

Abstract
Recent data show that colon cancer cells selectively overexpress cystathionine-β-synthase (CBS), which produces hydrogen sulfide (H2S), to maintain cellular bioenergetics, support tumor growth and stimulate angiogenesis and vasorelaxation in the tumor microenvironment. The purpose of the current study was to investigate the effect of the allosteric CBS activator S-adenosyl-L-methionine (SAM) on the proliferation and bioenergetics of the CBS-expressing colon cancer cell line HCT116. The non-transformed, non-tumorigenic colon epithelial cell line NCM356 was used as control. For assessment of cell proliferation, the xCELLigence system was used. Bioenergetic function was measured by Extracellular Flux Analysis. Experiments using human recombinant CBS or HCT116 homogenates complemented the cell-based studies. SAM markedly enhanced CBS-mediated H2S production in vitro, especially when a combination of cysteine and homocysteine was used as substrates. Addition of SAM (0.1 – 3 mM) to HCT116 cells induced a concentration-dependent increase H2S production. SAM exerted time-and concentration-dependent modulatory effects on cell proliferation. At 0.1–1 mM SAM increased HCT116 proliferation between 0–12 h, while the highest SAM concentration (3 mM) inhibited proliferation. Over a longer time period (12–24 h), only the lowest concentration of SAM used (0.1 mM) stimulated cell proliferation; higher SAM concentrations produced a concentration-dependent inhibition. The short-term stimulatory effects of SAM were attenuated by the CBS inhibitor aminooxyacetic acid (AOAA) or by stable silencing of CBS. In contrast, the inhibitory effects of SAM on cell proliferation was unaffected by CBS inhibition or CBS silencing. In contrast to HCT116 cells, the lower rate of proliferation of the low-CBS expressor NCM356 cells was unaffected by SAM. Short-term (1h) exposure of HCT116 cells to SAM induced a concentration-dependent increase in oxygen consumption and bioenergetic function at 0.1–1 mM, while 3 mM was inhibitory. Longer-term (72h) exposure of HCT116 cells to all concentrations of SAM tested suppressed mitochondrial oxygen consumption rate, cellular ATP content and cell viability. The stimulatory effect of SAM on bioenergetics was attenuated in cells with stable CBS silencing, while the inhibitory effects were unaffected. In NCM356 cells SAM exerted smaller effects on cellular bioenergetics than in HCT116 cells. We have also observed a downregulation of CBS in response to prolonged exposure of SAM both in HCT116 and NCM356 cells. Taken together, the results demonstrate that H2S production in HCT116 cells is stimulated by the allosteric CBS activator, SAM. At low-to intermediate levels and early time periods the resulting H2S serves as an endogenous cancer cell growth and bioenergetic factor. In contrast, the inhibition of cell proliferation and bioenergetic function by SAM does not appear to relate to adverse autocrine effects of H2S resulting from CBS over-stimulation but, rather to CBS-independent pharmacological effects.
Keywords: proliferation, bioenergetics, mitochondria, colorectal cancer, allosteric modulation
Introduction
Hydrogen sulfide (H2S) is an important signaling molecule involved in the regulation of vascular tone, angiogenesis and cellular bioenergetics [1–5]. With respect to its vascular effects, multiple pathways have been identified, including activation (opening) of potassium-dependent ATP (KATP) channels, and activation of cyclic GMP-dependent signaling via inhibition of phosphodiesterases [6–9]. Recent data show that H2S, at low physiological concentrations, serves as a physiological electron donor and inorganic source of energy in mammalian cells. Via these mechanisms, H2S supports mitochondrial electron transport and ATP generation [10–12].
Emerging data indicate that H2S plays an important role in the regulation of tumor cell biology. We have recently demonstrated that cystathionine-β-synthase (CBS), one of the H2S-producing enzymes, is abundantly expressed in human colon cancer cell lines and in human colon cancer tissue specimens, resulting in increased H2S production [13]. CBS-derived H2Sstimulates tumor cell bioenergetics, proliferation, migration and invasion. Moreover, by a paracrine action on peritumor/intratumor vascular endothelial cells, H2S promotes tumor angiogenesis [13]. Pharmacological inhibition or stable lentiviral-mediated silencing of CBS resulted in attenuated cellular energetic responses, suppressed cell proliferation and invasion in vitro, and inhibited tumor growth in vivo [13]. A follow-up paper by Bhattacharyya and colleagues [14] confirmed our findings related to bioenergetics, proliferation and intracellular localization of CBS in ovarian cancer cells and extended these observations to demonstrate that the downregulation/inhibition of CBS sensitizes the cancer cells to cisplatin. A substantial portion of CBS is localized to the mitochondria of the cancer cell, in stark contrast to non-transformed cells, where the low levels of CBS are predominantly cytosolic [13,14]. The intracellular levels and the mitochondrial translocation of CBS are regulated, at least in part, by proteolytic processes including the Lon protease [15,16]. In summary, the above-mentioned studies in colorectal and ovarian cancer cells [13,14], coupled with additional lines of evidence demonstrating the high expression of CBS in prostate cancer cells [17] and enhanced production of H2S in tumor-bearing experimental animals and cancer patients [18–21] suggest that cancer cell-derived H2S serves as an autocrine stimulator of tumor growth.
The purpose of the current study was to investigate the effect of the allosteric CBS activator S-adenosyl-L-methionine (SAM) on the proliferation and bioenergetics of the CBS-expressing colon cancer cell line HCT116. The non-tumorigenic colon epithelial cell line NCM356, which expresses low levels of CBS relative to HCT116 cells [13], was used as a control. We reasoned that, in accordance with the well-known bell-shaped character of the H2S dose-response curve (where low concentrations of H2S exert proliferative and positive bioenergetic effects, while high concentrations of H2S are inhibitory) SAM treatment would induce bell-shaped proliferative and bioenergetic responses in HCT116 cells. We further hypothesized that, if the cellular responses to SAM were primarily mediated by CBS activation and consequent H2S production, then the pharmacological responses to SAM would be more pronounced in HCT116 cells, when compared either to the responses of HCT116 cells with stable CBS silencing, or to NCM356 cells.
Material and methods
Materials
Aminooxyacetic acid (AOAA), antimycin A, 7-azido-4-methylcoumarin, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), Coomassie blue R-250, S-(5′-adenosyl)-L-methionine chloride dihydrochloride (SAM), d-aminolevulinic acid (d-ALA), N,N-dimethyl-p-phenylendiamine-sulfate (DPD), 2-deoxyglucose, glutathione (GSH), homocysteine, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride (INT), iron(III) chloride (FeCl3), lactic acid, L-cysteine, N-methylphenazonium methyl sulfate (PMS), nicotinamide adenine dinucleotide (NAD+), oligomycin, pyridoxal-5-phosphate (PLP), rotenone, sodium hydrosulfide hydrate (NaSH), zinc acetate (ZnAc) and trichloroacetic acid (TCA) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The human colorectal carcinoma cell line, HCT116 (ATCC, Manassas, VA) was cultured in McCoy’s 5A media and NCM356 cells, a non-tumorigenic colon epithelial cell line derived from the normal margin of a rectal cancer specimen, (Incell Corporation, San Antonio, TX) was cultured in DMEM media containing 1 g/l glucose. The culture medium were supplemented with 10% FBS, 100 IU/ml penicillin, and 100 mg/ml streptomycin and cells were grown in a 37 °C, 5% CO2 atmosphere.
ShRNA-mediated silencing of CBS
HCT116 cells were transduced with a lentiviral vector containing shRNA sequences targeting CBS (SHCLNV, clone TRCN0000045359) as previously described [13]. A non-targeting control shRNA sequence (shCTL) was used to control for off-target effects (SHC002V, MISSION shRNA, Sigma-Aldrich; St. Louis, MO). HCT116 cells were infected at a MOI of 3 with hexadimethrine bromide (8 μg/ml). Transduced cells were selected and maintained in McCoy’s 5A media supplemented with 10% FBS and puromycin (2 μg/ml). CBS silencing resulted in an approximately 50% inhibition of CBS expression, as determined by Western blotting [13].
Recombinant human CBS protein expression and purification
The expression and purification of human CBS was performed as described previously [26]. Briefly, E. Coli BL21(DE3) Codon Plus cells (Stratagene, La Jolla, CA, USA) containing the expression vector pGEX-Kg/GST-CBS were grown at 37 °C and 180 rpm in Luria–Bertani (LB) broth medium containing 100 μg/ml ampicillin to an absorption of 0.6–0.8 at 600 nm. Protein expression was induced by addition of 0.1 mM IPTG (isopropyl-b-D-thiogalactopyranoside) and cells were further incubated at 30 °C overnight. The bacteria were harvested and sonicated in lysis buffer PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4,1.8 mM KH2PO4, pH 7.8) containing a protease inhibitors cocktail (Sigma). The protein lysate was loaded onto a GSTrap FF 1 ml affinity column (Amersham Biosciences) and the GST-CBS recombinant protein was eluted with the elution buffer (50 mM Tris–HCl, 10 mM reduced glutathione, pH 8.0) and then dialyzed and concentrated in 10 mM sodium phosphate buffer (pH 8.2) and DTT (1 mM).
Measurement of H2S production by recombinant CBS
The measurement of H2S production by recombinant CBS enzyme was performed as described [26]. Briefly, each test consisted of a 100 μl reaction mixture in 50 mM sodium phosphate buffer pH 8.2 containing 1 μg of the purified human CBS enzyme, 0.01 mM pyridoxal-5′-phosphate (PLP), 10 mM L-cysteine in the absence or presence of 0.5 mM homocysteine. SAM (1 mM) was added to the reaction 15 min before the addition of L-cysteine to the solution. The reaction was initiated by transferring the Eppendorf tubes from ice to a 37°C shaking water bath. After 60 minutes of incubation at 37 °C, the reaction was terminated by adding 1% ZnAc followed by 10% trichloroacetic acid. Subsequently, N,N-dimethylphenylendiamine sulfate (20 mM in 7.2 M HCl) and FeCl3 (30 mM in 1.2 M HCl) were added and the optical absorbance of the solutions was measured at 650 nm. All samples were assayed in triplicate and H2S concentration was calculated against a calibration curve of standard NaHS solutions.
Detection of H2S production in HCT116 cell homogenates and live HCT116 cells
Proliferating HCT116 cells were washed twice with ice-cold PBS, scraped from flasks using ice-cold PBS, and centrifuged 700xg for 10 min at 4 °C. The cell pellet was lysed using non-denaturating lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 1% Triton X-100) on ice for 1h followed by centrifugation at 20,000g for 5 min at 4 °C to sediment un-lysed cells. The protein concentration was determined with DC Protein Assay (BioRad) with bovine serum albumin (Thermo Scientific) as a standard. The reaction mixture contained: 300 micrograms of protein cell extract, 100 mM Tris HCl pH 8.0, 50 μM PLP, 10 mM L-cysteine, 0.5 mM L-homocysteine, increasing concentration of SAM and 10 μM of the fluorescent H2S probe 7-azido-4-methylcoumarin [27]. After incubation for 2 h at 37 °C the fluorescence of H2S specific probe was measured using SpectraMax M2 (Molecular Devices) microplate reader using ex=365, em=450nm. Preliminary assays were carried out to ensure the linearity of fluorescence with respect to amount of protein extracts. Standard curve for H2S was generated by addition of NaHS to the fluorescent assay medium.
For detecting the effect of SAM on H2S production in live cells, 30,000 HCT116 or NCM356 cells were seeded in Lab-Tek II chamber coverglass system (Nalgen Nunc International) and incubated at 37 °C and 10% CO2 humidified incubator overnight. The cells were loaded with the fluorescent H2S probe 7-azido-4-methylcoumarin [27] at 10 μM final concentration for 30 min. Various concentrations of SAM were added to the cells and cells were further incubated for 1 hour. Cells were washed three times with PBS and dye’s specific fluorescence was visualized using Nikon eclipse 80i inverted microscope with Photometric CoolSNAP HQ2 camera and NIS-Elements BR 3.10 software.
Western blotting
Western blotting for CBS was performed as described [13]. Cells were washed with ice-cold phosphate-buffered saline (PBS, pH 7.4) and were lysed in 110 μl/well denaturing loading buffer (20 mM Tris-HCl pH=6.8, 2% SDS, 10% glycerol, 6 M urea, 100 mg/ml bromophenol blue, 200 mM β-mercaptoethanol), sonicated and boiled. Lysates (25 μg protein/10 μl/well) were resolved on 4–12% NuPage Bis–Tris acrylamide gels (Invitrogen Carlsbad, CA, USA) and transferred to PVDF membranes. Membranes were blocked in 10% non-fat dried milk or Starting BlockTM T20 (TBS) blocking buffer (Fischer Scientific, Pittsburgh, PA, USA). Then membranes were probed overnight with anti-CBS antibody (1:1000, Abnova, Walnut, CA, USA). On the following day, anti-rabbit-horseradish peroxidase conjugate antibody (HRP, 1:3000, Cell Signaling, Danvers, MA, USA) was applied and enhanced chemiluminescent substrate (ECL, Pierce) was used to detect the signal by high sensitivity films (Amersham Hyperfilm ECL). To normalize signals, membranes were re-probed with a HRP conjugated antibody against actin (anti-actin HRP linked, 1:2000, Santa Cruz Biotechnology Inc., Dallas, TX). CBS was detected at 60 and 45 kDa. β-actin was determined at 43 kDa.
Proliferation assays in vitro
For assessment of cell proliferation, the xCELLigence system (Roche) was used, as described [13]. Briefly, HCT116 or NCM356 cells were cultured until approximately 70% confluence in complete cell culture media. Cells were then detached by Trypsin-EDTA and re-suspended in fresh culture media at a concentration of 30,000 cells/ml. 200 μl of cell suspension was added to each well (6,000 cells/well) of a E-plate 96, a specially designed 96-well microtiter plate containing interdigitated microelectrodes to non-invasively monitor the cell proliferation by measuring the relative change in the electrical impedance of the cell monolayer, a unitless parameter named cell index (CI).
Bioenergetic analysis in cultured cells
The XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA) was used to measure bioenergetic function as previously described [12,13,28]. Oxygen consumption rate (OCR) after oligomycin (1.5 μg/ml) was used to assess ATP production rate and OCR after the addition of FCCP (0.5 μM) to assess maximal mitochondrial respiratory capacity. 2-deoxyglucose (100 mM) was used to estimate cellular glycolytic dependency and antimycin A (2 μg/ml) and rotenone (2 μM) were used to inhibit the flux of electrons through complex III and I, to detect residual non-mitochondrial oxygen consumption rate, which is mainly attributed to cytosolic oxidases.
Measurement of cellular ATP levels
ATP concentration was determined in NCM356 and HCT116 cell cultures using a commercially available kit (CellTiter-Glo® Luminescent Cell Viability Assay, Promega, Madison, WI, USA). The luminescent signal was recorded for 1 s utilizing the multimode reader SpectraMax M2 (Molecular Devices Corp., Sunnyvale, CA). Serial dilutions of ATP were used as calibration standards.
LDH assay
Lactate dehydrogenase (LDH) release was used to detect cytotoxicity/cell death, as described [29]. Briefly, 30 μl of cell culture supernatant was mixed with 100 μl freshly prepared LDH assay reagent to reach final concentrations of 85 mM lactic acid, 1040 mM nicotinamide adenine dinucleotide (NAD+), 224 mM N-methylphenazonium methyl sulfate (PMS), 528 mM 2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride (INT) and 200 mM Tris (pH 8.2). The changes in absorbance were read kinetically at 492 nm for 15 min (kinetic LDH assay) on a monochromator-based reader (Powerwave HT, Biotek) at 37 °C. The changes in absorbance were read kinetically at 492 nm for 15 min. LDH activity values are shown as Vmax for kinetic assays in mOD/min.
Statistics
Data are shown as means ± standard error of the mean (SEM). Student’s t tests, one-way and two-way ANOVA with Tukey’s post hoc test were used to detect differences between groups; *p < 0.05 and **p < 0.01 represent statistically significant variance versus control. All statistical calculations were performed using Graphpad Prism 5 analysis software.
Results
Full-length and truncated CBS are abundantly expressed in HCT116 cells
The human colon adenocarcinoma cell line HCT116 showed a marked increase of the CBS expression levels, as compared to the control non-malignant normal mucosa cell line NCM356 (Figure 1). In addition to the native (60 kDa) band, the presence of a truncated 45 kDa CBS isoform was also noted (Figure 1).
Figure 1. CBS is highly expressed in colon cancer cells and is downregulated by SAM.

Representative Western blot analysis of CBS expression in cultured NCM356 (left lanes) and HCT116 (right lanes) cells. Please note the high level of expression of a native (65 kDa) band and lower level of expression of an additional, lower intensity band at 45 kDa. The Western blot analysis was done at 72 hours after culturing in the presence of either vehicle or the indicated concentrations of SAM. A blot representative of three independent determinations is shown.
SAM enhances the activity of recombinant CBS in vitro
Incubation of human recombinant CBS with SAM (1 mM) caused a significant enhancement of H2S production, in the presence of L-cysteine (10 mM) and with the combination of L-cysteine (10 mM) and L-homocysteine (0.5 mM) (Figure 2a). When L-cysteine was added to CBS in the absence of SAM, a minor degree of H2S production was noted, while in the combined presence of SAM and L-cysteine, a low but detectable amount of H2S production was observed. In the presence of L-cysteine and L-homocysteine, CBS produced significantly higher amounts of H2S than with L-cysteine alone; in these latter conditions, once again, SAM caused a significant enhancement of H2S production. Overall, in the presence of SAM, in all of the experimental conditions tested, H2S production was markedly higher than in its absence (17.3 ± 3.6 versus 47.2 ± 0.8) (Figure 2a).
Figure 2. SAM significantly enhances the activity of CBS in vitro.
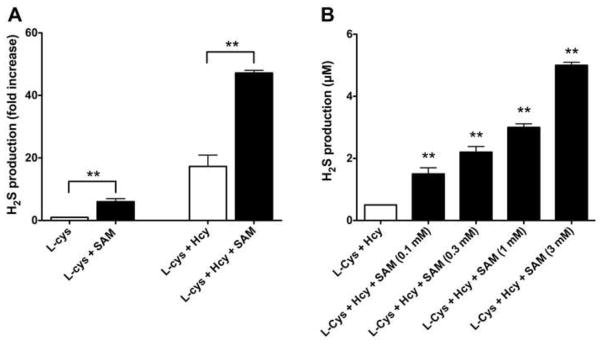
(A) The effect of SAM (1 mM) is shown on H2S production by the recombinant human CBS enzyme, as detected by the methylene blue method, in the presence of either L-cysteine (10 mM) or the combination of L-cysteine (10 mM) and L-homocysteine (0.5 mM). Data represent mean±SEM of n=3 experiments; **p<0.01 shows a significant enhancement of H2S production by SAM. (B) The effect of SAM (0.1–3 mM) is shown on H2S production in HCT116 cell homogenates, as detected by a fluorescent H2S method, in the presence of the combination of L-cysteine (10 mM) and L-homocysteine (0.5 mM). Data represent mean±SEM of n=3 experiments; **p<0.01 shows a significant enhancement of H2S production by SAM.
SAM significantly enhances H2S production in HCT116 cells
Incubation of HCT116 cell homogenates with SAM (0.1 – 3 mM) in the combined presence of L-cysteine (10 mM) and L-homocysteine (0.5 mM) caused a concentration- dependent, marked enhancement of H2S production (Figure 2b). The increase in H2S production was also visualized by fluorescent live cell imaging (Figure 3). SAM (3 mM) induced a marked increase in the H2S signal of HCT116 cells, while it only induced a slighter increase in the low-CBS-expressing NCM356 cells. Both the basal and the SAM-stimulated fluorescent signal was attenuated by the CBS inhibitor aminooxyacetic acid (AOAA, 1 mM) (Figure 3).
Figure 3. Visualization of the SAM-mediated increase in H2S production in HCT116 and NCM356 cells.

Cells were treated with SAM (3 mM) in the absence or presence of AOAA (1 mM) for 1 hour and intracellular H2S was detected using the AzMC fluorescent probe. Note the increase in H2S signal in response to SAM treatment, and the inhibition of both basal and SAM-stimulated H2S production by AOAA. The figures shown are representative of three independent experiments that were run in duplicates for each end-point.
Effects of SAM on the proliferation rate of HCT116 and NCM356 cells
Addition of SAM (0.1 – 1 mM) to HCT116 cells induced a concentration-dependent increase in cell proliferation between 0–12 hours, (Figures 4a, 4c, 5a, 5c). After a more prolonged time period (between 12–24 hours), intermediate to high concentrations of SAM (0.3–3 mM) induced cell death/suppression of cell proliferation (Figures 4a, 4c, 5a, 5c). NCM356 cells (which express low levels of CBS) proliferated at a slower rate compared to HCT116 with the growth being almost undetectable when seeded at the same density of 30,000 cells/ml. A cell titration experiment identified in 100,000 cells/ml the ideal initial density of the NCM356 monolayer, which resulted basal growth rates comparable to HCT116 and permitted further testing of the effect of SAM (Figure 4e). SAM failed to show any stimulatory effect on NCM356 cell proliferation in the early phase (0–12h) (Figures 4f, 4g) but maintained its toxic profile in the late phase (12–24h) (Figures 4f, 4h).
Figure 4. Dual effect of SAM on the proliferation of colon cancer cells (HCT116) and normal mucosa cells (NCM356).
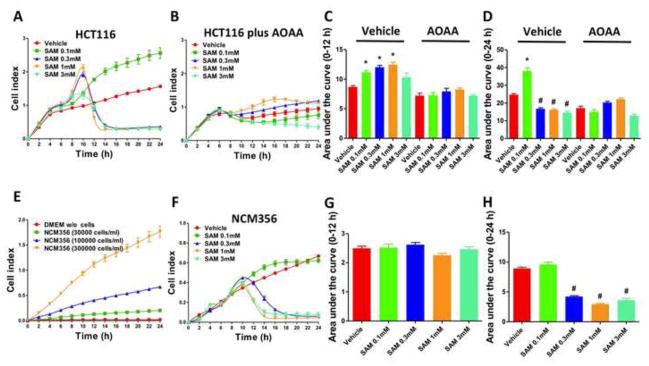
Colon cancer cell proliferation was monitored over a period of 24 hours (A and B). Results are also expressed as the area under the curve of the Cell Index (CI) over time (C and D). Short-time exposure (12 hours) to SAM (0.1–1 mM) significantly increased HCT116 cell proliferation rate in a concentration-dependent manner, but tended to decline as the concentration of SAM was further increased to 3 mM (A and C) (*p<0.05 vs corresponding vehicle). The stimulatory effect of SAM on HCT116 cell proliferation was abolished by AOAA (1 mM) (B and C). Long-term exposure (12 to 24 hours) of HCT 116 cells to relatively high concentrations of SAM caused a drop of the CI values, indicating inhibition of cell proliferation/cell death (#p<0.05 vs corresponding vehicle). Please also note that AOAA treatment reduced the basal HCT116 cell proliferation (C and D). NCM356 cells (which express relatively low CBS protein levels) grow at considerably slower rate compared to the HCT116 cells, showing almost no growth when seeded at the density of 30,000 cells/ml. A cell titration experiment (E) confirmed that the CI values depend on the density of the cell monolayer. The density of 100,000 cells/ml was then selected for further testing. Addition of SAM had no significant effect on NCM356 cell proliferation in the early phase (0–12 hours) indicating that the stimulatory effect of SAM is preferential for colon cancer cells which express high CBS levels (F and G). On the other hand, longer time exposure to SAM induced a concentration-dependent decrease in NCM356 cell proliferation (E) (#p<0.05 vs vehicle). Data represent mean±SEM of at least n=4.
Figure 5. Stable silencing of CBS in HCT116 cells abolishes the stimulatory effect of SAM on cell proliferation.

The effect of SAM on cell proliferation (0–24h) is shown in sham-silenced (shCTL) and CBS-silenced (shCBS) HCT116 cells in the presence of vehicle or low (0.3 mM), intermediate (1 mM) and high (3 mM) concentrations of SAM. (A and B). Results were also expressed as the area under the curve CI/time (C and D). *p<0.05 indicates significant stimulatory effects of low concentration of SAM (0.3 mM) on proliferation in non-targeted cells while #p<0.05 indicates significant inhibitory effect of long-term exposure to a high concentration of SAM (3 mM) on proliferation. Please note that shCBS cells proliferate slower than sham-silenced cells, and their proliferation rate is largely unaffected by SAM. Data represent mean±SEM of n=6.
Modulation of the pharmacological effects of SAM on cell proliferation after CBS silencing
Pharmacological inhibition of CBS by AOAA or its stable silencing with a lentiviral vector attenuated HCT116 cell proliferation (Figures 4, 5). Under these conditions, SAM failed to exert significant effects on cell proliferation: both the stimulation of the cell proliferation seen in wild-type cells between 0.1–1 mM SAM, and the relative inhibition of cell proliferation at 3 mM SAM (compared to the response seen at 1 mM SAM) were absent.
Effects of SAM on the cellular bioenergetics of HCT116 and NCM356 cells
Acute exposure of SAM at low concentrations (0.1 – 1 mM) stimulated the mitochondrial oxygen consumption rate (OCR) and bioenergetic functions of HCT116 cells in a dose-dependent fashion. In contrast, in NCM356 cells SAM resulted in a significantly lower degree of stimulation of the oxygen consumption rate and of the other bioenergetic parameters, when compared to the response seen in HCT116 cells (Figure 6).
Figure 6. Short-term exposure to SAM stimulates mitochondrial bioenergetics in HCT116 cells.
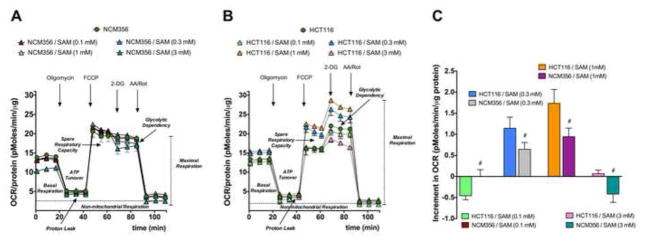
Extracellular Flux Analysis experiments assessing the effect of acute (1 h) exposure of (A) NCM356 or (B) HCT116 cells to SAM (0.1–3 mM). Panel (C) shows the mean±SEM of the increment in the FCCP-induced OCR in both cell types; #p<0.05 indicates significant differences between the responses of the NCM356 and the HCT116 cells. Data represent mean±SEM of n=9.
Longer-term exposure to SAM (0.3 – 1 mM for 72 hours) became cytotoxic in both cell lines (HCT116 and NCM356); this was evidenced by a significant suppression of mitochondrial respiration and cellular ATP content and a concomitant increase in LDH release (Figure 7). These effects tended to be more pronounced in HCT116 cells than in NCM356 cells. The lowest concentration of SAM used (0.1 mM) caused a slight suppression of cell respiration and viability in the HCT116 cells, but not in the NCM356 cells (Figure 7).
Figure 7. Long-term exposure to SAM suppresses oxygen consumption and cell viability, both in HCT116 and NCM356 cells.

Data represent the effect of 72 hours of incubation with SAM (0.1, 0.3 or 1 mM) on various parameters of cell respiration (A–C), LDH release, an index of cell death (D) and cellular ATP content (E). *,** indicates significant effects of SAM in NCM356 cells (p<0.05 and p<0.01, respectively), while #,## indicates significant effects of SAM in HCT116 cells (p<0.05 and p<0.01, respectively). Data represent mean±SEM of n=9.
Modulation of the pharmacological effects of SAM on cellular bioenergetics after CBS silencing
Stable silencing of CBS by a lentiviral vector attenuated the stimulatory effect of intermediate concentrations of SAM on HCT116 bioenergetics. The highest concentration of SAM tested (3 mM) inhibited cellular bioenergetics both in sham-silenced and CBS-silenced HCT116 cells (Figure 8).
Figure 8. Stable silencing of CBS in HCT116 cells abolishes the short-term stimulatory effect of SAM on cellular bioenergetics.

Oxygen consumption rate responses are shown in sham-silenced and CBS-silenced HCT116 cells in the presence of vehicle, and low (0.3 mM) and high (3 mM) concentrations of SAM at 1 hour. *p<0.05 indicates significant stimulatory effects of low concentration of SAM (0.3 mM) on cellular bioenergetics in wild-type cells, while #,## indicates significant inhibitory effect of SAM (0.3, 3 mM) on bioenergetics in cells with stable CBS silencing (p<0.05 and p<0.01, respectively). Please note that SAM no longer induces an increase in bioenergetics in shCBS cells. Data represent mean±SEM of n=9.
Effect of SAM on CBS expression in HCT116 and NCM356 cells
We have also tested whether exposure of the cells to SAM affects the expression of CBS at 72 hours of incubation. The results demonstrated that the expression of CBS in HCT116 cells is attenuated at highest concentrations used (1 and 3 mM) (Figure 1). SAM also induced CBS downregulation in the NCM356 cells (Figure 1).
Discussion
The main findings of the current study can be summarized as follows: (a) Using in vitro assays and human recombinant CBS, SAM markedly enhances H2S production, especially when a combination of cysteine and homocysteine are used as substrates; (b) Addition of SAM (0.1 – 1 mM) to HCT116 cells (a human colon cancer cell line, which exhibit high expression levels of CBS) concentration-dependently stimulates H2S production, but induces bell-shaped functional responses: lower concentrations and shorter incubation times stimulate proliferation, while higher concentrations/longer times inhibit it; (c) A similar, biphasic, concentration and time-dependent effect can also be observed on the bioenergetic responses of HCT116 cells; (d) Lower concentrations/shorter exposures to SAM stimulate the viability/energetics of HCT116 cells (e) Long-term incubation of the cells with SAM causes decreases the viability of HCT116 cells; (f) The stimulatory, but not the inhibitory effects of SAM are attenuated by CBS inhibition/CBS silencing; (g) The stimulatory effects of SAM on proliferation and bioenergetics are smaller in NCM356 cells (which express low levels of CBS) than in HCT116 cells. However the proliferation-inhibiting and cytotoxic effects of higher concentrations/longer exposures of SAM are comparable in NCM356 cells and in HCT116 cells; (h) Long-term incubation of the cells with SAM causes a downregulation of CBS both in HCT116 and NCM356 cells.
S-adenosyl-L-methionine (SAM, also termed AdoMet or AdoMetionine) was originally identified as a key intracellular regulator, an essential substrate facilitating the transfer of methyl groups (an effector of “transmethylation”). It is synthesized from methionine and ATP by methionine adenosyltransferase. Proteins, lipids and nucleic acids can all accept methyl groups from SAM. As reviewed by Lu and Mato [30], SAM serves important physiological roles as a broad regulator of cellular functions, especially in cell division, cell death, transcription, genetic stability, oxidant/antioxidant balance and polyamine homeostasis. It is also used therapeutically in humans as a nutritional supplement, and in some countries as a drug or nutraceutical agent for a variety of diseases ranging from osteoarthritis to liver injury [31–33].
In addition to regulating transmethylation reactions, SAM plays important roles in regulating transsulfuration reactions. Transsulfuration is traditionally viewed as a series of reactions whereby homocysteine is converted to cysteine (the rate-limiting precursor for GSH synthesis) via a two-step enzymatic process catalyzed by CBS and cystathionine γ-lyase (CSE). These two enzymes are now recognized as key contributors to the biosynthesis of H2S via a series of additional reactions that are collectively termed ‘alternative transsulfyration pathways’ [34–36]. SAM is a typical allosteric enzyme activator, that binds to the regulatory domain of CBS, which contains a hydrophobic tandem repeat of “CBS domains,” which is a secondary structure motif that is known to bind various adenine nucleotides and is believed to play a role in energy sensing. Upon binding, CBS undergoes a conformational change, resulting in a more open state of the active site of the enzyme, including a more accessible form of its prosthetic group, PLP, resulting in a 2–3 fold increase in its catalytic reaction velocity [23,25,37,38]. The relative contribution of CSE and CBS-mediated reactions to cellular H2S production, and the modulation of these reactions by SAM has been characterized by Banerjee and colleagues [35]. One of the conclusions of their study was that CBS contributes to cellular H2S generation primarily when it is sufficiently activated by SAM. The same authors have also noted that CBS produces much higher levels of H2S from cysteine+homocysteine than from cysteine alone [35]. These findings are in agreement with our current results (Figure 2a) showing that in the presence of cysteine+homocysteine both basal, and SAM-stimulated H2S production by human recombinant CBS is markedly higher than in the presence of cysteine alone. Although we have not conducted in vivo experiments with SAM, it is noteworthy that Jensen and colleagues have observed [39] in mice that ethionine (2-amino-4-(ethylthio)butyric acid, a methionine analog, which is converted to SAM in vivo [40]) induces a 3-fold increase in circulating H2S levels. These findings confirm the in vivo relevance of the activation of CBS by SAM, but also suggest that, even though cells contain substantial physiological amounts of SAM, CBS is not fully activated in vivo under physiological conditions.
The functional CBS enzyme is a tetramer of identical 60kDa units [38]. However, it can undergo proteolytic truncation to yield a 45kDa active form [41,42]. The 45kDa form is “hyperactive”, but cannot be further activated by SAM [42]. In addition to the predominant 60kDa form, we have found a smaller 45-kDa band in our cell homogenates (both in HCT116 and NCM356 cells) (Figure 1). Hence, our findings - also in line with data from Jensen and colleagues [39] reporting that native, cell-based CBS is more responsive to SAM than recombinant CBS - predicted that the cell types used in the present study remain functionally responsive to SAM. Therefore, after confirming the in vitro effect of SAM on H2S production by CBS, we proceeded to test the effect of SAM in colon cancer cells on two selected H2S-dependent cellular responses: cell proliferation and cellular bioenergetics.
With respect to cell proliferation, several lines of previous studies have already demonstrated that addition of exogenous H2S donors, or increasing endogenous H2S production by incubating the cells with substrates of H2S-producing enzymes can increase cell division and proliferation [2,13,43–46]. With respect to bioenergetics, several sets of studies demonstrated that addition of exogenous H2S donors, or increasing endogenous H2S production can increase mitochondrial function and cellular bioenergetics, in part via direct electron donation, and in part via inhibition of mitochondrial phosphodiesterases [10–13,47]. Given the bell-shaped pharmacological character of H2S in many biological processes (where low concentrations can be proliferative, antioxidant, cytoprotective and stimulatory on bioenergetics, while high concentrations can be antiproliferative, pro-oxidant, cytotoxic and inhibitory on bioenergetics) (reviewed in [48]), we hypothesized that a lower-level, optimal degree activation of CBS-dependent H2S production will stimulate proliferation, while higher levels of H2S may become inhibitory. Indeed, the results demonstrated the same: SAM, at low concentrations (below 1 mM) and at earlier time points (0–12 hours) tended to enhance proliferation in HCT116 cells, whereas higher concentrations (3 mM) and/or longer incubation times (12–24 hours) became inhibitory. The proliferation-stimulating effects of SAM were more prominent in HCT116 cells (which show high expression of CBS) than in NCM356 cells, which show low levels of CBS expression. This latter observation, coupled with the findings that the stimulatory effects of SAM are reduced in cells with CBS silencing or in the presence of the CBS inhibitor AOAA are consistent with the hypothesis that a significant portion of the stimulatory proliferative and bioenergetic effects of SAM seen in the current study are, in fact, related to intracellular H2S, and its downstream effects. In this context, it is also noteworthy that earlier studies found that intracellular SAM levels are highest during the proliferative phase of various cells [49] while recent studies demonstrated that depletion of endogenous SAM levels leads to a suppression of cell proliferation [50]. These findings are consistent with the hypothesis that SAM, at low/endogenous levels serves as a pro-proliferative, rather than antiproliferative molecule. However, the suppression of cell viability noted with long-term/higher concentrations of SAM (both in NCM356 and HCT116 cells) indicates that SAM also exerts cytotoxic effects in colon cancer cells via mechanisms unrelated to its CBS activating effect. In fact, SAM is known to regulate a variety of reactions in the cell, including the formation of powerful oxidizing agents (i.e. 5′-deoxyadenosyl radicals), the regulation of intracellular polyamine levels as well as the expression of a multitude of proteins, etc. [30,51]. These processes, directly or indirectly, may all contribute to processes that lead to a loss of cell viability in the presence of high concentrations of SAM. Some of these effects may also be responsible for the downregulation of CBS observed after long-term exposure to SAM.
SAM has been previously implicated as a negative regulator of tumor cell proliferation by affecting a variety of pathways including DNA methylation and polyamine biosynthesis [52–56]. What relevance, if any, do the current findings have to the anticancer effects of SAM? First of all, based on several sets of emerging data discussed in the Introduction section, endogenously produced CBS emerges as an endogenous tumor cell bioenergetic and tumor cell proliferation-producing factor. However, as mentioned above, we also know that elevation of cellular H2S levels beyond a certain level can turn into adverse (growth- inhibitory, cytotoxic) responses. Based on our in vitro findings - which, of course, represent a simple reductionist model of tumor biology - we conclude that low concentrations/short exposures to SAM, via stimulation of cellular H2S production, may not have antitumor effects (or, paradoxically, they may even be stimulatory for tumor cell growth and energetics), while longer exposures/higher concentrations may be, indeed of antitumor potential.
The current concentrations of SAM where the inhibition of proliferation of HCT116 cells was noted (1–3 mM), in fact, are comparable with the concentration range of SAM used in prior studies: the antiproliferative effects were typically reported at 1–10 mM [54,56]. When designing the experiments shown in the current report, one of the questions we wished to determine was whether that the antiproliferative effects of SAM are related to an ‘over-stimulation’ of CBS, followed by an elevation of intracellular H2S to cytotoxic levels. However, several findings speak against this possibility. (a) In CBS silenced cells the SAM-induced suppression of the proliferation is maintained. (b) Long-term exposure to SAM indiscriminately suppresses the proliferation and bioenergetics of both the high-CBS-expressing HCT116 cells and the low-CBS-expressing NCM356 cells (b). Long-term exposure to SAM results in a comparable degree of CBS downregulation in both cell types studied. The above findings are more consistent with the conclusion that the long-term growth inhibitory/antitumor effects of SAM involve CBS-independent mechanisms, as opposed to our original working model consisting of the ‘SAM-mediated CBS/H2S ‘overstimulation’. Because the inhibitory effects of SAM on proliferation and bioenergetics are not specific to HCT116 cells, and do not depend on CBS, we speculate that the specificity of SAM as an anticancer agent may be limited.
Highlights.
SAM markedly enhances CBS-mediated H2S production.
A combination of cysteine and homocysteine are the most effective substrates.
Lower concentrations of SAM stimulate HCT116 proliferation.
Shorter incubation times with SAM stimulate HCT116 proliferation.
Higher concentrations of SAM inhibit HCT116 proliferation.
Higher incubation times with SAM inhibit HCT116 proliferation.
A similar biphasic effect of SAM exists on bioenergetic responses.
In NCM356 cells the stimulating effects of SAM are minor.
In NCM356 cells the inhibitory effects of SAM are pronounced.
The stimulatory effects of SAM are attenuated in cells with CBS silencing.
The inhibitory effects of SAM are unaffected in cells with CBS silencing.
Long-term SAM exposure causes a downregulation of CBS in both cell types studied.
Acknowledgments
This work has been supported by the University of Texas (Institute for Translational Medicine) to M.R.H. and C.S. C.C. was supported by a grant from the American Heart Association. K.M. was supported by the University of Texas Medical Branch’s McLaughlin Fellowship. The project was co-financed by the European Union (European Social Fund - ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) - Research Funding Program: Thalis. Investing in knowledge society through the European Social Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–35. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 2.Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–65. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whiteman M, Le Trionnaire S, Chopra M, Fox B, Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci (Lond) 2011;121:459–88. doi: 10.1042/CS20110267. [DOI] [PubMed] [Google Scholar]
- 4.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 5.Módis K, Bos EM, Calzia E, van Goor H, Coletta C, Papapetropoulos A, Hellmich MR, Radermacher P, Bouillaud F, Szabo C. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br J Pharmacol. 2013 doi: 10.1111/bph.12368. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20:6008–16. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, Roviezzo F, Brancaleone V, Cirino G. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- 8.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–68. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I, Martin E, Szabo CC. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA. 2012;109:9161–6. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goubern M, Andriamihaja M, Nübel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21:1699–706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 11.Bouillaud F, Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid Redox Signal. 2011;15:379–91. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 12.Módis K, Coletta C, Erdélyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 13.Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A, Hellmich MR. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. 2013;110:12474–9. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, Rodriguez-Aguayo C, Lopez-Berestein G, Basal E, Weaver AL, Visscher DW, Cliby W, Sood AK, Bhattacharya R, Mukherjee P. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS One. 2013;8:e79167. doi: 10.1371/journal.pone.0079167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc Natl Acad Sci USA. 2013;110:12679–84. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta S, Wang L, Anderl J, Slifker MJ, Kirk C, Kruger WD. Correction of cystathionine β-synthase deficiency in mice by treatment with proteasome inhibitors. Hum Mutat. 2013;34:1085–93. doi: 10.1002/humu.22335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo H, Gai JW, Wang Y, Jin HF, Du JB, Jin J. Characterization of hydrogen sulfide and its synthases, cystathionine β-synthase and cystathionine γ-lyase, in human prostatic tissue and cells. Urology. 2012;79:483e1–5. doi: 10.1016/j.urology.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Yamagishi K, Onuma K, Chiba Y, Yagi S, Aoki S, Sato T, Sugawara Y, Hosoya N, Saeki Y, Takahashi M, Fuji M, Ohsaka T, Okajima T, Akita K, Suzuki T, Senawongse P, Urushiyama A, Kawai K, Shoun H, Ishii Y, Ishikawa H, Sugiyama S, Nakajima M, Tsuboi M, Yamanaka T. Generation of gaseous sulfur-containing compounds in tumour tissue and suppression of gas diffusion as an antitumour treatment. Gut. 2012;61:554–61. doi: 10.1136/gutjnl-2011-300721. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Huang J, Cushnir JR, Spanel P, Smith D, Hanna GB. Selected ion flow tube-MS analysis of headspace vapor from gastric content for the diagnosis of gastro-esophageal cancer. Anal Chem. 2012;84:9550–7. doi: 10.1021/ac302409a. [DOI] [PubMed] [Google Scholar]
- 20.Altomare DF, Di Lena M, Porcelli F, Trizio L, Travaglio E, Tutino M, Dragonieri S, Memeo V, de Gennaro G. Exhaled volatile organic compounds identify patients with colorectal cancer. Br J Surg. 2013;100:144–50. doi: 10.1002/bjs.8942. [DOI] [PubMed] [Google Scholar]
- 21.Chwatko G, Forma E, Wilkosz J, Głowacki R, Jóźwiak P, Różański W, Bryś M, Krześlak A. Thiosulfate in urine as a facilitator in the diagnosis of prostate cancer for patients with prostate-specific antigen less or equal 10 ng/mL. Clin Chem Lab Med. 2013;51:1825–31. doi: 10.1515/cclm-2013-0069. [DOI] [PubMed] [Google Scholar]
- 22.Miles EW, Kraus JP. Cystathionine beta-synthase: structure, function, regulation, and location of homocystinuria-causing mutations. J Biol Chem. 2004;279:29871–4. doi: 10.1074/jbc.R400005200. [DOI] [PubMed] [Google Scholar]
- 23.Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, Banerjee R. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Natl Acad Sci USA. 2006;103:6489–94. doi: 10.1073/pnas.0509531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–66. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koutmos M, Kabil O, Smith LJ, Banerjee R. Structural basis for substrate activation and regulation by cystathionine beta-synthase (CBS) domains in cystathionine β-synthase. Proc Natl Acad Sci USA. 2010;107:20958–63. doi: 10.1073/pnas.1011448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE) Br J Pharmacol. 2013;169:922–32. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorson MK, Majtan T, Kraus JP, Barrios AM. Identification of cystathionine β-synthase inhibitors using a hydrogen sulfide selective probe. Angew Chem Int Ed Engl. 2013;52:4641–4. doi: 10.1002/anie.201300841. [DOI] [PubMed] [Google Scholar]
- 28.Módis K, Gero D, Erdélyi K, Szoleczky P, DeWitt D, Szabo C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem Pharmacol. 2012;83:633–43. doi: 10.1016/j.bcp.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerö D, Módis DK, Nagy N, Szoleczky P, Tóth ZD, Dormán G, Szabo C. Oxidant-induced cardiomyocyte injury: identification of the cytoprotective effect of a dopamine 1 receptor agonist using a cell-based high-throughput assay. Int J Mol Med. 2007;20:749–61. [PubMed] [Google Scholar]
- 30.Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–42. doi: 10.1152/physrev.00047.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams AL, Girard C, Jui D, Sabina A, Katz DL. S-adenosylmethionine as treatment for depression: a systematic review. Clin Invest Med. 2005;28:132–9. [PubMed] [Google Scholar]
- 32.Lopez HL. Nutritional interventions to prevent and treat osteoarthritis. Part II: focus on micronutrients and supportive nutraceuticals. PM R. 2012;4:S155–68. doi: 10.1016/j.pmrj.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 33.Anstee QM, Day CP. S-adenosylmethionine therapy in liver disease: a review of current evidence and clinical utility. J Hepatol. 2012;57:1097–109. doi: 10.1016/j.jhep.2012.04.041. [DOI] [PubMed] [Google Scholar]
- 34.Jhee KH, Kruger WD. The role of cystathionine beta-synthase in homocysteine metabolism. Antioxid Redox Signal. 2005;7:813–22. doi: 10.1089/ars.2005.7.813. [DOI] [PubMed] [Google Scholar]
- 35.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–66. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabil O, Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5339. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oliveriusová J, Kery V, Maclean KN, Kraus JP. Deletion mutagenesis of human cystathionine beta-synthase. Impact on activity, oligomeric status, and S-adenosylmethionine regulation. J Biol Chem. 2002;277:48386–94. doi: 10.1074/jbc.M207087200. [DOI] [PubMed] [Google Scholar]
- 38.Ereño-Orbea J, Majtan T, Oyenarte I, Kraus JP, Martínez-Cruz LA. Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration. Proc Natl Acad Sci USA. 2013;110:E3790–9. doi: 10.1073/pnas.1313683110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jensen KK, Geoghagen NS, Jin L, Holt TG, Luo Q, Malkowitz L, Ni W, Quan S, Waters MG, Zhang A, Zhou HH, Cheng K, Luo MJ. Pharmacological activation and genetic manipulation of cystathionine beta-synthase alter circulating levels of homocysteine and hydrogen sulfide in mice. Eur J Pharmacol. 2011;650:86–93. doi: 10.1016/j.ejphar.2010.09.080. [DOI] [PubMed] [Google Scholar]
- 40.Finkelstein JD, Kyle WE, Martin JL, Pick AM. Activation of cystathionine synthase by adenosylmethionine and adenosylethionine. Biochem Biophys Res Commun. 1975;66:81–7. doi: 10.1016/s0006-291x(75)80297-x. [DOI] [PubMed] [Google Scholar]
- 41.Dong A, Kery V, Matsuura J, Manning MC, Kraus JP, Carpenter JF. Secondary structure of recombinant human cystathionine beta-synthase in aqueous solution: effect of ligand binding and proteolytic truncation. Arch Biochem Biophys. 1997;344:125–32. doi: 10.1006/abbi.1997.0202. [DOI] [PubMed] [Google Scholar]
- 42.Kery V, Poneleit L, Kraus JP. Trypsin cleavage of human cystathionine beta-synthase into an evolutionarily conserved active core: structural and functional consequences. Arch Biochem Biophys. 1998;355:222–32. doi: 10.1006/abbi.1998.0723. [DOI] [PubMed] [Google Scholar]
- 43.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabo C. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA. 2009;106:21972–7. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai WJ, Wang MJ, Ju LH, Wang C, Zhu YC. Hydrogen sulfide induces human colon cancer cell proliferation: role of Akt, ERK and p21. Cell Biol Int. 2010;34:565–72. doi: 10.1042/CBI20090368. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Wei H, Kong G, Shim W, Zhang G. Hydrogen sulfide augments the proliferation and survival of human induced pluripotent stem cell-derived mesenchymal stromal cells through inhibition of BKCa. Cytotherapy. 2013;15:1395–405. doi: 10.1016/j.jcyt.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 46.Szabo C, Hellmich MR. Endogenously produced hydrogen sulfide supports tumor cell growth and proliferation. Cell Cycle. 2013;12:2915–6. doi: 10.4161/cc.26064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Módis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun. 2013;433:401–7. doi: 10.1016/j.bbrc.2013.02.131. [DOI] [PubMed] [Google Scholar]
- 48.Szabo C, Ransy C, Módis K, Andriamihaja M, Murghes B, Coletta C, Olah G, Yanagi K, Bouillaud F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2013 doi: 10.1111/bph.12369. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maclean KN, Janosík M, Kraus E, Kozich V, Allen RH, Raab BK, Kraus JP. Cystathionine beta-synthase is coordinately regulated with proliferation through a redox-sensitive mechanism in cultured human cells and Saccharomyces cerevisiae. J Cell Physiol. 2002;192:81–92. doi: 10.1002/jcp.10118. [DOI] [PubMed] [Google Scholar]
- 50.Lin DW, Chung BP, Kaiser P. S-adenosylmethionine limitation induces p38 mitogen activated protein kinase and triggers cell cycle arrest in G1. J Cell Sci. 2013 doi: 10.1242/jcs.127811. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atta M, Mulliez E, Arragain S, Forouhar F, Hunt JF, Fontecave M. S-Adenosylmethionine-dependent radical-based modification of biological macromolecules. Curr Opin Struct Biol. 2010;20:684–92. doi: 10.1016/j.sbi.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 52.Pascale RM, Simile MM, Satta G, Seddaiu MA, Daino L, Pinna G, Vinci MA, Gaspa L, Feo F. Comparative effects of L-methionine, S-adenosyl-L-methionine and 5′-methylthioadenosine on the growth of preneoplastic lesions and DNA methylation in rat liver during the early stages of hepatocarcinogenesis. Anticancer Res. 1991;11:1617–24. [PubMed] [Google Scholar]
- 53.Regenass U, Caravatti G, Mett H, Stanek J, Schneider P, Müller M, Matter A, Vertino P, Porter CW. New S-adenosylmethionine decarboxylase inhibitors with potent antitumor activity. Cancer Res. 1992;52:4712–8. [PubMed] [Google Scholar]
- 54.Zhao Y, Li JS, Guo MZ, Feng BS, Zhang JP. Inhibitory effect of S-adenosylmethionine on the growth of human gastric cancer cells in vivo and in vitro. Chin J Cancer. 2010;29:752–60. doi: 10.5732/cjc.010.10046. [DOI] [PubMed] [Google Scholar]
- 55.Luo J, Li YN, Wang F, Zhang WM, Geng X. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int J Biol Sci. 2010;6:784–95. doi: 10.7150/ijbs.6.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hussain Z, Khan MI, Shahid M, Almajhdi FN. S-adenosylmethionine, a methyl donor, up regulates tissue inhibitor of metalloproteinase-2 in colorectal cancer. Genet Mol Res. 2013;12:1106–18. doi: 10.4238/2013.April.10.6. [DOI] [PubMed] [Google Scholar]