerization of Homoallyl-Modified Alanine 3.
| yielda (%) |
Z selectivityb (%) |
||||
|---|---|---|---|---|---|
| entry | solvent | 1 | 2 | 1 | 2 |
| 1 | THF | 74 | 76 | 91 | 94 |
| 2 | MeCN | 51 | 49 | 88 | 91 |
| 3 | DCE | 72 | 72 | 88 | 92 |
| 4 | DMF | 55 | 59 | 84 | 87 |
| 5 | DMSO | 57 | 55 | 90 | 90 |
| 6 | NMP | 67 | 65 | 87 | 87 |
| 7 | MeOH | 65 | 70 | 85 | 88 |
| 8 | EtOH | 68 | 64 | 88 | 90 |
| 9 | H2O/t-BuOH (1:1) | 64 | 70 | 90 | 92 |
| 10 | MeNO2 | <10 | <10 | n.d. | n.d. |
Isolated yields.
Determined by 1H or 13C NMR spectroscopy.
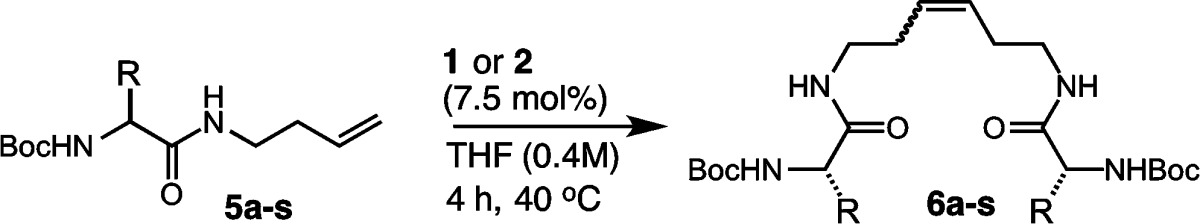
To probe the activities of catalysts 1 and 2 further, we investigated the homodimerization of other canonical amino acids (Table 3). Boc-protected amino acids bearing aliphatic or aromatic side chains were active in Z-selective metathesis (entries 1–4). Branched side chains from amino acids including valine (5a), isoleucine (5b), and leucine (5c) afforded products 6a–c in yields approaching 74% with 94% Z selectivity (entries 1–3). Aromatic side chains from amino acids including phenylalanine 5d afforded product 6d in 75% yield and 93% Z selectivity in the presence of catalyst 2 (entry 4). Exceptions include glycine68 and proline,69 as these are inactive in homodimerization (entries 5 and 6). Amino acids bearing heterocycles had variable activity. Homodimerization of tryptophan afforded product 6g in 64% yield with 90% Z selectivity (entry 7). In contrast, histidine was deemed incompatible with catalysts 1 and 2 (entry 8).70 Unprotected alcohols from the side chains of serine 5i or threonine 5j were tolerated by catalysts 1 and 2 but required protection to reach acceptable conversions, providing products 6i and 6j in 72% and 73% yields, respectively (entries 9 and 10). Homodimerization of tyrosine afforded product 6k in 64% yield and 87% Z selectivity in the presence of catalyst 1 and 68% yield and 90% Z selectivity using catalyst 2 (entry 11). In contrast to alcohols, side chains bearing thiols (i.e., cysteine) or thioethers (i.e., methionine 5l) generally led to catalyst deactivation (entry 12).71 Protecting the side chain of cysteine could overcome catalyst inactivity and afford product 6m in 55% yield and 87% Z selectivity (entry 13). Polar side chains bearing carboxylate (5n,o), carboxamide (5p,q), or amine (5r) functionality required protection prior to homodimerization. In these cases, yields from 70% to 80% could be achieved with high Z selectivity (entries 14–18). Side chains bearing a protected guanidinium functionality (i.e., arginine 5s) were less active in homodimerization, affording product 6s in 34% and 33% yields using catalysts 1 and 2, respectively. These findings reveal that the identity of the amino acid side chain can profoundly influence the activity of cyclometalated ruthenium catalysts.
Table 3. Homodimerization of Canonical Amino Acids for Investigating Side-Chain Influence on Catalytic Activity.
| yielda (%) |
Z selectivityb (%) |
|||||
|---|---|---|---|---|---|---|
| entry | amino acid | product | 1 | 2 | 1 | 2 |
| 1 | valine (5a) | 6a | 74 | 71 | 90 | 94 |
| 2 | isoleucine (5b) | 6b | 68 | 72 | 88 | 92 |
| 3 | leucine (5c) | 6c | 70 | 71 | 88 | 91 |
| 4 | phenylalanine (5d) | 6d | 73 | 75 | 89 | 93 |
| 5 | glycine (5e) | 6e | <10 | <10 | n.d. | n.d. |
| 6 | proline (5f) | 6f | <10 | <5 | n.d. | n.d. |
| 7 | tryptophan (5g) | 6g | 66 | 64 | 85 | 90 |
| 8 | histidine (5h) | 6h | <5 | <5 | n.d. | n.d. |
| 9 | serine (5i) | 6i | 72 | 70 | 84 | 90 |
| 10 | threonine (5j) | 6i | 73 | 70 | 88 | 92 |
| 11 | tyrosine (5k) | 6k | 64 | 68 | 87 | 90 |
| 12 | methionine (51) | 61 | <5 | <10 | n.d. | n.d. |
| 13 | cysteine (5m)c | 6m | 55 | 53 | 87 | 92 |
| 14 | aspartic acid (5n)d | 6n | 61 | 60 | 87 | 90 |
| 15 | glutamic acid (5o)d | 6o | 74 | 71 | 88 | 91 |
| 16 | asparagine (5p)c | 6p | 70 | 71 | 88 | 91 |
| 17 | glutamine (5q)c | 6q | 74 | 74 | 88 | 91 |
| 18 | lysine (5r)e | 6r | 78 | 81 | 81 | 89 |
| 19 | arginine (5s)e | 6s | 34 | 33 | 81 | 89 |
Isolated yields.
Determined by 1H or 13C NMR spectroscopy.
Side chain protected with a trityl group.
Side chain protected as the t-butyl ester.
Side chain protected as the t-butyl carbamate.
Side chain protected with 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf).
Assessing Side-Chain Influence and Stoichiometry on Cross Metathesis of Amino Acids
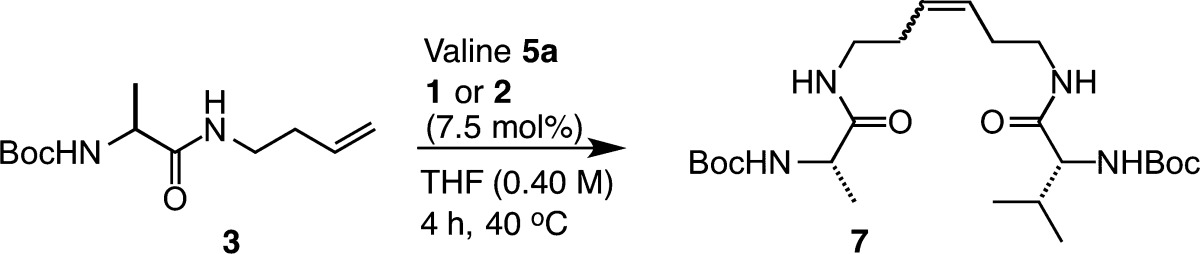
Emboldened by the success of Z-selective homodimerization of amino acids, we next evaluated the effects of varying the metathesis partner to achieve Z-selective cross metathesis (CM) (Table 4). For our studies, we chose cross partners that showed variable activity in homodimerization. In this way, we could begin to classify substrates on the basis of their propensity for achieving productive CM.72 We explored CM using homoallyl-modified alanine 3 and the similarly reactive substrate valine 5a as the cross partner. Cross metathesis in the presence of an equimolar ratio of 3 to 5a afforded the desired heterocross product 7 in 41% yield with 90% Z selectivity (entry 1). Increasing the stoichiometry of 5a relative to 3 led to a modest increase in yield, providing product 7 in 48% yield with 91% Z selectivity (entry 2). Further increases in 5a afforded similar trends, with yields approaching 60% and 90% Z selectivity (entries 3 and 4). To demonstrate the versatility of this method, we explored conditions in which 5a was used as the limiting reagent (entries 5–7). Under these conditions, an incremental increase in the yield of product 7 was observed upon increasing the ratio of 3 to 5a. As in the case of excess 5a, modest improvements in the yield of 7 were achieved with increasing equivalents of 3, demonstrating the statistical nature of CM using similar reactive substrates.73 Given the modest conversions to product and high Z selectivities, we can conclude that the products of CM are sparingly consumable using catalysts 1 and 2, ensuring that the Z selectivity can remain high throughout the course of the reaction.
Table 4. Cross Metathesis of Amino Acids 3 and 5a.
| yielda (%) |
Z selectivityb (%) |
|||||
|---|---|---|---|---|---|---|
| entry | equiv of 3 | equiv of 5a | 1 | 2 | 1 | 2 |
| 1 | 1 | 1 | 41 | 47 | 90 | 93 |
| 2 | 1 | 2 | 44 | 48 | 86 | 91 |
| 3 | 1 | 4 | 48 | 41 | 91 | 90 |
| 4 | 1 | 6 | 58 | 60 | 88 | 90 |
| 5 | 2 | 1 | 44 | 58 | 90 | 93 |
| 6 | 4 | 1 | 52 | 57 | 91 | 91 |
| 7 | 6 | 1 | 51 | 60 | 87 | 90 |
Isolated yields.
Determined by 1H or 13C NMR spectroscopy.
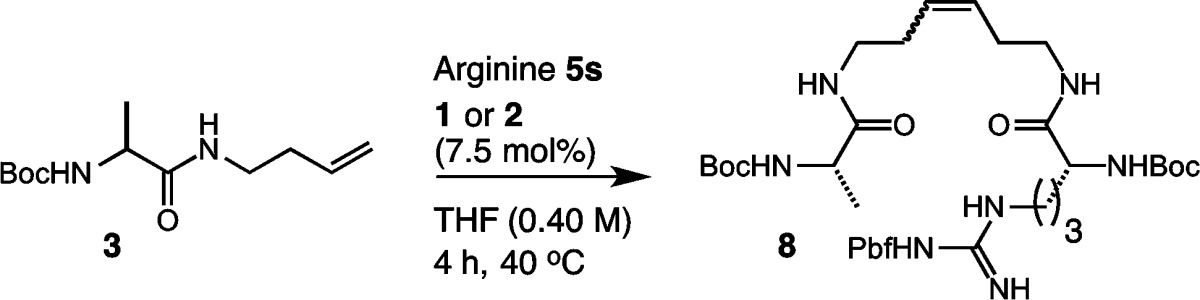
We next evaluated whether amino acids of differing reactivity profiles could be used for selective CM (Table 5). In choosing the requisite cross partners, we focused on substrate 3 and homoallyl-modified arginine 5s, as they both showed relatively low reactivity in homodimerization. We began our studies using an equimolar ratio of 3 and 5s, and this afforded 8 in 46% yield and 93% Z selectivity (entry 1). Increasing the ratio of 5s to 3 led to slightly diminished yields and Z selectivities (entries 2–4).74 By contrast, reversing the order such that 3 was in excess of 5s afforded 8 in yields of 47% and 58% using catalysts 1 and 2, respectively (entry 5). Further increasing the ratio of 3 to 5s could achieve the heterocross product 8 in improved yields and high Z selectivities (entries 6 and 7). Taken together, these findings reveal that the intrinsic reactivity differences between homoallyl-modified amino acids can be used for productive Z-selective cross metathesis.
Table 5. Cross Metathesis of Amino Acids 3 and 5s.
| yielda (%) |
Z selectivityb (%) |
|||||
|---|---|---|---|---|---|---|
| entry | equiv of 3 | equiv of 5s | 1 | 2 | 1 | 2 |
| 1 | 1 | 1 | 46 | 47 | 90 | 93 |
| 2 | 1 | 2 | 43 | 48 | 84 | 90 |
| 3 | 1 | 4 | 38 | 41 | 84 | 91 |
| 4 | 1 | 6 | 34 | 38 | 72 | 88 |
| 5 | 2 | 1 | 47 | 58 | 88 | 91 |
| 6 | 4 | 1 | 58 | 60 | 90 | 92 |
| 7 | 6 | 1 | 62 | 66 | 87 | 90 |
Isolated yields.
Determined by 1H or 13C NMR spectroscopy.
Influence of Allylic Heteroatoms on Homodimerization and Cross Metathesis of Noncanonical Amino Acids
The unique reactivity profiles of canonical amino acids in Z-selective homodimerization and CM prompted investigation of a subset of non-natural amino acids that have shown promise in peptide and protein modification using olefin metathesis. Allyl-protected amino acids including serine, cysteine, and selenocysteine have been shown to enhance the rate of metathesis when incorporated into peptides and proteins.38,75 Studies by Davis and co-workers ascribed the unique chemical reactivity of such amino acids through a chelation-assisted mechanism whereby precoordination of the heteroatom to ruthenium increases the effective concentrations of the alkylidene and alkene without detrimental chelation.38 Moreover, softer nucleophiles such as sulfur and selenium were found to have an activating effect relative to oxygen for enhancing the rate of CM. These findings are intriguing considering that sulfur can have a deactivating effect on olefin metathesis,76−78 and our results suggest that sulfur-containing amino acids (e.g., compound 5l) lead to catalyst inactivity. Nonetheless, a wealth of information suggests that heteroatoms can modulate metathesis activity;79−81 however, this phenomenon was unexplored using Z-selective cyclometalated catalysts.
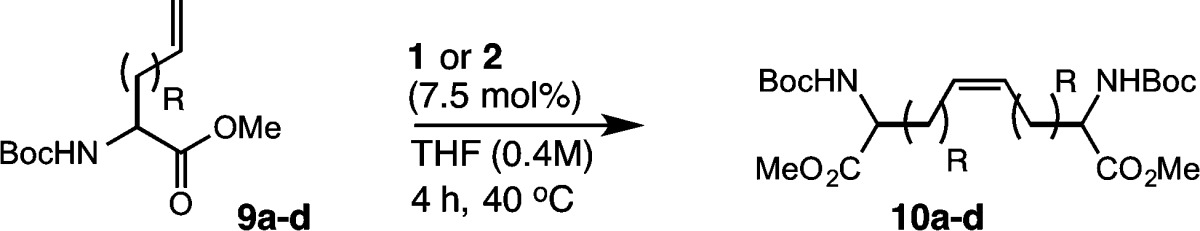
To investigate the influence of allylic heteroatoms on the activities of catalysts 1 and 2, we examined a series of allyl-protected amino acids in homodimerization (Table 6). We chose allylglycine (9a) and homoallylglycine (9b), as well as allyl-protected serine (9c) and cysteine (9d), for our studies. In this sense, 9a–d would reveal both the role of sterics (i.e., comparison of 9a and 9b) and the effect of chelation by heteroatoms (i.e., comparison of 9b to 9c,d) in facilitating metathesis. For these tests, we investigated homodimerization of 9a and compared its reactivity relative to that of substrate 9b. Conversion of 9a to the dimerized product 10a was low using catalyst 1 or 2, occurring in 45% yield and 90% Z selectivity (entry 1). By comparison, dimerization of homoallylglycine 9b afforded product 10b in 59% yield and 90% Z selectivity (entry 2). This corroborates our earlier findings that the steric environment around the alkene can influence the efficiency of Z-selective metathesis.58 We next evaluated the effects of heteroatoms in facilitating homodimerization. Allyl-protected serine 9c afforded product 10c in 67% yield with 92% Z selectivity (entry 3). By comparison, cysteine 9d was more active in homodimerization, leading to 71% yield and 93% Z selectivity in the presence of catalyst 2 (entry 4). These results suggest that allylic heteroatoms can facilitate Z-selective metathesis in the presence of cyclometalated ruthenium catalysts.
Table 6. Influence of Heteroatoms on Homodimerization of Noncanonical Amino Acids.
| yielda (%) |
Z selectivityb (%) |
|||||
|---|---|---|---|---|---|---|
| entry | R | product | 1 | 2 | 1 | 2 |
| 1 | CH2 (9a) | 10a | 44 | 46 | 88 | 93 |
| 2 | CH2CH2 (9b) | 10b | 59 | 58 | 90 | 92 |
| 3 | CH2OCH2 (9c) | 10c | 67 | 69 | 92 | 94 |
| 4 | CH2SCH2 (9d) | 10d | 74 | 71 | 90 | 90 |
Isolated yields.
Determined by 1H or 13C NMR spectroscopy.
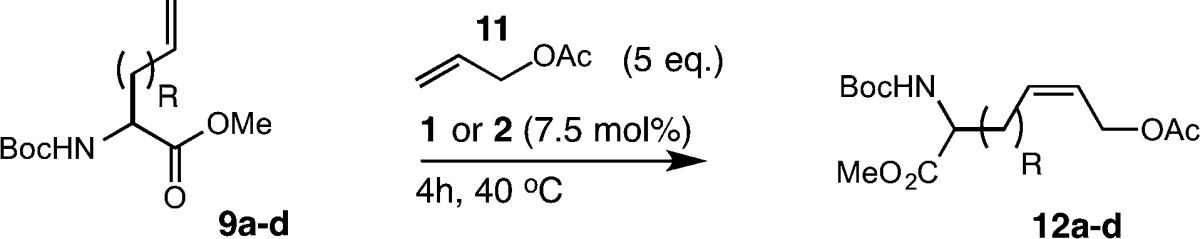
The insights garnered from the homodimerization of substrates 9a–d revealed that the identity of the alkene can influence the activities of catalysts 1 and 2 in Z-selective metathesis. Although these studies provide important insight for assessing the intrinsic reactivity of allyl-modified amino acids with cyclometalated ruthenium catalysts, the general utility of such catalysts could be further illuminated by their use in CM. As such, we examined catalysts 1 and 2 in CM using allyl-modified amino acids 9a–d (Table 7). To assess the relative activities of substrates 9a–d in CM, we chose allyl acetate 11 as the cross partner, as we previously showed that 11 is highly active in Z-selective CM.82 We explored a variety of conditions to test the generality of CM including the use of solvents that are compatible with native peptides and proteins. For our initial studies, we examined CM between allylglycine 9a and 11 under our previously optimized conditions, and this afforded the heterocross product 12a in 42% yield and 90% Z selectivity. We next explored CM under aqueous conditions, including the use of additives shown to enhance the efficiency of CM on peptides and proteins.83 In the presence of aqueous tert-butanol, the heterocross product 12a was achieved in 38% yield with 90% Z selectivity (entry 2). Inclusion of salts such as LiCl or MgCl2, which has been shown to be beneficial for enhancing methathesis on peptides,84,85 afforded product 12a in 31% yield but with diminished Z selectivity (entries 3 and 4). These trends were also observed using homoallylglycine 9b as the cross partner (entries 5–8). To investigate whether amino acids bearing allylic heteroatoms influence the efficiency of Z-selective CM, we exposed allylserine 9c and allylcysteine 9d to similar reaction conditions. Synthesis of the heterocross product was improved, affording 12c in 63% yield with 92% Z selectivity using catalyst 2 (entry 9). The use of aqueous conditions (entry 10) or inclusion of additives (entries 11 and 12) led to slightly diminished yields and Z selectivity. By comparison, the use of allylcysteine 9d as the cross partner afforded the desired product 12d in 60% yield with 90% Z selectivity (entry 13). As observed with substrates 9a–c, a decrease in catalyst activity occurred under aqueous conditions and in the presence of salt additives (entries 14–16).86 Collectively, these results suggest that the activities of catalysts 1 and 2 are highly dependent on the reaction conditions. In general, the trends observed in CM with 9a–d parallel those observed using nonchelated ruthenium catalysts;87 however, the use of salts as additives appears to have a deactivating effect on the activity of cyclometalated ruthenium catalysts. These results lend support to the importance of the chelating ligand (i.e., bidentate versus mondentate ligand coordination) in the activities and selectivities of catalysts 1 and 2.
Table 7. Cross Metathesis of Allyl-Modified Amino Acids with Allyl Acetate.
| yielda (%) |
Z selectivityb (%) |
|||||
|---|---|---|---|---|---|---|
| entry | substrate | product | 1 | 2 | 1 | 2 |
| 1c | 9a | 12a | 40 | 42 | 88 | 90 |
| 2d | 38 | 36 | 90 | 92 | ||
| 3e | 31 | 31 | 76 | 84 | ||
| 4f | 30 | 34 | 72 | 83 | ||
| 5c | 9b | 12b | 56 | 55 | 90 | 95 |
| 6d | 53 | 51 | 90 | 93 | ||
| 7e | 48 | 50 | 84 | 87 | ||
| 8f | 44 | 46 | 79 | 84 | ||
| 9c | 9c | 12c | 63 | 66 | 88 | 92 |
| 10d | 64 | 67 | 87 | 93 | ||
| 11e | 54 | 61 | 67 | 79 | ||
| 12f | 56 | 61 | 63 | 84 | ||
| 13c | 9d | 12d | 62 | 61 | 88 | 90 |
| 14d | 63 | 67 | 86 | 92 | ||
| 15e | 55 | 60 | 74 | 88 | ||
| 16f | 58 | 60 | 76 | 82 | ||
Isolated yields.
Determined by 1H or 13C NMR spectroscopy.
Reaction conditions: 1 mmol of 9a–d in THF.
Reaction conditions: 1 mmol of 9a–d in 1:1 H2O/t-BuOH.
Reaction conditions: 1 mmol of 9a–d in 1:1 H2O/t-BuOH + 2 mM LiCl.
Reaction conditions: 1 mmol of 9a–d in 1:1 H2O/t-BuOH + 2 mM MgCl2.
Z-Selective Cross Metathesis on Linear Peptides
Investigation of both canonical and noncanonical amino acids in homodimerization and cross metathesis revealed that the choice of cross partner is critical to the success of ruthenium-catalyzed Z-selective metathesis. In this regard, amino acid side chains bearing aliphatic, aromatic, or protected polar functionalities were highly active in metathesis. Those amino acids that generally lead to lower conversion (i.e., arginine) could undergo selective cross metathesis in the presence of a more highly reactive substrate. Moreover, the steric environments around the olefin and allylic heteroatoms were shown to impact the efficiency of Z-selective CM. Whereas these observations apply generally to amino acids that are commonly used in olefin metathesis, we wanted to investigate whether cyclometalated ruthenium catalysts could be used in CM on more complex substrates, including peptides. In choosing the requisite cross partners, we took inspiration from peptides known to adopt defined β-sheet secondary structures when tethered through a turn-promoting moiety or as part of a macrocycle.88−90 Such structures hold promise in applications ranging from supramolecular chemistry91,92 to biology93−95 and offer challenging substrates for catalysts 1 and 2.
A wealth of information regarding the use of peptides and small molecules as β-sheet mimics has revealed that both hydrogen bonding and amino acid side-chain pairing preferences can be used to dictate the stability of β-sheet formation.88,96,97 To this end, we synthesized peptides 13 and 14, which represent typical sequences found in parallel β-sheets and whose structures are known to rely on the sequence of amino acids (Scheme 1).88 For our studies, we used an excess of peptide 13, as we had shown that less reactive amino acids, including arginine, could be used for selective cross metathesis in the presence of more highly active amino acids. Under these conditions, conversion reached 60% for the desired peptide 15 with greater than 90% Z selectivity.98 Attempts to improve the yield of the heterocross product by conducting CM in solvents that promote hydrogen bonding and thereby preorganize the peptides to facilitate metathesis99−101 led to similar conversions.102 These results attest to the functional-group tolerability of cyclometalated ruthenium catalysts and point to further strategies aimed at accessing complex olefinic substrates bearing multiple functionalities.
Scheme 1. Z-Selective Cross Metathesis on Peptides.

Determined by analytical high-performance liquid chromatography mass spectrometry (HPLC-MS).
Z-Selective Ring-Closing Metathesis on α-Helical Peptides
Our results regarding the activity of cyclometalated ruthenium catalysts in cross metathesis of amino acids and peptides revealed that the kinetic selectivities of catalysts 1 and 2 can be used to synthesize peptides highly enriched in Z-olefins. Although these studies provide a framework for promoting metathesis through judicious choice of the amino acid sequence, we observed that conversions in CM were optimal at relatively high concentrations (∼0.3–0.4 M), which, for many substrates, might be a limitation. We sought a more general strategy such that a variety of reaction conditions could be used to promote Z-selective metathesis on a diverse collection of peptides. To this end, we investigated ring-closing metathesis (RCM) of olefinic amino acids for the synthesis of “stapled” peptides. Such peptides hold promise as novel therapeutics by virtue of their enhanced α-helicity;41,103 proteolytic stability;34,42,104−108 and ability to target intracellular proteins involved in cancer,32,39,47,104 infectious disease,44−46 and metabolism.109 As such, we set about to broaden the available catalysts used to synthesize this important class of peptides under conditions that would be amenable to comprehensive screening of catalyst activity in the presence of varying peptide sequences.
Traditional methods for the synthesis of stapled peptides by RCM have relied on the use of O-allyl serine28,29,110 or Cα-tetrasubstituted amino acids41,111−113 to install macrocyclic cross-links into synthetic peptides. Most strategies incorporate non-natural amino acids at positions spanning one (i, i + 4) or two (i, i + 7) turns of a helix that serve to preorganize the reactive side chains on the same helical face. A wealth of knowledge derived from computational114,115 and experimental47,116−118 approaches has illuminated the minimal constraints necessary for achieving RCM on peptides using first- and second-generation or Grubbs–Hoveyda ruthenium catalysts.119,120 An unmet challenge in the synthesis of stapled peptides has been the ability to control olefin geometry in the product, as the use of noncyclometalated ruthenium catalysts typically gives rise to both E and Z isomers that are often inseparable. This imposes challenges for examining the role of olefin geometry in the stability and biological activity of stapled peptides, which, to date, has not been thoroughly explored.106,121−123 To this end, we employed catalysts 1 and 2 in Z-selective RCM for stapling α-helical peptides that encompass the vast majority of peptides used for biological studies.
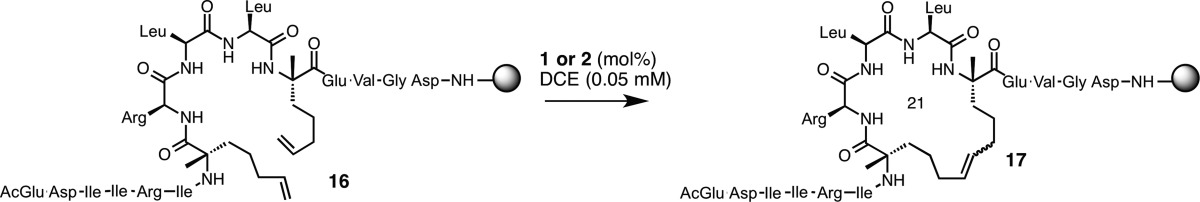
As part of our ongoing effort to expand the utility of catalysts 1 and 2, we chose to conduct RCM on resin-supported peptides. This would streamline the synthesis of peptides and offer a modular platform to test the activities of cyclometalated ruthenium catalysts. Our goal was to compare the activities of Z-selective catalysts with those of noncyclometalated ruthenium catalysts in RCM, and we focused our efforts on peptides with known biological activity. The sequence we chose is derived129 from an α-helical peptide known to target the BCL-2 family of proteins involved in the regulation of apoptosis and whose activity is modulated by constraining the peptide through hydrocarbon stapling (Table 8).34 The chemical features of peptide 16 consist of two stereochemically defined α,α-disubstituted olefinic amino acids124 separated by one turn of a helix (i.e., olefins positioned at residues i and i + 4) that, upon ring closure, would generate a 21-membered macrocycle.
Table 8. Z-Selective RCM to form i, i + 4 Stapled Peptides.
| conversionb,c (%) |
Z selectivityd (%) |
||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst (mol %) | resina | time (h) | 1 | 2 | 1 | 2 |
| 1 | 10 | Wang | 2 | 25 | 20 | n.d. | n.d. |
| 2 | 10 | TentaGel | 2 | 40 | 30 | n.d. | n.d. |
| 3 | 10 | MBHA | 2 | 60 | 55 | n.d. | n.d. |
| 4 | 10 | MBHA | 4 | 70 | 60 | >85 | >90 |
| 5 | 10 (×2) | MBHA | 4 | 75 | 75 | >85 | >90 |
| 6 | 10 (×2) | MBHA | 4 | 80e | 70 | >85 | >90 |
Loading capacities for resin: Wang, 0.5 mmol/g; TentaGel, 0.25 mmol/g; MBHA, 0.5 mmol/g.
Conversions determined by analytical HPLC of cleaved peptide.
Amino acids were protected prior to RCM.
Determined by analytical HPLC-MS.
Reaction carried out at 40 °C.
The stabilities and activities of catalysts 1 and 2 in the presence of resins were unexplored, and we examined commonly used resins for solid-phase peptide synthesis that varied based on composition and loading capacity. Throughout our studies, we performed RCM in solvents that promote α-helicity [e.g., dicholorethane (DCE)] at concentrations that favor macrocyclic ring closure. Initial screening revealed that the choice of resin influenced the activities of catalysts 1 and 2 in RCM. Conversions to the desired RCM product 17 were typically low on Wang resin using 10 mol % catalyst at room temperature for 2 h (entry 1). Resins bearing hydrophilic linkers proved beneficial, affording conversions approaching 40% under the same reaction conditions (entry 2). The use of MBHA resin led to 60% conversion (entry 3), and we focused further optimization using this resin. We next explored the effects of reaction time and catalyst loading on RCM. Prolonging the reaction led to modest improvements, generating product 17 in 70% conversion with greater than 90% Z selectivity (entry 4), and subjecting the resin-bound peptide to successive rounds of catalyst111 resulted in conversions of 75% using two cycles of catalyst addition (entry 5). Increasing the temperature to 40 °C afforded 17 in 80% yield and with greater than 90% Z selectivity (entry 6).125
To probe the generality of the method, we investigated peptides bearing olefinic amino acids spanning two turns (i, i + 7) of a helix with varying amino acid sequences (Scheme 2).130 To span the distance of two helical turns, we modified the N-terminal olefinic amino acid by increasing the tether length (from five to eight carbon atoms) and inverting the stereochemical configuration (from S to R), both of which were predicted to facilitate RCM.41,104,126 Under our optimized conditions, conversions of 85% for the desired RCM product 19 could be achieved after two cycles of catalyst addition with greater than 90% Z selectivity. These results demonstrate that cyclometalated ruthenium catalysts can promote Z-selective RCM on solid support for the synthesis of stapled peptides bearing all hydrocarbon cross-links.
Scheme 2. Z-Selective RCM to form i, i + 7 Stapled Peptides.

Determined by analytical HPLC-MS.
i, i + 3 Z-Selective Ring-Closing Metathesis Attempts on Aib-Containing Peptides
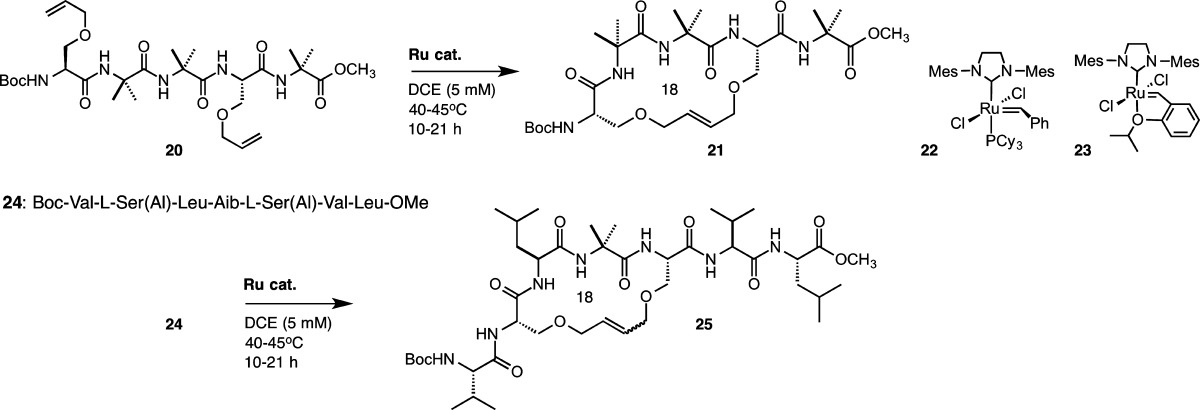
We earlier demonstrated that Aib-rich peptides bearing i, i + 3 l-serine O-allyl residues afford highly E-selective RCM products,110 in studies motivated, in part, by a theoretical prediction that suggested an RCM-derived 18-membered ring using these side chains would serve as a minimal constraint for the 310-helix.127 We were curious to see whether a Z-selective catalyst could overcome any substrate bias favoring the E-olefin geometry. To this end, we studied the RCM conversions of pentapeptide Boc-l-Ser(Al)-Aib-Aib-l-Ser(Al)-Aib-OMe (20) and heptapeptide Boc-l-Val-l-Ser(Al)-l-Leu-Aib-l-Ser(Al)-l-Val-l-Leu-OMe (24) to macrocycles 21 and 25, respectively, using second-generation catalysts 22 and 23 and Z-selective catalyst 1 (Table 9). As expected from our earlier studies, peptide 20 readily cyclized to the E macrocycle in the presence of 10 mol % 22 or 23 in DCE held at 45 °C for 10 h (entries 1 and 2). Under similar reaction conditions, no macrocyclization was observed with catalyst 1, even with a 3-fold increase in catalyst loading and extended reaction time (entry 3). The same behavior was observed for heptapeptide 24, although, in this case, the Z macrocycle did form with 22 or 23 to the extent of ca. 8% (entries 4 and 5). Peptide 24 rapidly cyclized with the second-generation catalysts in refluxing dichloromethane, but it was unreactive toward catalyst 1 under identical conditions (entry 6). Whereas most peptide side-chain RCM reactions produce E/Z mixtures, the minimal i, i + 3 cross-link in these Aib-containing systems seems reluctant to form the Z-olefin, probably a consequence of the conformational restrictions imposed by the Cα-tetrasubstituted α-amino residues.128
Table 9. RCM to Form i, i + 3 Stapled Peptides.
| entry | substrate | product | catalyst (mol %) | temperature (°C) | time (h) | conversiona (%) | selectivity |
|---|---|---|---|---|---|---|---|
| 1 | 20 | 21 | 22 (10) | 45 | 10 | 86 | >20:1 E/Zb |
| 2 | 23 (10) | 45 | 10 | 85 | >20:1 E/Zb | ||
| 3 | 1 (30) | 40 | 21 | <5 | n.d. | ||
| 4 | 24 | 25 | 22 (10) | 45 | 3.5 | 100 | 13:1 E/Za |
| 5 | 23 (10) | 45 | 3.5 | 94 | 11:1 E/Za | ||
| 6 | 1 (10) | 40 | 4 | <5 | n.d. |
Determined by analytical HPLC-MS.
Determined by 1H NMR spectroscopy.
These results demonstrate that the i + 1 and i + 2 Aib residues play a role in controlling the E-selective RCM behavior with catalysts 22 and 23 and this conformational bias cannot be overcome by Z-selective catalysts. These studies highlight the importance of peptide sequence and conformation in controlling olefin geometry in the macrocyclic product and point to further strategies aimed at understanding how these parameters affect the efficiency of Z-selective RCM.
Conclusions
In summary, we have reported the first examples of Z-selective metathesis on peptides using cyclometalated ruthenium catalysts. By examining a broad range of canonical and noncanonical amino acids in cross metathesis, homodimerization, and ring-closing metathesis, we have gleaned important criteria for achieving high conversion while maintaining excellent Z selectivity. The following insights based on our results are summarized:
The side-chain identity of an amino acid can dictate the activities of catalysts 1 and 2 in cross metathesis and homodimerization. In general, amino acids bearing aliphatic or aromatic side chains (e.g., alanine, leucine, and phenylalanine) are highly active in metathesis, with yields approaching 85% and 94% Z selectivity. Exceptions include glycine and proline, as these are inactive in metathesis. Sterically hindered side chains (e.g., valine or isoleucine) and amino acids bearing bulky protecting groups lead to lower conversions but without degradation of Z selectivity. Amino acids bearing carboxylate functionalities (i.e., glutamic acid and aspartic acid) require protection, as substrates bearing acidic functionalities can lead to catalyst decomposition and diminished Z selectivity. Amino acids bearing thiols or thioethers generally deactivate cyclometalated ruthenium catalysts; however, in some cases, the use of protecting groups can lead to productive turnovers. Side chains bearing hydroxyl groups (e.g., serine or threonine) are generally tolerated by catalysts 1 and 2, whereas amino acids bearing heterocycles have variable activity. Tryptophan is active in Z-selective homodimerization and cross metathesis, whereas histidine is generally inactive. Polar side chains bearing carboxamide (i.e., glutamine or asparagine) or guanidinium (i.e., arginine) functionalities are generally intolerant of cyclometalated ruthenium catalysts. Protection of these side chains can restore catalyst activity, leading to products highly enriched in Z-olefins.
Cross metathesis and homodimerzation of amino acids and peptides using Z-selective ruthenium catalysts can be performed in a variety of solvents, provided that the acidity of the reaction medium is kept minimal. The use of solvents such as MeCN, DMSO, and DMF generally leads to lower conversions than the use of noncoordinating solvents (e.g., DCE). The use of protic solvents (e.g., MeOH, EtOH, or H2O) can lead to products enriched in Z-olefins. Prolonged reaction times in protic solvents can, in some cases, lead to decomposition of catalysts 1 and 2.
Amino acids bearing allylic or homoallylic functionality are active in Z-selective metathesis. In general, higher conversions in homodimerization and CM can be achieved using homoallylic functionalities, particularly for sterically hindered substrates. Noncanonical amino acids containing allylic heteroatoms including those that could be incorporated into peptides and proteins are active in Z-selective cross metathesis and follow trends similar to those of noncyclometalated ruthenium catalysts. The use of aqueous conditions in the presence of salts as additives appears to diminish the activities of catalysts 1 and 2.
Cyclometalated ruthenium catalysts can be used to synthesize stapled peptides bearing hydrocarbon olefinic cross-links positioned at residues i, i + 4 or i, i + 7. To probe the limits of these catalysts in peptide stapling, we exposed Z-selective catalysts to Aib-rich peptides bearing O-allyl serine cross-links positioned at residues i and i + 3 that predominantly give rise to highly E-selective macrocyclic products. In these cases, catalysts such as 1 failed to undergo Z-selective RCM, suggesting that conformational restrictions imposed by substrates such as 20 can influence the activities of cyclometalated ruthenium catalysts 1 and 2 in RCM.
Notably, compounds 15, 17, and 19 represent the most complex substrates synthesized by cyclometalated ruthenium catalysts, and this bodes well for further studies aimed at applying Z-selective metathesis on substrates bearing multiple functionalities. It is envisioned that the studies reported herein will serve as a guideline in choosing appropriate alkene cross partners or promoting RCM on peptides. We anticipate that cyclometalated ruthenium catalysts could access new structures and provide insight into the role of alkene geometry in the biological activities of stapled peptides. Moreover, installation of Z-alkenes into peptides and proteins could allow sites for further modifications. Progress in the design and development of stereoselective ruthenium catalysts will continue to broaden the application of olefin metathesis in natural-product synthesis and in biology.
Acknowledgments
This work was financially supported by the NIH (NIH R01-GM031332). Research at Pomona College was funded in part by the Gordon and Betty Moore Foundation. NMR spectra were obtained on instruments supported by the NIH (RR027690). Materia, Inc., is acknowledged for the generous donation of catalysts 1 and 2. The authors also thank Scott Virgil (Caltech Center for Catalysis and Chemical Synthesis), Samir Das, and Arundhati Nag for helpful discussions.
Supporting Information Available
Experimental details and characterization data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Furstner A. Angew. Chem., Int. Ed. 2000, 39, 3012. [PubMed] [Google Scholar]
- Mol J. J. Mol. Catal. A: Chem. 2004, 213, 39. [Google Scholar]
- Samojłowicz C.; Grela K. ARKIVOC 2011, 4, 71. [Google Scholar]
- Schrock R. R. Chem. Rev. 2002, 102, 145. [DOI] [PubMed] [Google Scholar]
- Trnka T. M.; Grubbs R. H. Acc. Chem. Res. 2001, 34, 18. [DOI] [PubMed] [Google Scholar]
- Mangold S. L.; Prost L. R.; Kiessling L. L. Chem. Sci. 2012, 3, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe T. J.; Fishlock L. P.; Procopiou P. A. Chem.—Eur. J. 2008, 14, 5716. [DOI] [PubMed] [Google Scholar]
- Donohoe T. J.; Orr A. J.; Bingham M. Angew. Chem., Int. Ed. 2006, 45, 2664. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. Science 2000, 287, 1964. [DOI] [PubMed] [Google Scholar]
- Schuster M.; Blechert S. Angew. Chem., Int. Ed. 1997, 36, 2036. [Google Scholar]
- Tatton M. R. S.; I. Donohoe T. J. Org. Lett. 2014, 16, 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoveyda A. H.; Malcolmson S. J.; Meek S. J.; Zhugralin A. R. Angew. Chem., Int. Ed. 2010, 49, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Bulger P. G.; Sarlah D. Angew. Chem., Int. Ed. 2005, 44, 4490. [DOI] [PubMed] [Google Scholar]
- Kim N. Y.; Jeon N. L.; Choi I. S.; Takami S.; Harada Y.; Finnie K. R.; Girolami G. S.; Nuzzo R. G.; Whitesides G. M.; Laibinis P. E. Macromolecules 2000, 33, 2793. [Google Scholar]
- Leitgeb A.; Wappel J.; Slugovc C. Polymer 2010, 51, 2927. [Google Scholar]
- Liu X.; Basu A. J. Organomet. Chem. 2006, 691, 5148. [Google Scholar]
- Sveinbjörnsson B. R.; Weitekamp R. A.; Miyake G. M.; Xia Y.; Atwater H. A.; Grubbs R. H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitekamp R. A.; Atwater H. A.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 16817. [DOI] [PubMed] [Google Scholar]
- Binder J. B.; Raines R. T. Curr. Opin. Chem. Biol. 2008, 12, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. A.; Chalker J. M.; Davis B. G. ChemBioChem. 2009, 10, 959. [DOI] [PubMed] [Google Scholar]
- Song W.; Wang Y.; Qu J.; Lin Q. J. Am. Chem. Soc. 2008, 130, 9654. [DOI] [PubMed] [Google Scholar]
- van Hest J. C. M.; Kiick K. L.; Tirrell D. A. J. Am. Chem. Soc. 2000, 122, 1282. [PubMed] [Google Scholar]
- Zhang Z.; Wang L.; Brock A.; Schultz P. G. Angew. Chem., Int. Ed. 2002, 41, 2840. [DOI] [PubMed] [Google Scholar]
- Bernardes G. J. L.; Chalker J. M.; Errey J. C.; Davis B. G. J. Am. Chem. Soc. 2008, 130, 5052. [DOI] [PubMed] [Google Scholar]
- Lin Y. A.; Boutureira O.; Lercher L.; Bhushan B.; Paton R. S.; Davis B. G. J. Am. Chem. Soc. 2013, 135, 12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; van der Donk W. A. Org. Lett. 2001, 3, 1189. [DOI] [PubMed] [Google Scholar]
- Miller S. J.; Blackwell H. E.; Grubbs R. H. J. Am. Chem. Soc. 1996, 118, 9606. [Google Scholar]
- Blackwell H. B.; Grubbs R. H. Angew. Chem., Int. Ed. 1998, 37, 3281. [DOI] [PubMed] [Google Scholar]
- Blackwell H. B.; Sadowsky J. D.; Howard R. J.; Sampson N. S.; Chao J. A.; Steinmetz W. E.; O’Leary D. J.; Grubbs R. H. J. Org. Chem. 2001, 66, 5291. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Quah S. T.; Jong J.; Goh A. M.; Chiam P. C.; Khoo K. H.; Choong M. L.; Lee M. A.; Yurlova L.; Zolghadr K.; Joseph T. L.; Verma C. S.; Lane D. P. ACS Chem. Biol. 2013, 8, 506. [DOI] [PubMed] [Google Scholar]
- Gionnet-Estieu K.; Guichard G. Exp. Opin. Drug Discovery 2011, 6, 937. [DOI] [PubMed] [Google Scholar]
- Verdine G. L.; Hilinski G. J. Methods Enzymol. 2012, 503, 3. [DOI] [PubMed] [Google Scholar]
- Verdine G. L.; Walensky L. D. Clin. Cancer Res. 2007, 13, 7264. [DOI] [PubMed] [Google Scholar]
- Walensky L. D.; Kung A. L.; Escher I.; Malia T. J.; Barbuto S.; Wright R. D.; Wagner G.; Verdine G. L.; Korsmeyer S. J. Science 2004, 305, 1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchey L. K.; Jochim A. L.; Arora P. S. Curr. Opin. Chem. Biol. 2008, 12, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Wang D.; Zheng Q.; Lu M.; Arora P. S. J. Am. Chem. Soc. 2008, 130, 4334. [DOI] [PubMed] [Google Scholar]
- Patgiri A.; Jochim A. L.; Arora P. S. Acc. Chem. Res. 2008, 41, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. A.; Chalker J. M.; Davis B. G. J. Am. Chem. Soc. 2010, 132, 16805. [DOI] [PubMed] [Google Scholar]
- Bernal F.; Wade M.; Godes M.; Davis T. N.; Whitehead D. G.; Kung A. L.; Wahl G. M.; Walensky L. D. Cancer Cell 2010, 18, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips C.; Roberts L. R.; Schade M.; Bazin R.; Bent A.; Davies N. L.; Moore R.; Pannifer A. D.; Pickford A. R.; Prior S. H.; Read C. M.; Scott A.; Brown D. G.; Xu B.; Irving S. L. J. Am. Chem. Soc. 2011, 133, 9696. [DOI] [PubMed] [Google Scholar]
- Schafmeister C. E.; Po J.; Verdine G. L. J. Am. Chem. Soc. 2000, 122, 5891. [Google Scholar]
- Bird G. H.; Madani N.; Perry A. F.; Princiotto A. M.; Supko J. G.; He X.; Gavathiotis E.; Sodroski J. G.; Walensky L. D. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S.; Zhang H.; Debnath A. K.; Cowburn D. J. Biol. Chem. 2008, 283, 16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y. Q.; Huang S. X.; Zawahir Z.; Xu Z. L.; Li H.; Sanchez T. W.; Zhi Y.; DeHouwer S.; Christ F.; Debyser Z.; Neamati N. J. Med. Chem. 2013, 56, 5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Curreli F.; Waheed A. A.; Mercredi D. Y.; Mehta M.; Bhargava P.; Scacalossi D.; Tong X.; Lee S.; Cooper A.; Summers M. F.; Freed E. O.; Debnath A. K. Retrovirology 2013, 10, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Zhao Q.; Bhattacharya S.; Waheed A. A.; Tong X.; Hong A.; Heck S.; Curreli F.; Goger M.; Cowburn D.; Freed E. O.; Debnath A. K. J. Mol. Biol. 2008, 378, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. S.; Gravesb B.; Guerlavaisa V.; Tovarb C.; Packmanb K.; Tob K.-H.; Olsona K. A.; Kesavana K.; Gangurdea P.; Mukherjeea A.; Bakera T.; Darlaka K.; Elkina C.; Filipovich Z.; Qureshib F. Z.; Caia H.; Berry P.; Feyfanta E.; Shia X. E.; Horsticka J.; Annisa D. A.; Manninga A. M.; Fotouhib N.; Nasha H.; Vassilev L. T.; Sawyer T. K. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, e3445.23946421 [Google Scholar]
- Takada K.; Zhu D.; Bird G. H.; Sukhdeo K.; Zhao J. J.; Mani M.; Lemieux M.; Carrasco D. E.; Ryan J.; Horst D.; Fulciniti M.; Munshi N. C.; Xu W.; Kung A. L.; Shivdasani R. A.; Walensky L. D.; Carrasco D. R. Sci. Transl. Med. 2012, 4, 148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y.; Wang C.; Cao B.; Hirsch B. M.; Song J.; Markowitz S. D.; Ewing R. M.; Sedwick D.; Liu L.; Zheng W.; Wang Z. Cancer Cell 2013, 23, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann T. N.; Yeh J. T. H.; Bowman B. R.; Chu Q.; Moellering R. E.; Verdine G. L. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial N. N.; Walensky L. D.; Zhang C.-Y.; Choi C. S.; Fisher J. K.; Molina A. J. A.; Datta S. R.; Pitter K. L.; Bird G. H.; Wikstrom J. D.; Deeney J. T.; Robertson K.; Morash J.; Kulkarni A.; Neschen S.; Kim S.; Greenberg M. E.; Corkey B. E.; Shirihai O. S.; Shulman G. I.; Lowell B. B.; Korsmeyer S. J. Nat. Med. 2008, 14, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szlyk B.; Braun C. R.; Ljubicic S.; Patton E.; Bird G. H.; Osundiji M. A.; Matschinsky F. M.; Walensky L. D.; Danial N. N. Nat. Struct. Mol. Biol. 2014, 21, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. W.; Grubbs R. H. Org. Lett. 2000, 2, 2145. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Jimenez M.; Hansen A.; Raiber E. A.; Schreiber S. L.; Young D. W. J. Am. Chem. Soc. 2011, 133, 9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunet J. Angew. Chem., Int. Ed. 2003, 42, 2826. [DOI] [PubMed] [Google Scholar]
- Grubbs R. H., Ed. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1. [Google Scholar]
- Keitz B. K.; Endo K.; Patel P. R.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley B. L.; Grubbs R. H. Chem. Sci. 2014, 5, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosebrugh L. E.; Herbert M. B.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo K.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. S.; Grubbs R. H. Angew. Chem., Int. Ed. 2013, 52, 9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert M. B.; Marx V. M.; Pederson R. L.; Grubbs R. H. Angew. Chem., Int. Ed. 2013, 52, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Xu X.; Dong X.; Keitz B. K.; Herbert M. B.; Grubbs R. H.; Houk K. J. Am. Chem. Soc. 2012, 134, 1464. [DOI] [PubMed] [Google Scholar]
- Homoallyl-modified amino acids were used to benchmark the intrinsic reactivity of amino acids in homodimerization using cyclometalated ruthenium catalysts. We favored this approach over the analogous allyl-modified amino acids to reduce the chance of competing olefin isomerization that has been observed with cyclometalated ruthenium catalysts.
- A reaction time of 4 h was deemed optimal under the reported conditions. Extended reaction times led to minor amounts of Z degradation
- Prolonged heating in the presence of protic solvents can lead to decomposition of catalysts 1 and 2 with concomitant Z degradation.
- Herbert M. B.; Lan Y.; Keitz B. K.; Liu P.; Endo K.; Day M. W.; Houk K.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We note that chelation could account for the inactivity of glycine toward homodimerization.
- We have yet to fully explain the low activity of homoallyl-modified proline in Z-selective homodimerization. Further insight into this observation is the subject of ongoing investigations.
- Protecting the side chain of histidine did not improve the yield of homodimerization
- These results are consistent with observations that sulfur-containing substrates can have deleterious effects on catalyst activity.
- Chatterjee A. K.; Choi T.-L.; Sanders D. P.; Grubbs R. H. J. Am. Chem. Soc. 2003, 125, 11360. [DOI] [PubMed] [Google Scholar]
- Exposing the dimer of substrate 3 or 5a to the reaction conditions did not improve the yield of product 7, nor did it lead to significant Z degradation. This supports the observation that secondary metathesis events occur slowly with homoallyl-modified amino acids.
- Using an excess of arginine 5s can lead to minor amounts of catalyst decomposition and Z degradation over extended reaction times.
- Chalker J. M.; Goncalo J. L. B.; Davis B. D. Acc. Chem. Res. 2011, 44, 730. [DOI] [PubMed] [Google Scholar]
- Ben-Asuly A.; Tzur E.; Diesendruck C. E.; Sigalov M.; Goldberg I.; Lemcoff N. G. Organometallics 2008, 27, 811. [Google Scholar]
- McReynolds M. D.; Dougherty J. M.; Hanson P. R. Chem. Rev. 2004, 104, 2239. [DOI] [PubMed] [Google Scholar]
- Schwab P.; Grubbs R. H.; Ziller J. W. J. Am. Chem. Soc. 1996, 118, 100. [Google Scholar]
- Hoveyda A. H.; Lombardi P. J.; O’Brien R. V.; Zhugralin A. R. J. Am. Chem. Soc. 2009, 131, 8378. [DOI] [PubMed] [Google Scholar]
- Hoye T. R.; Zhao H. Org. Lett. 1999, 1, 1123. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Leung G. Y. C.; Dethe D. H.; Guduru R.; Sun Y.-P.; Lim C. S.; Chen D. Y. K. J. Am. Chem. Soc. 2008, 130, 10019. [DOI] [PubMed] [Google Scholar]
- Hartung J.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. A.; Chalker J. M.; Davis B. G. J. Am. Chem. Soc. 2010, 132, 16805. [DOI] [PubMed] [Google Scholar]
- Roberts K. S.; Sampson N. S. J. Org. Chem. 2003, 68, 2020. [DOI] [PubMed] [Google Scholar]
- Whelan A. N.; Elaridi J.; Mulder R. J.; Robinson A. J.; Jackson W. R. Can. J. Chem. 2005, 83, 875. [Google Scholar]
- We believe that salt metathesis could account for the lower activity of catalysts 1 and 2 in the presence of lithium chloride or magnesium chloride. Exchange of a bidentate nitrato ligand to a monodentate chloride ligand has been observed to decrease the catalytic activity of cyclometalated ruthenium complexes.
- Lin Y. A.; Chalker J. M.; Floyd N.; Bernardes C. J. L.; Davis B. G. J. Am. Chem. Soc. 2008, 130, 9642. [DOI] [PubMed] [Google Scholar]
- Almeida A. M.; Li R.; Gellman S. H. J. Am. Chem. Soc. 2012, 134, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire F.; Almeida A. M.; Fisk J. D.; Steinkruger J. D.; Gellman S. H. Angew. Chem. Int. .Ed. 2011, 50, 8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods R. J.; Brower J. O.; Castellanos E.; Hashemzadeh M.; Khakshoor O.; Russu W. A.; Nowick J. S. J. Am. Chem. Soc. 2007, 129, 2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P. N.; Pham J. D.; Nowick J. S. J. Am. Chem. Soc. 2013, 135, 5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowick J. S. Acc. Chem. Res. 2008, 41, 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P. N.; Liu C.; Zhao M.; Eisenberg D.; Nowick J. S. Nat. Chem. 2012, 4, 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Annu. Rev. Biochem. 2006, 75, 333. [DOI] [PubMed] [Google Scholar]
- Karran E.; Mercken M.; De Strooper B. Nat. Rev. Drug Discovery 2011, 10, 698. [DOI] [PubMed] [Google Scholar]
- Fisk J. D.; Gellman S. H. J. Am. Chem. Soc. 2001, 123, 343. [DOI] [PubMed] [Google Scholar]
- Fooks H. M.; Martin A. C.; Woolfson D. N.; Sessions R. B.; Hutchinson E. G. J. Mol. Biol. 2006, 356, 32. [DOI] [PubMed] [Google Scholar]
- The remaining mass balance in the Z-selective CM of peptides 13 and 14 can be attributed to unreacted starting material and byproducts resulting from homodimerization.
- Clark T. D.; Ghadiri M. R. J. Am. Chem. Soc. 1995, 117, 12364. [Google Scholar]
- Yang X.; Gong B. Angew. Chem., Int. Ed. 2005, 44, 1352. [DOI] [PubMed] [Google Scholar]
- Zeng J.; Wang W.; Deng P.; Feng W.; Zhou J.; Yang Y.; Yuan L.; Yamato K.; Gong B. Org. Lett. 2011, 13, 3798. [DOI] [PubMed] [Google Scholar]
- Performing cross metathesis in the presence of solvents that can influence interstrand hydrogen bonding including DMSO, DCE, and aqueous tert-butanol mixtures showed no appreciable increase in the yield of the heterocross product.
- Guo Z.; Mohanty U.; Noehre J.; Sawyer T. K.; Sherman W.; Krilov G. Chem. Biol. Drug Des. 2010, 75, 348. [DOI] [PubMed] [Google Scholar]
- Bernal F.; Tyler A. F.; Korsmeyer S. J.; Walensky L. D.; Verdine G. L. J. Am. Chem. Soc. 2007, 129, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird G. H.; Bernal F.; Pitter K.; Walensky L. D. Methods Enzymol. 2008, 446, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis H.; Slaninova J.; Bednarova L.; Monincova L.; Budesinsky M.; Cerovsky V. Amino Acids 2012, 43, 2047. [DOI] [PubMed] [Google Scholar]
- Green B. R.; Klein B. D.; Lee H. K.; Smith M. D.; White S. H.; Bulaj G. Bioorg. Med. Chem. 2013, 21, 303. [DOI] [PubMed] [Google Scholar]
- Sviridov D. O.; Ikpot I. Z.; Stonik J.; Drake S. K.; Amar M.; Osei-Hwedieh D. O.; Piszczek G.; Turner S.; Remaley A. T. Biochem. Biophys. Res. Commun. 2011, 410, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird G. H.; Gavathiotis E.; Labelle J. L.; Katz S. G.; Walensky L. D. ACS Chem. Biol. 2014, 9, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boal A. K.; Guryanov I.; Moretto A.; Crisma M.; Lanni E. L.; Toniolo C.; Grubbs R. H.; O’Leary D. J. J. Am. Chem. Soc. 2007, 129, 6986. [DOI] [PubMed] [Google Scholar]
- Kim Y. W.; Grossmann T. N.; Verdine G. L. Nat. Protoc. 2011, 6, 761. [DOI] [PubMed] [Google Scholar]
- Toniolo C.; Benedetti E. Trends Biochem. Sci. 1991, 16, 350. [DOI] [PubMed] [Google Scholar]
- Bird G. H.; Crannell W. C.; Walensky L. D. Curr. Protoc. Chem. Biol. 2011, 3, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutchukian P. S.; Yang J. S.; Verdine G. L.; Shakhnovich E. I. J. Am. Chem. Soc. 2009, 131, 4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhee P.; van der Sloot A. M.; Verschueren E.; Serrano L.; Rousseau F.; Schymkowitz J. Trends Biotechnol. 2011, 29, 231. [DOI] [PubMed] [Google Scholar]
- Boal A. K.; Guryanov I.; Moretto A.; Crisma M.; Lanni E. L.; Toniolo C.; Grubbs R. H.; O’Leary D. J. J. Am. Chem. Soc. 2007, 129, 6986. [DOI] [PubMed] [Google Scholar]
- Kim Y.-W.; Kutchukian P. S.; Verdine G. L. Org. Lett. 2010, 12, 3046. [DOI] [PubMed] [Google Scholar]
- Kim Y. W.; Verdine G. L. Bioorg. Med. Chem. Lett. 2009, 19, 2533. [DOI] [PubMed] [Google Scholar]
- Bergman Y. E.; Del Borgo M. P.; Gopalan R. D.; Jalal S.; Unabia S. E.; Ciampini M.; Clayton D. J.; Fletcher J. M.; Mulder R. J.; Wilce J. A.; Aguilar M.-I.; Perlmutter P. Org. Lett. 2009, 11, 4438. [DOI] [PubMed] [Google Scholar]
- Shim S. Y.; Kim Y. W.; Verdine G. L. Chem. Biol. Drug Des. 2013, 82, 635. [DOI] [PubMed] [Google Scholar]
- Cianni A. D.; Carotenuto A.; Brancaccio D.; Novellino E.; Reubi J. C.; Beetschen K.; Papini A. M.; Ginanneschi M. J. Med. Chem. 2010, 53, 6188. [DOI] [PubMed] [Google Scholar]
- Stymiest J. L.; Mitchell B. F.; Wong S.; Vederas J. C. J. Org. Chem. 2005, 70, 7799. [DOI] [PubMed] [Google Scholar]
- van Lierop B. J.; Robinson S. D.; Kompella S. N.; Belgi A.; McArthur J. R.; Hung A.; MacRaild C. A.; Adams D. J.; Norton R. S.; Robinson A. J. ACS Chem. Biol. 2013, 8, 1815. [DOI] [PubMed] [Google Scholar]
- Peptide 16 is a modified sequence of the known BID peptide used to target the BCL-2 family of proteins. We chose to modify the original peptide sequence to facilate synthesis.
- The non-natural amino acid Fmoc-(S)-2-(4-pentenyl)alanine was incorporated at both residues i and i + 4
- Using higher temperatures or conducting RCM under microwave conditions led to catalyst decomposition
- Peptide sequence 18 is derived from a sequence of the tumor suppressor protein p53.
- Baek S.; Kutchukian P. S.; Verdine G. L.; Huber R.; Holak T. A.; Lee K. W.; Popowicz G. M. J. Am. Chem. Soc. 2012, 134, 103. [DOI] [PubMed] [Google Scholar]
- Saviano M.; Benedetti E.; Vitale R. M.; Kaptein B.; Broxterman Q. B.; Crisma M.; Formaggio F.; Toniolo C. Macromolecules 2002, 35, 4204. [Google Scholar]
- Our earlier study focused mainly on the octapeptide Boc-Aib-Aib-Aib-Ser(O-allyl)-Aib-Aib-Ser(O-allyl)-Aib-OMe that afforded highly E-selective (>20:1 E/Z) macrocycles. From these studies, we can conclude that Aib residues at positions i + 1 and i + 2 control the E/Z ratio of the RCM product.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.