

Abstract
Disease phenotypes and defects in function can be traced to nonsynonymous single nucleotide polymorphisms (nsSNPs), which are important indicators of action sites and effective potential therapeutic approaches. Identification of deleterious nsSNPs is crucial to characterize the genetic basis of diseases, assess individual susceptibility to disease, determinate molecular and therapeutic targets, and predict clinical phenotypes. In this study using PolyPhen2 and MutPred in silico algorithms, we analyzed the genetic variations that can alter the expression and function of the ABCA1 gene that causes the allelic disorders familial hypoalphalipoproteinemia and Tangier disease. Predictions were validated with published results from in vitro, in vivo, and human studies. Out of a total of 233 nsSNPs, 80 (34.33%) were found deleterious by both methods. Among these 80 deleterious nsSNPs found, 29 (12.44%) rare variants resulted highly deleterious with a probability >0.8. We have observed that mostly variants with verified functional effect in experimental studies are correctly predicted as damage variants by MutPred and PolyPhen2 tools. Still, the controversial results of experimental approaches correspond to nsSNPs predicted as neutral by both methods, or contradictory predictions are obtained for them. A total of seventeen nsSNPs were predicted as deleterious by PolyPhen2, which resulted neutral by MutPred. Otherwise, forty two nsSNPs were predicted as deleterious by MutPred, which resulted neutral by PolyPhen2.
1. Introduction
Nonsynonymous single nucleotide polymorphisms (nsSNPs) are single base changes in coding regions that cause an amino acid substitution in the correspondent proteins. These missense variants constitute the most identifiable group of SNPs represented by a small (<1%) proportion [1]. The nsSNPs might alter structure, stability, and function of proteins and produce the least conservative substitutions with drastic phenotypic consequences [2–5]. Studies suggest that about 60% of Mendelian diseases are caused by amino acid exchanges [6]. Thousands of associations between Mendelian and complex diseases reveal a phenotypic code that links each complex disorder to a unique set of Mendelian loci [7]. Discriminating disease-associated from neutral variants would help to understand the genotype/phenotype relation and to develop diagnosis and treatment strategies for diseases. Nonetheless, the most important application is the evaluation of functional effect and impact of genomic variation, relating interactions with phenotypes translating the finding into medical practices.
ATP-binding cassette transporter ABCA1 gene also known as the cholesterol efflux regulatory protein (CERP) encodes a 220 kDa protein [8]. This protein is crucial for reverse cholesterol transport and is considered as an important target in antiatherosclerosis treatment. ABCA1 mediates the efflux of cholesterol and phospholipids to lipid-poor apolipoproteins (apoA1 and apoE), which form nascent high-density lipoproteins (HDL). ABCA1 resides on the cell membrane and has an extensive intracellular pathway, with rapid movement of the transporter between the cell membrane and intracellular vesicles [9]. ABCA1 is present in higher quantities in tissues that transfer or are involved in the turnover of lipids such as the liver, the small intestine, and adipose tissue [10–12]. As well, lipid export activity of ABCA1 improves the function of pancreatic cells and ameliorates insulin release [13], reduces biliary cholesterol content protecting against gallstone [14], and plays a key role in lipid homeostasis in the lung [15]. Besides, evidence suggest a causal link between ABCA1 as cholesterol transporter and its antitumor activity [16, 17], as well as its implication in brain cholesterol homeostasis [15–20] founding lipid and myelin abnormalities in schizophrenia and Alzheimer's disease [18–20].
Although the entire ABCA1 protein 3D-structure remains unknown electron microscopic studies suggest a structural model consisting of a transmembrane domain (TMD) and a nucleotide-binding domain (NBD) (Figure 1) [14, 21, 22], where an NBD-TMD dimer is the minimum unit required for transport function [22, 23]. Also, X-ray structures are available for different domains in the C-terminus protein essential for lipid efflux activity [24, 25]. Many variants disrupting the normal ABCA1 protein function result in modest or no circulating HDL [26–32]. Cholesterol accumulated within cells produces a toxicity that impairs cell function leading to a diversity of phenotypes, from severe disease states to mild impacts on health. In fact, the ABCA1 variability is associated with myocardial infarction, cancer, type 2 diabetes, and metabolic syndrome [33]. Heterozygous states, nearly one-third of them, are associated with hypoalphalipoproteinemia, known as familial HDL deficiency syndrome (FHA). Two copies cause a more severe syndrome Tangier disease (TD) [34–38] described by reduced HDL-c plasma level (<5%), impaired cholesterol efflux, and a trend to accumulate intracellular cholesterol [34–43]. Indeed, loss of function of ABCA1 mutations in TD patients has a major impact on lipoprotein metabolism. A failure to acquire apolipoproteins leads to a rapid catabolism of lipid-poor apoA1 and accumulation of lipids in macrophages, intestinal cells, platelet, and hepatocytes [34–38, 44]. Compared with unaffected family members, heterozygotes and homozygotes have a more prevalent, premature, and severe atherosclerosis [42].
Figure 1.
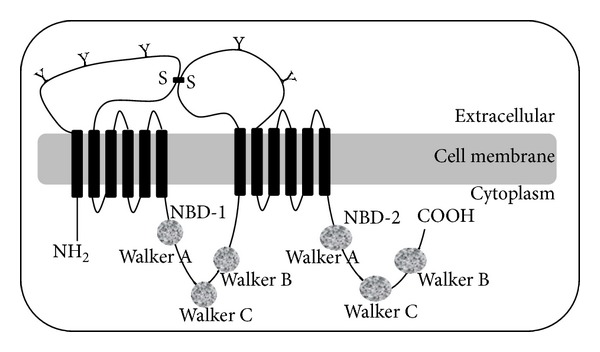
Predicted structure of ABCA1 [21, 23, 105]. The protein consists of 2261 amino acids and comprises 2 halves of similar structure. Each half encodes a transmembrane domain containing 6 helices (1–6 and 7–12) and 1 nucleotide binding domain (NBD-1 and NBD-2), where ATP is bound and utilized as energy for substrate transport across the membrane. Each NBD contains 2 highly conserved peptide motifs known as Walker A and Walker B, which are present in many proteins that use ATP and a Walker C signature unique to ABC transporters. ABCA1 is predicted to have an N terminus oriented into the cytosol and two large extracellular loops that are highly glycosylated and linked by cysteine bonds (Y indicates the glycosylation sites, and S–S indicates predicted disulfide bonds).
Because high levels of HDL-c are atheroprotective there is considerable interest in developing agents that act to increase ABCA1 expression and thereby raise plasma HDL-c levels. The nsSNPs are important indicators of action sites and effective potential therapeutic approaches. Therefore, it is crucial to identify deleterious nsSNPs to characterize the genetic basis of diseases, assess individual susceptibility to these diseases, determinate molecular and therapeutic targets, and predict clinical phenotypes. Beyond the genetic level, a disease depends on the sequence and the structural location of the nsSNPs of the protein. While the nsSNPs occur all through the ABCA1 gene, they tend to cluster in the extracellular loops, the NBD, and the COOH-terminal region (Figure 1). In fact, three structural motifs have been functionally associated with disease: the ARA motif, an interface between NBD and TMD that forms a partially buried α-helix able to interact with the transmembrane helices, the conserved-loop 1, a allosteric loop between the membrane and globular domains, and the conserved-loop 2, an interaction surface for intracellular partners, critical in ATP-binding.
Even though many of nsSNP (rare or common) found in human ABCA1 have been identified, mainly in the HapMap project (http://hapmap.ncbi.nlm.nih.gov/), the molecular bases relating these variants and the caused phenotypes have not been studied in detail. To explore the effect of the large number of nsSNPs ABCA1 by experimental approaches would be extremely time-consuming and with low statistical chance. Alternatively, bioinformatic approaches, based on the biophysical severity of the amino acid exchange and the protein sequence and structural information, can offer a more feasible phenotype prediction. As such, MutPred (mutation prediction) [45] and PolyPhen2 (polymorphism phenotyping 2) algorithms [46], were used in this study to investigate the impact of all known nsSNPs on ABCA1 protein function. Besides, based on the results of in vitro, in vivo, and human studies of this gene in the literature we validated the predictions made by reviewing the effect of the most critical nsSNPs in ABCA1 gene and its pathological consequences.
2. Methods
Figure 2 shows the workflow designed to predict the nsSNPs effect on ABCA1 protein. ABCA1 human gene variants including SNPs, short insertions, and deletions were retrieved from Ensemble Variation 72 database 3141. Mutations were annotated using the SnpEff v3.2 toolbox [47] based on the human genome assembly GRCh37.68. Only variants found on the canonical transcript were considered for functional effect prediction, for what we used two different algorithms. PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) algorithm uses a naive Bayesian classifier to predict allele function based on a combination of sequence and structure-based attributes (if available) [46]. It calculates the probability for a given mutation to be benign, possibly damaging, or probably damaging. Then, we used MutPred (http://mutpred.mutdb.org/) [45] based upon SIFT algorithm [48] and a gain/loss of 14 predicted structural and functional properties. The predicted mutation outcome is based on a random forest (RF) classifier. The MutPred output includes the top 5 property scores and a general score (RF) equal to the probability of amino acid exchange is either deleterious or disease-associated. The ROC curves for both methods were generated using R programming language and the ROCR package (Figure 3) by a variation dataset obtained from VariBench (http://structure.bmc.lu.se/VariBench/) that contains mutations affecting protein tolerance including a neutral set of mutations comprising 17393 human coding nsSNPs and a pathogenic set of 14610 missense mutations obtained by manual curation from the PhenCode database.
Figure 2.
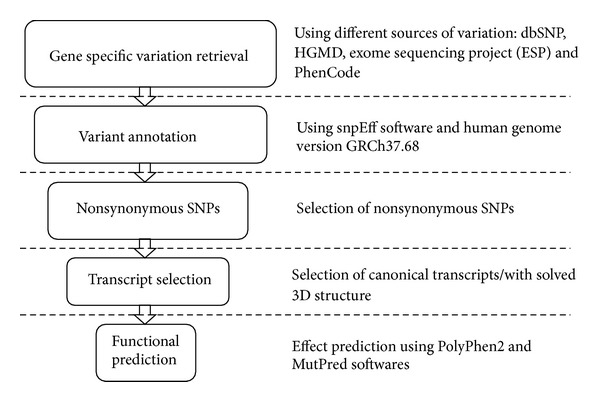
Workflow for predicting the effect of nsSNPs on ABCA1 protein. Gene specific variations were retrieved from the main variation databases. Mutations were annotated and nsSNPs belonging to the canonical transcript were selected in order to run the algorithms for functional prediction.
Figure 3.
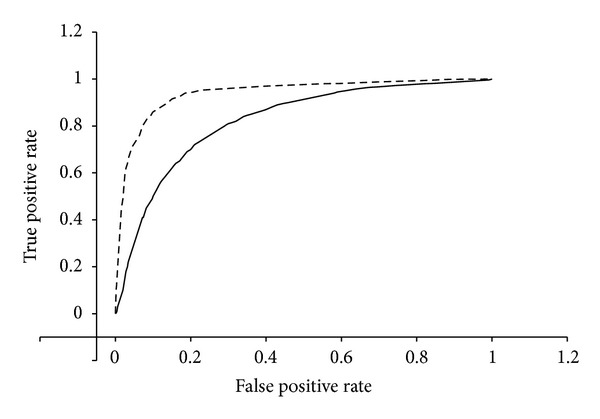
ROC (receiver operating characteristics) curve comparing the performance of MutPred and PolyPhen2 methods in predicting the outcome of nsSNPs. The dashed curve corresponds to MutPred predictions. As the area under the curve (AUC) for the MutPred method predictions is larger than the areas over the curve (AOC) which corresponds to the PolyPhen2 predictions curve, we can confirm that MutPred outperforms PolyPhen2 in predicting the outcome of nsSNPs. The dataset used for the performance comparison was obtained from Varibench as stated in Methods section.
Prediction accuracy accomplished by MutPred and PolyPhen2 depends on their specific criterion. Twelve structural and six sequence-based properties were used in this study (Table 1). About 28% of validated nsSNPs in the Human Genome Variation Database are predicted to affect protein function [49]. Similarly, about 25% of nsSNPs affecting protein activity was predicted by PolyPhen2 [49]. MutPred offers classification accuracy with respect to human disease mutations. Considering conservative thresholds on the predicted disruption of molecular function, MutPred generates accurate and reliable hypotheses on the molecular basis of disease for about 11% of known inherited disease-causing mutations [45].
Table 1.
Structural and functional properties used by MutPred and PolyPhen2.
| PolyPhen2 | MutPred | |
|---|---|---|
| Sequence based properties | ||
| Bond annotation | X | |
| Functional site annotation | X | X |
| Region annotation | X | X |
| PHAT score | X | |
| PSIC score | X | |
| SIFT score | X | |
| Evolutionary attributes | X | X |
| Structural properties | ||
| Secondary structure | X | X |
| Solvent-accessible surface area | X | X |
| Phi-psi dihedral angles | X | |
| Normalized accessible surface area | X | |
| Change in accessible surface propensity | X | |
| Change in residue side chain volume | X | |
| Region of the phi-psi map (Ramachandran map) | X | |
| Normalized B-factor | X | X |
| Ligand contacts | X | |
| Interchain contacts | X | |
| Functional site contacts | X | |
| Molecular Recognition Fragments (MoRFs) | X |
Our MutPred and PolyPhen2 predictions were validated by comparing them with previously obtained results from in vitro, in vivo, and human studies of ABCA1 gene in the databases and literature. When a given nsSNP found experimentally to be associated with a remarkable change of phenotype such as altered transporting activity or a disease was predicted by in silico methods as deleterious, it was considered that the prediction on this nsSNPs was correct. The prediction was defined as an error if such a deleterious nsSNP was predicted as tolerant.
3. Results and Discussion
The importance of ABCA1 in cholesterol efflux was demonstrated by the identification of ABCA1 mutations in TD and FHA families [34–38]. This has produced extensive research into the possibility to provide protection from atherosclerosis by increasing ABCA1 expression and thereby to raise plasma HDL-c levels. The identification of the large number of alleles for this transporter gene as target directly involved in HDL-c regulation constitutes a significant therapeutic strategy in reducing the risk for atherosclerosis.
3.1. Accuracy of the Prediction of the Functional Impact of nsSNPs
Out of a total of 3141 SNPs in ABCA1 gene retrieved from dbSNP, we found 233 nsSNPs, 126 sSNPs, 59 mRNA 3′-UTR SNPs, 12 mRNA 5′-UTR SNPs, and 2543 intronic SNPs (Figure 4). Among the 233 nsSNPs, MutPred (RF score > 0.5) predicted 122 (52.36%) as deleterious whereas PolyPhen2 (pph2_prob > 0.5) identified 97 (41.63%) as potentially damaging and damaging. Then, once that MutPred was used to predict the nsSNP disease-association probability, the damaging probability of nsSNPs was validated by PolyPhen2. A total of 80 (34.33%) nsSNPs were found to be deleterious by both methods. Among these 80 deleterious nsSNPs, the 29 (12.44%) targeted (MAF/NA) that resulted with high pathological phenotype (probability > 0.8) are C1477R, W590L, W590S, A1046D, N1611D, M1091T, F2009S, N935H, R2081W, R1068H, N935S, R1068C, D1099Y, D1099N, W1699C, W840R, A937V, I1517R, C1660R, R1680W, P1065S, R1615P, T929I, Y2206D, L1379F, T940M, G1216V, Y2178H, and R1680Q. As shown in Table 2, a good correlation index was obtained between the scores observed from the evolutionary-based approach MutPred and the structural-based approach PolyPhen2. As shown in Figure 5, the overall correlation of the predictions made by both methods is high (~0.57). The majority of mutations classified as pathogenic by PolyPhen2 with the highest score (=1) are also classified as pathogenic by MutPred but within a score range between 0.51 and 1. The prediction accuracy depends not only by limitations of the in silico algorithms such as false positive error and interference of redundant motifs but also by the phenotype data from experimental studies [3].
Figure 4.
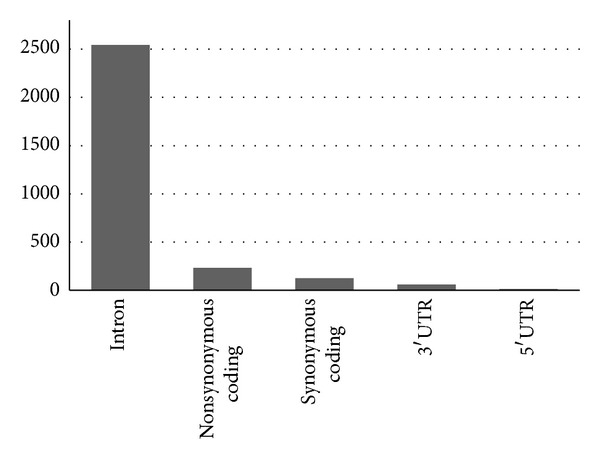
Distribution of ABCA1 nonsynonymous SNPs (nsSNPs), synonymous SNPs (sSNPs), 3′-UTR and 5′-UTR, and intronic SNPs.
Table 2.
Functional effects of nsSNPs for ABCA1 gene (UniProt: O95477) predicted by PolyPhen2 and MutPred. A score >0.5 means that the mutation is classified as damaging by the algorithms. In addition, MutPred algorithm formulates hypotheses about structural and functional impact of the mutation; the most statistically significant hypothesis is reported in this table. The MAP column includes Minor Allele Frequency. The deviation column represents how close the two predicted scores are; a value of 0 corresponding to identical values.
| Mutation | pph2_prob | Polyphen class | RFscore | Mutpred hypothesis | MAF | Deviation |
|---|---|---|---|---|---|---|
| A1010V | 0,104 | neutral | 0,816 | Loss of ubiquitination at K1009 | NA | 0,50346003 |
| A1046D | 0,999 | deleterious | 0,938 | Loss of MoRF binding | NA | 0,04313351 |
| A1182T | 0,001 | neutral | 0,425 | Loss of sheet | 0,00045914 | 0,29981328 |
| A1407T | 0,119 | neutral | 0,277 | Gain of helix | 0,00045914 | 0,11172287 |
| A1631V | 0,401 | neutral | 0,321 | Loss of sheet | NA | 0,05656854 |
| A1670T | 0,997 | deleterious | 0,566 | Gain of glycosylation at A1670 | NA | 0,30476302 |
| A1756T | 0,81 | deleterious | 0,418 | Gain of glycosylation at A1756 | NA | 0,27718586 |
| A2028V | 0,146 | neutral | 0,564 | Gain of methylation at K2023 | 0 | 0,29557064 |
| A255T | 0 | neutral | 0,755 | Loss of helix | NA | 0,53386562 |
| A343V | 0,001 | neutral | 0,227 | Loss of disorder | NA | 0,15980613 |
| A391S | 0,318 | neutral | 0,34 | Gain of disorder | NA | 0,01555635 |
| A594T | 0,02 | neutral | 0,406 | Loss of catalytic residue at A594 | NA | 0,27294322 |
| A697T | 0,416 | neutral | 0,508 | Gain of sheet | 0,00045914 | 0,06505382 |
| A746G | 1 | deleterious | 0,608 | Loss of stability | 0,00045914 | 0,27718586 |
| A779T | 0,411 | neutral | 0,468 | Loss of helix | NA | 0,04030509 |
| A795S | 0,07 | neutral | 0,494 | Loss of helix | NA | 0,29981328 |
| A937V | 0,991 | deleterious | 0,921 | Loss of disorder | NA | 0,04949748 |
| C1477F | 1 | deleterious | 0,496 | Loss of catalytic residue at C1477 | NA | 0,35638182 |
| C1477R | 1 | deleterious | 0,852 | Gain of methylation at C1477 | NA | 0,10465181 |
| C1660R | 0,954 | deleterious | 0,917 | Gain of methylation at C1660 | NA | 0,02616295 |
| C3S | 0,005 | neutral | 0,48 | Gain of disorder | NA | 0,33587572 |
| C887F | 0,002 | neutral | 0,604 | Gain of phosphorylation at S884 | 0,00137741 | 0,42567828 |
| D1018G | 0,992 | deleterious | 0,535 | Loss of catalytic residue at D1018 | NA | 0,3231478 |
| D1099N | 1 | deleterious | 0,906 | Gain of sheet | NA | 0,06646804 |
| D1099Y | 1 | deleterious | 0,946 | Loss of disorder | NA | 0,03818377 |
| D1263E | 0,003 | neutral | 0,214 | Gain of helix | NA | 0,14919953 |
| D1289N | 0,339 | neutral | 0,573 | Gain of sheet | NA | 0,16546299 |
| D1553E | 0,001 | neutral | 0,273 | Loss of helix | NA | 0,19233304 |
| D1706N | 1 | deleterious | 0,611 | Loss of helix | NA | 0,27506454 |
| D2243E | 0,078 | neutral | 0,491 | Gain of relative solvent accessibility | 0,00459137 | 0,2920351 |
| D434E | 0,005 | neutral | 0,413 | Gain of disorder | NA | 0,28849957 |
| D441N | 0 | neutral | 0,348 | Loss of helix | NA | 0,24607316 |
| D444E | 0 | neutral | 0,338 | Loss of helix | NA | 0,23900209 |
| D446E | 0 | neutral | 0,301 | Loss of helix | NA | 0,21283914 |
| D457E | 0,001 | neutral | 0,566 | Loss of helix | NA | 0,39951533 |
| D462G | 0,071 | neutral | 0,555 | Loss of catalytic residue at D462 | NA | 0,34223968 |
| D581E | 0,38 | neutral | 0,346 | Loss of helix | NA | 0,02404163 |
| D677E | 0,158 | neutral | 0,846 | Loss of loop | NA | 0,48648947 |
| D831N | 0,999 | deleterious | 0,589 | Gain of helix | NA | 0,28991378 |
| D917N | 0,1 | neutral | 0,577 | Gain of MoRF binding | NA | 0,33728994 |
| E1005K | 0,026 | neutral | 0,542 | Gain of MoRF binding | NA | 0,3648671 |
| E1172D | 0,003 | neutral | 0,253 | Loss of sheet | 0,0509642 | 0,1767767 |
| E1172G | 0,003 | neutral | 0,281 | Loss of sheet | 0,00091827 | 0,19657569 |
| E1253K | 0,048 | neutral | 0,453 | Gain of ubiquitination at E1253 | NA | 0,28637825 |
| E1916A | 0,027 | neutral | 0,34 | Gain of MoRF binding | 0,00183655 | 0,22132442 |
| E226G | 0,019 | neutral | 0,47 | Loss of stability | NA | 0,31890516 |
| E284K | 0,206 | neutral | 0,842 | Gain of methylation at E284 | NA | 0,44971991 |
| E815G | 0,072 | neutral | 0,361 | Loss of disorder | NA | 0,20435386 |
| E868K | 0,051 | neutral | 0,69 | Gain of methylation at E868 | 0,00275482 | 0,45184123 |
| F1285L | 0,001 | neutral | 0,235 | Loss of loop | NA | 0,16546299 |
| F1573S | 0,065 | neutral | 0,532 | Gain of disorder | NA | 0,33021887 |
| F2009S | 0,811 | deleterious | 0,932 | Gain of disorder | NA | 0,08555992 |
| F2082V | 0,266 | neutral | 0,442 | Gain of MoRF binding | NA | 0,12445079 |
| F2163S | 0,939 | deleterious | 0,727 | Loss of sheet | NA | 0,14990664 |
| F346L | 0 | neutral | 0,239 | Gain of disorder | NA | 0,16899852 |
| F426L | 0,94 | deleterious | 0,549 | Gain of helix | 0,00045914 | 0,27647875 |
| F632C | 0,998 | deleterious | 0,694 | Loss of stability | 0,00045914 | 0,21496046 |
| F950S | 0,995 | deleterious | 0,664 | Gain of disorder | NA | 0,23405235 |
| G1049R | 1 | deleterious | 0,638 | Gain of MoRF binding | NA | 0,25597266 |
| G1216V | 0,951 | deleterious | 0,844 | Loss of ubiquitination at K1214 | NA | 0,07566043 |
| G1321A | 0,936 | deleterious | 0,715 | Gain of catalytic residue at G1321 | NA | 0,15627061 |
| G156V | 0,017 | neutral | 0,389 | Loss of helix | NA | 0,26304372 |
| G2147E | 0,49 | neutral | 0,397 | Loss of catalytic residue at S2148 | NA | 0,06576093 |
| G315W | 0,997 | deleterious | 0,526 | Loss of disorder | NA | 0,33304729 |
| G616V | 0,998 | deleterious | 0,444 | Loss of disorder | NA | 0,39173716 |
| G788W | 1 | deleterious | 0,664 | Loss of catalytic residue at P784 | NA | 0,23758788 |
| G851R | 0,998 | deleterious | 0,71 | Gain of catalytic residue at G851 | NA | 0,20364675 |
| H263D | 0 | neutral | 0,399 | Loss of helix | NA | 0,28213561 |
| H551D | 0,613 | deleterious | 0,468 | Gain of sheet | NA | 0,10253048 |
| I1084V | 0,241 | neutral | 0,511 | Gain of MoRF binding | NA | 0,19091883 |
| I1239V | 0,078 | neutral | 0,376 | Loss of catalytic residue at L1244 | NA | 0,21071782 |
| I1517R | 0,962 | deleterious | 0,917 | Gain of catalytic residue at I1517 | NA | 0,03181981 |
| I1555T | 0,439 | neutral | 0,484 | Loss of stability | NA | 0,03181981 |
| I1911T | 0,006 | neutral | 0,437 | Loss of stability | 0,00045914 | 0,30476302 |
| I35V | 0,066 | neutral | 0,31 | Loss of helix | 0,00045914 | 0,17253406 |
| I546V | 0 | neutral | 0,31 | Gain of MoRF binding | NA | 0,2192031 |
| I560T | 0,829 | deleterious | 0,496 | Loss of stability | NA | 0,23546656 |
| I649F | 0,97 | deleterious | 0,669 | Loss of catalytic residue at I649 | NA | 0,21283914 |
| I659V | 0,034 | neutral | 0,405 | Loss of methylation at K663 | NA | 0,26233662 |
| I883M | 0,002 | neutral | 0,296 | Gain of disorder | 0,365473 | 0,20788939 |
| K1250R | 0,118 | neutral | 0,377 | Loss of methylation at K1250 | 0,00045914 | 0,18314066 |
| K1587R | 0,034 | neutral | 0,377 | Gain of helix | 0,410927 | 0,24253763 |
| K166R | 0 | neutral | 0,251 | Loss of ubiquitination at K166 | NA | 0,1774838 |
| K1761T | 0,038 | neutral | 0,545 | Loss of methylation at K1761 | 0,00045914 | 0,35850314 |
| K1974R | 0,158 | neutral | 0,679 | Loss of ubiquitination at K1974 | NA | 0,36840263 |
| K2040E | 0,243 | neutral | 0,459 | Loss of MoRF binding | 0,00045914 | 0,15273507 |
| K401Q | 0,007 | neutral | 0,322 | Loss of ubiquitination at K401 | NA | 0,22273864 |
| K613E | 0,002 | neutral | 0,428 | Loss of ubiquitination at K613 | 0,00045914 | 0,30122749 |
| K776N | 0,985 | deleterious | 0,568 | Loss of methylation at K776 | 0,00183655 | 0,29486353 |
| L1041V | 0,999 | deleterious | 0,625 | Loss of ubiquitination at K1040 | 0,00137741 | 0,26445794 |
| L1379F | 0,987 | deleterious | 0,866 | Loss of catalytic residue at L1379 | NA | 0,08555992 |
| L1408F | 0,443 | neutral | 0,418 | Loss of stability | 0,00045914 | 0,01767767 |
| L1648P | 0,961 | deleterious | 0,749 | Loss of stability | NA | 0,14990664 |
| L184S | 0,007 | neutral | 0,576 | Loss of sheet | 0,00045914 | 0,40234376 |
| L184W | 0,928 | deleterious | 0,54 | Gain of helix | 0,00045914 | 0,27435743 |
| L2032M | 0,897 | deleterious | 0,688 | Gain of methylation at K2036 | NA | 0,14778532 |
| L2168P | 0,984 | deleterious | 0,693 | Gain of disorder | NA | 0,20576807 |
| L2187Q | 0,995 | deleterious | 0,698 | Gain of disorder | 0,00045914 | 0,21001071 |
| M1012I | 0,008 | neutral | 0,595 | Loss of catalytic residue at V1008 | NA | 0,41507168 |
| M1091T | 0,98 | deleterious | 0,955 | Gain of disorder | NA | 0,01767767 |
| M233V | 0 | neutral | 0,417 | Loss of stability | NA | 0,29486353 |
| M415L | 0,002 | neutral | 0,423 | Gain of relative solvent accessibility | NA | 0,29769196 |
| M674L | 0,534 | deleterious | 0,599 | Loss of catalytic residue at M674 | 0,00045914 | 0,04596194 |
| N1185K | 0,01 | neutral | 0,583 | Gain of MoRF binding | NA | 0,40517219 |
| N1185S | 0,002 | neutral | 0,358 | Gain of helix | NA | 0,25173001 |
| N1406K | 0,006 | neutral | 0,408 | Gain of ubiquitination at N1406 | NA | 0,28425693 |
| N1611D | 0,968 | deleterious | 0,921 | Loss of MoRF binding | NA | 0,03323402 |
| N1800H | 0,758 | deleterious | 0,826 | Loss of stability | NA | 0,04808326 |
| N1800S | 0,241 | neutral | 0,84 | Loss of stability | NA | 0,42355696 |
| N2119Y | 0,96 | deleterious | 0,727 | Loss of MoRF binding | NA | 0,16475588 |
| N820S | 0,001 | neutral | 0,348 | Loss of catalytic residue at N820 | NA | 0,24536605 |
| N935H | 0,996 | deleterious | 0,966 | Gain of disorder | NA | 0,02121321 |
| N935S | 0,831 | deleterious | 0,969 | Gain of disorder | NA | 0,09758074 |
| P1065S | 0,998 | deleterious | 0,906 | Gain of MoRF binding | NA | 0,06505382 |
| P1475S | 1 | deleterious | 0,235 | Gain of phosphorylation at P1475 | NA | 0,54093669 |
| P1878T | 0,019 | neutral | 0,482 | Gain of helix | NA | 0,32739044 |
| P2150L | 0,068 | neutral | 0,826 | Loss of disorder | NA | 0,53598694 |
| P248A | 0 | neutral | 0,207 | Loss of helix | NA | 0,1463711 |
| P250L | 0 | neutral | 0,244 | Loss of glycosylation at P250 | NA | 0,17253406 |
| P641L | 1 | deleterious | 0,603 | Gain of MoRF binding | NA | 0,28072139 |
| P855S | 0,97 | deleterious | 0,59 | Loss of catalytic residue at W856 | NA | 0,26870058 |
| P85L | 0,394 | neutral | 0,618 | Loss of disorder | 0,00045914 | 0,15839192 |
| Q1279K | 0,032 | neutral | 0,715 | Gain of MoRF binding | NA | 0,48295393 |
| Q188K | 0,001 | neutral | 0,543 | Gain of ubiquitination at Q188 | 0 | 0,38325188 |
| Q205E | 0,01 | neutral | 0,255 | Loss of loop | NA | 0,17324116 |
| Q205R | 0 | neutral | 0,279 | Gain of helix | NA | 0,19728279 |
| Q2196H | 0,011 | neutral | 0,798 | Loss of MoRF binding | NA | 0,55649304 |
| Q2210H | 0,984 | deleterious | 0,388 | Gain of catalytic residue at D2214 | NA | 0,42143564 |
| Q597R | 1 | deleterious | 0,71 | Gain of MoRF binding | NA | 0,20506097 |
| Q849R | 0,689 | deleterious | 0,662 | Gain of methylation at Q849 | NA | 0,01909188 |
| R104C | 0,985 | deleterious | 0,589 | Loss of MoRF binding | NA | 0,28001429 |
| R1068C | 1 | deleterious | 0,962 | Loss of MoRF binding | NA | 0,02687006 |
| R1068H | 1 | deleterious | 0,972 | Loss of MoRF binding | NA | 0,01979899 |
| R1082C | 1 | deleterious | 0,694 | Loss of MoRF binding | NA | 0,21637468 |
| R1195Q | 0,044 | neutral | 0,364 | Loss of loop | NA | 0,22627417 |
| R1195W | 0,952 | deleterious | 0,589 | Gain of catalytic residue at A1194 | NA | 0,25667976 |
| R126H | 0 | neutral | 0,467 | Loss of MoRF binding | NA | 0,33021887 |
| R1273L | 0,007 | neutral | 0,35 | Loss of loop | NA | 0,24253763 |
| R1283C | 0,001 | neutral | 0,449 | Gain of ubiquitination at K1278 | NA | 0,31678384 |
| R1341T | 0,016 | neutral | 0,604 | Loss of methylation at R1341 | ≤0,001 | 0,41577879 |
| R1344W | 0,993 | deleterious | 0,64 | Loss of methylation at K1345 | 0,00045914 | 0,24960869 |
| R1417H | 0,837 | deleterious | 0,457 | Loss of phosphorylation at T1416 | 0,00183655 | 0,26870058 |
| R1615P | 0,947 | deleterious | 0,895 | Loss of methylation at R1615 | ≤0,001 | 0,03676955 |
| R1615Q | 0,187 | neutral | 0,679 | Gain of helix | NA | 0,34789654 |
| R1680Q | 0,997 | deleterious | 0,807 | Gain of ubiquitination at K1683 | NA | 0,13435029 |
| R1680W | 1 | deleterious | 0,911 | Loss of disorder | NA | 0,06293251 |
| R1839H | 0,006 | neutral | 0,48 | Loss of phosphorylation at S1842 | 0,00045914 | 0,33516861 |
| R1851Q | 0,015 | neutral | 0,897 | Gain of catalytic residue at R1851 | NA | 0,62366818 |
| R1897W | 0,009 | neutral | 0,408 | Probably damaging | NA | 0,28213561 |
| R1901S | 0,091 | neutral | 0,81 | Gain of phosphorylation at R1901 | NA | 0,50840978 |
| R1925Q | 0,006 | neutral | 0,69 | Loss of MoRF binding | 0,00229568 | 0,48366104 |
| R2004K | 0,911 | deleterious | 0,699 | Gain of ubiquitination at R2004 | NA | 0,14990664 |
| R2030Q | 0,032 | neutral | 0,442 | Loss of MoRF binding | NA | 0,28991378 |
| R2081W | 0,998 | deleterious | 0,936 | Loss of MoRF binding | NA | 0,04384062 |
| R2173Q | 0,008 | neutral | 0,382 | Gain of sheet | NA | 0,26445794 |
| R2189G | 0,142 | neutral | 0,443 | Loss of MoRF binding | NA | 0,21283914 |
| R219K | 0 | neutral | 0,41 | Gain of ubiquitination at R219 | 0,419192 | 0,28991378 |
| R230C | 0 | neutral | 0,68 | Loss of MoRF binding | 0,00734619 | 0,48083261 |
| R306G | 0,04 | neutral | 0,463 | Loss of helix | ≤0,001 | 0,29910617 |
| R306H | 0,899 | deleterious | 0,431 | Loss of helix | NA | 0,33092597 |
| R369H | 0,937 | deleterious | 0,433 | Loss of MoRF binding | NA | 0,35638182 |
| R437Q | 0,088 | neutral | 0,48 | Loss of helix | NA | 0,27718586 |
| R437W | 0,991 | deleterious | 0,562 | Gain of catalytic residue at L435 | NA | 0,30334881 |
| R443K | 0 | neutral | 0,44 | Loss of stability | 0,00045914 | 0,31112698 |
| R496W | 0,209 | neutral | 0,576 | Loss of loop | NA | 0,25950819 |
| R500H | 0,013 | neutral | 0,519 | Loss of helix | NA | 0,35779603 |
| R587W | 1 | deleterious | 0,688 | Loss of disorder | NA | 0,22061732 |
| R638Q | 0,957 | deleterious | 0,664 | Loss of methylation at R638 | NA | 0,20718229 |
| R638W | 0,999 | deleterious | 0,671 | Loss of methylation at R638 | 0,00045914 | 0,23193102 |
| R666Q | 1 | deleterious | 0,437 | Gain of ubiquitination at K668 | NA | 0,39810112 |
| R666W | 1 | deleterious | 0,56 | Gain of catalytic residue at R666 | NA | 0,31112698 |
| R672Q | 0,832 | deleterious | 0,501 | Loss of MoRF binding | NA | 0,23405235 |
| R965C | 0,302 | neutral | 0,608 | Loss of disorder | ≤0,001 | 0,21637468 |
| R999C | 0,127 | neutral | 0,766 | Loss of solvent accessibility | NA | 0,45184123 |
| R999L | 0,208 | neutral | 0,748 | Loss of solvent accessibility | NA | 0,38183766 |
| S107A | 0,002 | neutral | 0,342 | Gain of helix | NA | 0,24041631 |
| S1141Y | 0,805 | deleterious | 0,306 | Loss of phosphorylation at S1141 | NA | 0,35284628 |
| S1157N | 0,022 | neutral | 0,355 | Gain of sheet | 0,00045914 | 0,23546656 |
| S116N | 0,196 | neutral | 0,241 | Loss of phosphorylation at S116 | NA | 0,03181981 |
| S1181F | 0,042 | neutral | 0,542 | Loss of disorder | 0,00045914 | 0,35355339 |
| S1255R | 0,027 | neutral | 0,326 | Loss of glicosilation at S1255 | 0,00183655 | 0,21142493 |
| S1280R | 0,009 | neutral | 0,351 | Gain of sheet | 0,00045914 | 0,24183052 |
| S1376G | 0,007 | neutral | 0,407 | Gain of methylation at K1373 | ≤0,001 | 0,28284271 |
| S139G | 0 | neutral | 0,284 | Gain of helix | NA | 0,20081833 |
| S1506L | 0,996 | deleterious | 0,733 | Loss of helix | NA | 0,18596908 |
| S1536F | 0,998 | deleterious | 0,594 | Loss of disorder | NA | 0,28567114 |
| S1731C | 0,893 | deleterious | 0,526 | Loss of helix | ≤0,001 | 0,25950819 |
| S212T | 0,028 | neutral | 0,375 | Gain of helix | 0,00459137 | 0,24536605 |
| S2182F | 0,578 | deleterious | 0,498 | Loss of disorder | NA | 0,05656854 |
| S2186F | 0,862 | deleterious | 0,458 | Loss of disorder | NA | 0,28567114 |
| S442R | 0,001 | neutral | 0,449 | Loss of helix | NA | 0,31678384 |
| S713G | 1 | deleterious | 0,576 | Loss of stability | NA | 0,29981328 |
| S780N | 0,996 | deleterious | 0,559 | Loss of stability | NA | 0,30900566 |
| T1175M | 0,432 | neutral | 0,331 | Loss of sheet | 0,00091827 | 0,07141779 |
| T1242M | 0,993 | deleterious | 0,62 | Loss of catalytic residue at T1242 | NA | 0,26375083 |
| T1399M | 0,003 | neutral | 0,279 | Loss of glycosylation at T1399 | NA | 0,19516147 |
| T1401I | 0,003 | neutral | 0,368 | Loss of glycosylation at T1401 | NA | 0,25809398 |
| T1427M | 0,005 | neutral | 0,352 | Gain of catalytic residue at T1427 | 0,00091827 | 0,24536605 |
| T2073A | 0,852 | deleterious | 0,657 | Loss of phosphorylation at T2073 | ≤0,001 | 0,13788582 |
| T459P | 0,03 | neutral | 0,534 | Gain of helix | ≤0,001 | 0,35638182 |
| T515A | 0,04 | neutral | 0,449 | Gain of helix | NA | 0,28920667 |
| T774P | 0,003 | neutral | 0,61 | Gain of methylation at K776 | 0,00183655 | 0,42921382 |
| T929I | 0,946 | deleterious | 0,889 | Loss of disorder | NA | 0,04030509 |
| T940M | 1 | deleterious | 0,844 | Loss of methylation at K939 | NA | 0,11030866 |
| V1054I | 0,951 | deleterious | 0,51 | Loss of ubiquitination at K1052 | NA | 0,31183409 |
| V1096I | 0 | neutral | 0,467 | Gain of helix | NA | 0,33021887 |
| V1158I | 0,065 | neutral | 0,226 | Loss of sheet | NA | 0,11384419 |
| V1674I | 0,053 | neutral | 0,376 | Loss of catalytic residue at V1674 | 0,00137741 | 0,22839549 |
| V1704D | 0,752 | deleterious | 0,876 | Loss of helix | NA | 0,08768124 |
| V1806M | 0,98 | deleterious | 0,417 | Gain of ubiquitination at K1804 | NA | 0,39810112 |
| V1858M | 0,305 | neutral | 0,472 | Loss of catalytic residue at V1858 | NA | 0,11808683 |
| V2035M | 0,042 | neutral | 0,38 | Loss of methylation at K2036 | NA | 0,23900209 |
| V2244I | 0,002 | neutral | 0,386 | Gain of sheet | NA | 0,271529 |
| V304M | 0,823 | deleterious | 0,387 | Gain of disorder | NA | 0,30829856 |
| V380I | 0,001 | neutral | 0,353 | Gain of methylation at K376 | NA | 0,24890159 |
| V399A | 0,071 | neutral | 0,563 | Gain of disorder | 0,00183655 | 0,34789654 |
| V408G | 0,101 | neutral | 0,431 | Loss of stability | NA | 0,23334524 |
| V481L | 0,001 | neutral | 0,374 | Gain of sheet | 0,00045914 | 0,26375083 |
| V589I | 0,004 | neutral | 0,334 | Gain of catalytic residue at V589 | 0,00091827 | 0,23334524 |
| V654G | 0,433 | neutral | 0,438 | Loss of stability | NA | 0,00353553 |
| V702A | 0,002 | neutral | 0,408 | Loss of stability | NA | 0,28708535 |
| V724M | 0,833 | deleterious | 0,462 | Loss of catalytic residue at V724 | 0,00045914 | 0,26233662 |
| V771M | 0,034 | neutral | 0,421 | Loss of ubiquitination at K776 | 0,0606061 | 0,27365032 |
| V825I | 0,001 | neutral | 0,401 | Loss of catalytic residue at V825 | 0,128099 | 0,28284271 |
| W1699C | 1 | deleterious | 0,915 | Gain of catalytic residue at L1700 | NA | 0,06010408 |
| W590L | 0,841 | deleterious | 0,888 | Loss of helix | ≤0,001 | 0,03323402 |
| W590S | 0,889 | deleterious | 0,857 | Gain of disorder | ≤0,001 | 0,02262742 |
| W840R | 0,998 | deleterious | 0,923 | Gain of methylation at W840 | NA | 0,05303301 |
| Y1921H | 0,986 | deleterious | 0,752 | Gain of disorder | NA | 0,16546299 |
| Y2178H | 0,982 | deleterious | 0,826 | Gain of disorder | NA | 0,11030866 |
| Y2206D | 0,992 | deleterious | 0,878 | Gain of sheet | NA | 0,08061017 |
| Y482C | 0,03 | neutral | 0,826 | Gain of loop | NA | 0,56285691 |
| Y835H | 0,967 | deleterious | 0,653 | Loss of stability | NA | 0,22203153 |
Figure 5.
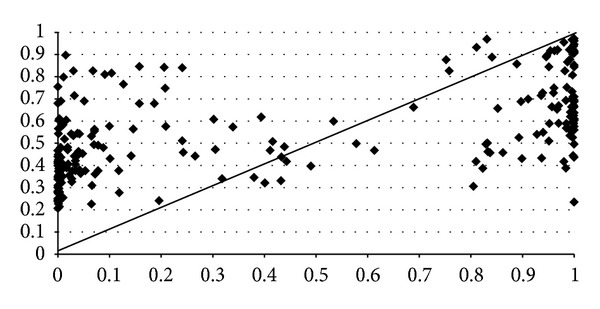
Scatter plot shows the correlation between the predictions made by PolyPhen2 (x-axis) and MutPred (y-axis) for 233 amino acid substitutions on ABCA1. The diagonal line represents a perfect correlation (=1) between both prediction methods for every mutation. The overall correlation of the predictions made by both methods is high (0.57). The majority of mutations classified as pathogenic by PolyPhen2 with the highest score (=1) are also classified as pathogenic by MutPred but within a score range between 0.51 and 1.
Equally important is to consider the incorrect predictions in order to know the limitations of both algorithms and to suggest how they might be improved. Where MutPred predicts P2150L variant as deleterious, PolyPhen2 indicates a benign amino acid exchange. Conversely, MutPred predicted P85L to be probably damaging, while PolyPhen2 indicates it as neutral. Conflicting results were observed for a few other nsSNPs included in Table 2. A total of seventeen deleterious nsSNPs predicted by PolyPhen2 resulted neutral by MutPred. In contrast, forty two deleterious nsSNPs by MutPred result neutral by PolyPhen2. We have observed (Figure 5, Table 2) that some mutational characteristics of nsSNPs such as C1477F, R666Q, P1475S, G616V, Q2210H, V1806M, and V304M show high PolyPhen2 values but very low MutPred scores due, at least in part, to loss or gain of catalytic residues and disorder and gain of ubiquitination and phosphorylation to the protein. On the other hand, some mutational characteristics of nsSNPs included T459P, A2028V, T774P, Q1279K, N1185K, D917N, E1005K, C887F, D1289N, Q188K, D462G, M1012I, R965C, S1181F, A255T, D457E, R496W, R1341T, R1925Q, R230C, L184S, R999L, and K1974R with low PolyPhen2 values but high MutPred scores produce, however loss of solvent accessibility and of disorder, gain of phosphorylation, and both loss and gain of molecular recognition features (MoRFs) binding, loss and gain of methylation, and loss and gain of helix structure. Both, loss and gain of catalytic residues are actively involved in human inherited disease. Also, the small ubiquitin—a 76 residue β-grasp protein—is about 95% conserved from yeast to human. Overall, both gain and loss of a phosphorylation site in a target protein may be important features for predicting cancer-causing mutations and may represent a molecular cause of disease for a number of inherited and somatic mutations. Changes in secondary structure impair large functional alterations, as well as the solvent accessibility degree. Therefore, inaccurate predictions occurred at these sites could be explicated not only for the limited effects of genetic variant but also for gene-environment interactions. Since the MutPred is based on a predicted structure of the protein under study rather than a solved structure as PolyPhen2 and considering the fact that nowadays the ABCA1 protein structure is only partially solved, it makes sense to prioritize the MutPred predictions. This fact was confirmed after evaluating the performance of both methods using a curated nsSNPs dataset with known outcome as a benchmark as shown in Figure 3. We have also observed that mostly, nsSNPs with verified functional effect in experimental studies are correctly predicted as damage variants by MutPred and PolyPhen2 tools. Still, controversial experimental data are obtained for those nsSNPs predicted as neutral by both of these methods.
3.2. Functional Assessment of ABCA1 Variants
Disease-causing variants are under strong selective constraints, which determines if mutation frequency will increase, decrease, or change randomly during evolution. Most alterations are deleterious and so are finally removed during purifying selection. Benign mutations can sweep through the population and become fixed contributing to species differentiation. The ABCA1 gene is highly conserved between species. Human ABCA1 is 95.2% identical to mouse, 85.3% to chicken, 25.5% to drosophila, 21.6% to C. elegans, and 10.2% identical to fugu. In humans, there is an abundance of common nsSNPs that disrupt sites highly conserved across species and likely to be deleterious [50]. The information of nsSNPs can be used to outline the migration patterns of ancient humans and the ancestry of modern humans. Causal nsSNPs in single gene disorders are sufficient to impart large effects. Instead, complex traits are due to a much more complicated system of causative mechanisms that in aggregate increase the probability of disease. Genome-wide association studies reveal common genetic variants effects (common disease/common variant hypothesis) in complex traits. However, where common nsSNPs account for a relatively slight heritability of the traits, rare variants might produce large effects on the phenotype (rare variant/common disease hypothesis). The frequency range includes alleles that are exceptionally rare and even unique to an individual genome to be extremely common. Most deleterious nsSNPs are retained at low-population frequencies due to negative selection. Thus, variants with large effect tend to be rare and those that exert weak effects are more common. It is worthy to note that rare alleles can also have weak effect or no effect. A specific locus may contain numerous rare alleles, so there may be many rare variants with large effect and a few common variants with weak effects. Although it has not yet been possible to determine whether other variables are associated with specific nsSNPs frequencies, variants within metabolic genes are not randomly distributed along the human population but follow diverse ethnic and/or geographic-specific patterns. It has been reported [51] that a significant proportion (~16%) of individuals with low HDL-c from the general population has the rare sequence of 25 variants in ABCA1 gene (Table 2, MAF ≤ 0.01). However, consistent with MutPred and PolyPhen2 only nine of them, N1800H, W590L, S1731C, C1477R, D1706N, R1615P, R638Q, T2073A, and A1670T, are predicted as functionally impaired. Some deleterious mutations from some other genes have reached intermediate to high frequencies. Specifically, the ancestral APOE4 allele, remains higher in populations like Pygmies (0.41), Khoi San (0.37), Papuans (0.37), some Native Americans (0.28), Lapps (0.31) and aborigines of Malaysia (0.24), and Australia (0.26) [52]. The exposure of APOE4 to the current environmental conditions could have rendered it a susceptibility allele for cardiovascular and Alzheimer diseases. However, the prediction for variant within ABCA1 gene indicates lack of harmful alleles to MAF ≥ 0.01. Therefore we have evaluated and contrasted the predictions made for nsSNPs (rare/common) most widely studied for their role in cholesterol pathway by reviewing the effect of the most significant nsSNPs in ABCA1 gene and its pathological consequences.
3.2.1. Accurate Prediction of the Functionally Deleterious nsSNPs in the ABCA1 Gene
The N1800H ABCA1 has been fully characterized showing a complete lack of protein function in terms of cholesterol efflux and HDL production [53, 54]. Unlike the WT (wild-type), which is found at the endoplasmic reticulum and plasma membrane, N1800H is accumulated intracellularly [54]. Even similar physicochemical properties (polar, medium size) of exchanged residues the N1800H nsSNP, located between transmembrane domains [54], is a critical site for protein function. Scores from in silico methods predict the N1800H variant as highly deleterious.
The W590L was never studied, but the W590S ABCA1 variant affecting the same position is extensively known [54]. Distribution of W590S is identical to WT [55] as well as apoA1 binding activity [54–57]; however it shows defective lipid transport [54, 56, 58, 59]. Since multiple alignments often show a leucine residue in this position, it could be assumed that W590L had a similar behavior or even a lower impact than W590S on the protein function. Both W590S and W590L were predicted as deleterious nsSNPs with loss of functionality.
Studies indicate that S1731C variant alters the activity of ABCA1 protein [27, 51, 60]. This allele is present in French-Canadian families with low HDL-c levels [27] but not in subjects with normal [60] or high [51] HDL-c levels. Compared with WT, heterozygous show decreased ~60% the cholesterol efflux activity [27, 51, 61]. Interestingly, some but not all families harboring S1731C also carried the 2144X stop mutation [60] able to produce the most severe effects on HDL-c levels and on cholesterol efflux [62]. These data along with our in silico predictions indicate that conserved S1731C is highly likely to affect protein function.
S1506L, Q597R, and C1477R variants are linked to TD and FHA and found in tumor cancer [54]. Normal function of ABCA1 inhibits tumor growth in human cancer cells [54]. However, although expressed to similar levels as WT, these alleles show deficient cellular cholesterol efflux and HDL production and do not decrease tumor growth [17]. The three are located intracellularly but C1477R is also found in membrane [54], which indicates that membrane localization is essential but not sufficient for apoA1 binding [54, 63]. In fact, ApoA1 binds to ABCA1 protein oligomers but not with monomers [64]. Thus, conformation changes in binding sites might be produced by these nsSNPs found as deleterious by our in silico analysis.
The R587W reaches the cell surface but reduces the apoA1 binding efficiency ~50% [56]. Others studies indicate that this allele is mainly retained intracellularly decreasing cholesterol efflux and apoA1 binding ~75% [54]. Severe HDL deficiency [34] and premature CVD is caused by R587W [65]. This variant is highly conserved during evolution, and the in silico analysis predicts it as strongly damaging and disease-associated. Besides to be related with TD, the R587W as well as W590S variants are linked to AD. As the WT, these mutants significantly reduce Aβ-peptide synthesis ~45% [66], but increase by ~2-fold (R587W) and by 25% (W590S) amyloid precursor protein intracellular domain, a major cytotoxic of AD [66].
The A1046D, localized between conserved motifs [54, 67], shows an intermediate phenotype caused by its limited presence in the plasma membrane. This variant shows reduced apoA1 binding efficacy, poor HDL-c, and folding protein alteration. Both in silico methods predict A1046D as a functional residue with a probability to be deleterious very close to 1.
According to literature, N1611D is associated to probable atherosclerosis [62, 68]. The mutated protein expression was comparable to WT although cholesterol efflux from the cells was markedly reduced. Our theoretical results indicate very high probabilities of this nsSNP being deleterious, which indicate an adverse and potential harmful effect on ABCA1 function.
The M1091T variant exerts a dominant-negative impact on ABCA1 function with severe phenotype observed in subjects carrying this variant [42, 54, 62]. It is retained intracellularly preventing the protein from reaching the membrane [54]. In heterozygous, M1091T is lowered by 50% HDL and inhibits apoA1 binding and cholesterol efflux [54]. From evolutionary path, the inherited residue at this position has been methionine. Among related homologues ABCA2 and ABCA4 share a methionine at this position, while ABCA7 substitutes a leucine. Consistent with this fact, ABCA1 and ABCA7 are functionally divergent, with ABCA7 easing the efflux of phospholipids but not cholesterol [69]. Despite the modest conservation at this position, located in a critical cluster at the C-terminal region, in silico data suggest a severe-negative impact.
The F2009S is conserved between human and mouse, that along with the exchange from large size and aromatic (F) to small size and polar (S) explicates its reduced cholesterol efflux, low HDL-c, and apoA1 levels [70]. The functional effect produced by F2009S variant is consistent with our prediction made by PolyPhen2 and MutPred indicating a deleterious mutant.
The N935S variant is found intracellularly [54] in subjects without risk of premature atherosclerosis but with extremely low levels of HDL and signs of severe dementia and amyloid depositions in the brain [71, 72]. This variant was predicted as deleterious by the used methods.
R1068H mutation is located within the first ATP-binding domain. It is identified in TD homozygous [73]. Since the R1068H mutation is likely to produce a dysfunctional protein, one would expect it to be associated with FHA in the heterozygous state [73]. Residue R1068 is located in an α-helix of the Walker B motif in the NBD, vulnerable to interaction with D1092 and E1093 [74]. Homology modeling of the ABCA1 protein showed that the R1068H mutation disrupts the conformation of NBD. Functional studies of R1068H showed a lack of cholesterol efflux activity due to defective transference to the plasma membrane, mainly caused by impaired oligomerization [74]. The in silico analysis predicts a high possibility for R1068H to be damaging. Besides, a different mutation of this position, R1068C, predicted as a deleterious by our methods, has been reported in a compound heterozygote with almost no HDL [31].
D1099Y is located at possible interaction site and exchanges the medium size and acidic residue to the large and aromatic tyrosine. Surface residues not at defined interfaces are usually preserved. Still, a moderate to highly conserved domain on the surface of the structure includes this nsSNP, which is associated to familial HDL deficiency [70, 75] and predicted as deleterious in our analysis.
The W1699C, located within the transmembrane domain, is accumulated within the cytoplasm and a small proportion reaches the plasma membrane [76]. It introduces a cysteine residue, which stimulates the formation of a new disulphide bridge able to disrupt the ABCA1 protein structure preventing its oligomerization and transference to the plasma membrane. Probably, W1699C retains some residual functions, as shown by the plasma HDL-c levels found in members carrying this mutation which were not as low as might be expected in carriers [76]. In silico analysis with PolyPhen2 and MutPred indicate a deleterious effect of this nsSNP on ABCA1 function.
3.2.2. Controversial Results for Prediction of the Neutral nsSNPs in the ABCA1 Gene
The mutant R1897W that induces a change from basic (R) to aromatic (W) is predicted functionally neutral in this analysis. This variant was identified in the mother and the brother of an FHA patient, who had plasma HDL levels in the lower range of the normal values [77].
Both the D1289N and P2150L variants identified in TD patients are considered as disease causative [42, 78, 79]. Further experimental evidences disagreeing with these results suggest that both could be nonfunctional variants [51, 54, 80]. Indeed, they showed a lipid transport activity, apoA1 binding, and distribution similar to WT [54, 80]. Interestingly, P2150L is only found in patients who also harbor a second variant, the deleterious R587W described above [54]. Besides, TD patients with D1289N variant were homozygous for a second mutation R2081W that could cause the shown pathological phenotype [79]. R2081W is missed at the plasma membrane and instead accumulated intracellularly [54]. Our results suggest that mutations R2081W and R587W are highly deleterious. For D1289N and P2150L variants, PolyPhen2 predicts a neutral impact on protein function contrary to MutPred predictions that indicates a high probability for these mutants to be deleterious. The positions 1289 and 2150 are conserved among all ABCA1 orthologs but with the close-related ABCA7 and ABCA4. Since conservation patterns in ABCA1 protein endure for a relatively short time in evolutionary path, it is hard to determine if the conservation at these positions is due to functional constraint or simply reflects random chance. Along with experimental data, this suggests that R2081W is a major responsible of ABCA1 protein dysfunction found in TD patients.
The rare R219K polymorphism is located on an N-terminal extracellular loop, which mediates ABCA1 protein interaction with apoA1 [39, 56, 58, 59]. Despite high number of case-control studies conducted to investigate the functionality of R219K variant the results have been inconclusive [60, 81–83]. While some reports suggest an association of R219K is with risk of CVD [84], other research indicates a decreased atherosclerosis progression in general population [60, 85]. Conversely, large prospective studies found no association with HDL-c levels or atherosclerosis susceptibility [82, 86]. A meta-analysis indicates that R219K polymorphism is protective against CVD in Asians but not in Caucasians [87]. Unexpectedly, the K219 allele was associated with a decreased risk of myocardial infarction [18, 60, 84]. Also, this variant has effect on triglycerides [60] but not with HDL-c [84] or with apoA1 levels [85]. Otherwise, a study indicates that blood lipid levels do not seem to be R219K dependent [88]. Whether this variant confers major susceptibility to CVD is for clarification. The association of R219K variant to risk of AD has been studied in diverse ethnic groups [18–20, 89, 90]. Although conflicting results were noted, a study observed a protective dependence in delaying the risk of late-onset AD [18]. Equal to other cases, experimental results inconclusive and contradictories result prediction of the R219K polymorphism was predicted to be neutral in our in silico analysis.
Some studies [60, 91–93] but not all [39, 94] indicate that I883M variant severely increases the risk of atherosclerosis and AD [20]. The I883M has been reported as a milder phenotype with a significant reduction of HDL-c and cholesterol efflux (~70% of WT) [51]. In contrast, others studies [28, 60, 82, 83, 88] did not find any difference in lipid levels in I883M carriers. Studies among different healthy people [95, 96] as well as population with T2D [97] correlated the I883M variant with higher HDL-c concentration. Also, a stepwise regression approach identified I883M as one the key predictors of ischemic heart disease, whereas additive effects were found for V771M/I883M and I883M/E1172D pairs [82, 98]. As well, several studies have reported associations between V825I/I883M and increased plasma HDL-c levels [39, 67, 99]. Despite the controversial experimental results on the influence on cholesterol efflux activity observed of this polymorphism, our data predict that the I883M variant is functionally neutral. Interestingly, both alleles are found in the human population and the minor allele, methionine, is likely to be the ancestral allele at this position. Along with the human ABCA1orthologs, murine aligns valine at this position and the chimpanzee sequence aligns methionine. This divergence could explain why a simple conservation-based approach predicts the I883M change as neutral.
The R1851Q variant exchanges the large size and basic arginine (R) residue to medium size and polar glutamine predicted deleterious by MutPred and neutral by PolyPhen2. R1851Q occurs within the extracellular loop proximal to the transmembrane [68, 100]. Heterozygotes states show low HDL-c and apoA1 levels compared with those related to WT protein.
The R230C variant, found in Native American groups but not in European, Asian, or African individuals, has been associated with low levels of HDL-c and apoA1 [101]. These results are confirmed after adjusting for gender, BMI, and waist circumference [102]. Besides, the C230 allele is associated with obesity, metabolic syndrome, and T2D in Mexican population [101]. Still, R230C may have conferred resistance against certain infectious diseases [101]. R230C has been reported as a rare variant causing FHA in an Oji-Cree individual [67]. MutPred predicts a high probability of functional impairment of R230C, while that the PolyPhen2 program predicts the variant as neutral. Other facts that suggest functionally damage are (1) R230C occurs at the first extracellular loop, where TD and FHA mutations are clustered; (2) the arginine at position 230 is conserved between species; and (3) very different nature of residues involved; whereas arginine is basic and hydrophilic, the hydrophobic cysteine is vulnerable to disulfide bond.
The variants, R1901S that induces a change from large size and basic (R) to small size and polar (S); Q2196H that exchanges residues with similar physicochemical property (medium size, polar); and E284K that exchanges a medium size and acidic (E) to large size and basic (K), are predicted to be deleterious by MutPred and neutral by PolyPhen2. The R1901S and Q2196H variants occur within the C-terminal domain, close to the NBD, and E284K was located in the first extracellular loop, all of them associated to FHA [76]. The A594T, I659V, T1512M, and R2004K polymorphisms display different degrees of mislocalization to the plasma membrane and slight impacts on cholesterol efflux [103]. These nsSNPs were identified in low-HDL subjects [29]. The A594T, I659V, and T1512M were predicted to be functionally neutral and the R2004K mutation possibly damaging [29]. Finally, the novel mutation (P85L) in ABCA1 was identified in one family with low HDL but was not detected in over 400 chromosomes of healthy subjects [104]. Our in silico prediction indicated this variant as possible damaging by MutPred and neutral by PolyPhen2.
4. Conclusion and Future Directions
The practice of medicine, including health promotion and disease prevention, is primarily based on phenotype-based approaches. Most of them are proximal phenotypes achieved through biochemical markers. Finding genetic determinants of the phenotypes could not only clarify biological and functional consequence of variants but also might translate and extend to clinical phenotype. This focus would consider the large locus heterogeneity and numerous nongenetic factors to contribute to the phenotype. Since high levels of HDL-c are atheroprotective, there is extensive interest in developing agents that enhance ABCA1 expression and thereby raise plasma HDL-c levels. Amino acid exchange variants are crucial indicators of action sites and effective potential therapeutic approaches. In fact, nsSNPs represent disease modifiers capable of altering drug/nutrient response and potential targets vulnerable to environmental factors.
Evaluation of 233 nsSNPs (rare or common) found in ABCA1 transporter indicates that the rare 29 (12.44%) of them resulted to be highly deleterious with a probability >0.8. From 20 sequence variants found in about 16% of individuals with low HDL cholesterol only nine of them, D1706N, R1615P, W590L, C1477R, N1800H, R638Q, T2073A, A1670T, and S1731C, were predicted by MutPred and PolyPhen2 as functionally impaired. We have observed that mostly nsSNPs with verified functional effect in all experimental studies made are correctly predicted as damage variants by MutPred and PolyPhen2 tools. However, controversial experimental data are obtained for those nsSNPs predicted as neutral by both methods. Presumably clinical phenotype is the result of the additive effects and interactions among multiple alleles with different effect degree. Multiple rare alleles in ABCA1 contribute to plasma HDL-c levels in the general population.
Predicting the phenotypic consequence of nsSNPs using computational algorithms provides a better understanding of genetic differences in susceptibility to diseases and drug/nutrient response. These methods predict whether an amino acid altering mutation is deleterious or disease-causing based on physicochemical properties, population frequency, protein structure, and cross-species conservation. However, computational prediction tools are generally based on machine learning algorithms, which need to be trained before classifying a mutation as either neutral or deleterious. A major obstacle of these approaches is the lack of experimentally validated and impartial data sets. A further complication is that mutations in highly conserved sequences do not always produce phenotypes that are easily noticeable. Besides, knowledge of protein structure is crucial to accurately predict functional nsSNPs and understand their linkage with disease. Severe limitation arises thus when protein 3D-structure is not available as the ABCA1 case. Thus, an accurate, efficient, and generally applicable approach is needed to establish a genotype/phenotype correlation. Whole genome sequencing is likely to become a commodity that could be readily available at a reasonable cost and be easily accommodated into the decision making tree of health care of every individual. The challenging task will be to identify variants that are disease-causing or likely disease-causing and develop strategies to prevent and attenuate the evolving phenotype. Likewise, various complementary studies, genetic and biological, would be necessary to discern the associated alleles from the true disease-causing variants. Moreover a better understanding of genome components, such as functional, large intergenic noncoding RNAs, small noncoding RNAs, and primary transcripts, would be essential. An integrated approach that utilizes genomics, transcriptomic, proteomics, and metabolomic would be expected to facilitate identification and characterization of the mechanisms involved in the pathogenesis of the phenotype.
Acknowledgment
This work has been supported by European Union Structural Funds.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publishing of this paper.
References
- 1.Pennisi E. ENCODE project writes eulogy for junk DNA. Science. 2012;337(6099):1159–1161. doi: 10.1126/science.337.6099.1159. [DOI] [PubMed] [Google Scholar]
- 2.Yates CM, Sternberg MJE. The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein-protein interactions. Journal of Molecular Biology. 2013;425(21):3949–3969. doi: 10.1016/j.jmb.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annual Review of Genomics and Human Genetics. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 4.González-Castejón M, Marín F, Soler-Rivas C, Reglero G, Visioli F, Rodríguez-Casado A. Functional non-synonymous polymorphisms prediction methods: current approaches and future developments. Current Medicinal Chemistry. 2011;18(33):5095–5103. doi: 10.2174/092986711797636081. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Casado A. In silico investigation of functional nsSNPs an approach to rational drug design. Research and Reports in Medicinal Chemistry. 2012;2:31–42. [Google Scholar]
- 6.Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nature Genetics. 2003;33:228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 7.Blair DR, Lyttle CS, Mortensen JM. A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell. 2013;155(1):70–80. doi: 10.1016/j.cell.2013.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oram JF. HDL apolipoproteins and ABCA1 partners in the removal of excess cellular cholesterol. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(5):720–727. doi: 10.1161/01.ATV.0000054662.44688.9A. [DOI] [PubMed] [Google Scholar]
- 9.Santamarina-Fojo S, Remaley AT, Neufeld EB, Brewer HB., Jr. Regulation and intracellular trafficking of the ABCA1 transporter. Journal of Lipid Research. 2001;42(9):1339–1345. [PubMed] [Google Scholar]
- 10.Brunham LR, Kruit JK, Iqbal J, et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. Journal of Clinical Investigation. 2006;116(4):1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Timmins JM, Lee J, Boudyguina E, et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. Journal of Clinical Investigation. 2005;115(5):1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunham LR, Singaraja RR, Duong M, et al. Tissue-specific roles of ABCA1 influence susceptibility to atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(4):548–554. doi: 10.1161/ATVBAHA.108.182303. [DOI] [PubMed] [Google Scholar]
- 13.Brunham LR, Kruit JK, Pape TD, et al. β-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nature Medicine. 2007;13(3):340–347. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Shirk A, Oram JF, Lee SP, Kuver R. Polarized cholesterol and phospholipid efflux in cultured gall-bladder epithelial cells: evidence for an ABCA1-mediated pathway. Biochemical Journal. 2002;364(2):475–484. doi: 10.1042/BJ20011493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McNeish J, Aiello RJ, Guyot D, et al. High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATp-binding cassette transporter-1. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(8):4245–4250. doi: 10.1073/pnas.97.8.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sjoblom T, Jones S, Wood LD. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 17.Smith B, Land H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Reports. 2012;2(3):580–590. doi: 10.1016/j.celrep.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wollmer MA, Streffer JR, Lütjohann D, et al. ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimerߣs disease. Neurobiology of Aging. 2003;24(3):421–426. doi: 10.1016/s0197-4580(02)00094-5. [DOI] [PubMed] [Google Scholar]
- 19.Sundar PD, Feingold E, Minster RL, DeKosky ST, Kamboh MI. Gender-specific association of ATP-binding cassette transporter 1 (ABCA1) polymorphisms with the risk of late-onset Alzheimer’s disease. Neurobiology of Aging. 2007;28(6):856–862. doi: 10.1016/j.neurobiolaging.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Ota M, Fujii T, Nemoto K, et al. A polymorphism of the ABCA1 gene confers susceptibility to schizophrenia and related brain changes. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(8):1877–1883. doi: 10.1016/j.pnpbp.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg MF, Callaghan R, Ford RC, Higgins CF. Structure of the multidrug resistance p-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. The Journal of Biological Chemistry. 1997;272(16):10685–10694. doi: 10.1074/jbc.272.16.10685. [DOI] [PubMed] [Google Scholar]
- 22.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. Journal of Lipid Research. 2001;42(7):1007–1017. [PubMed] [Google Scholar]
- 23.Fitzgerald ML, Mendez AJ, Moore KJ, Andersson LP, Panjeton HA, Freeman MW. ATP-binding cassette transporter A1 Contains an NH2-terminal signal anchor sequence that translocates the protein's first hydrophilic domain to the exoplasmic space. Journal of Biological Chemistry. 2001;276(18):15137–15145. doi: 10.1074/jbc.m100474200. [DOI] [PubMed] [Google Scholar]
- 24.Scheffel F, Demmer U, Warkentin E, Hülsmann A, Schneider E, Ermler U. Structure of the ATPase subunit CysA of the putative sulfate ATP-binding cassette (ABC) transporter from Alicyclobacillus acidocaldarius. FEBS Letters. 2005;579(13):2953–2958. doi: 10.1016/j.febslet.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Reyes CL, Chang G. Structure of the ABC transporter MsbA in complex with ADP.vanadate and lipopolysaccharide. Science. 2005;308(5724):1028–1031. doi: 10.1126/science.1107733. [DOI] [PubMed] [Google Scholar]
- 26.Kiss RS, Kavaslar N, Okuhira K, et al. Genetic etiology of isolated low HDL syndrome: incidence and heterogeneity of efflux defects. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(5):1139–1145. doi: 10.1161/ATVBAHA.106.137646. [DOI] [PubMed] [Google Scholar]
- 27.Alrasadi K, Ruel IL, Marcil M, Genest J. Functional mutations of the ABCA1 gene in subjects of French-Canadian descent with HDL deficiency. Atherosclerosis. 2006;188(2):281–291. doi: 10.1016/j.atherosclerosis.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 28.Mantaring M, Rhyne J, Ho Hong S, Miller M. Genotypic variation in ATP-binding cassette transporter-1 (ABCA1) as contributors to the high and low high-density lipoprotein-cholesterol (HDL-C) phenotype. Translational Research. 2007;149(4):205–210. doi: 10.1016/j.trsl.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 29.Slatter TL, Jones GT, Williams MJA, van Rij AM, McCormick SPA. Novel rare mutations and promoter haplotypes in ABCA1 contribute to low-HDL-C levels. Clinical Genetics. 2008;73(2):179–184. doi: 10.1111/j.1399-0004.2007.00940.x. [DOI] [PubMed] [Google Scholar]
- 30.Chhabria MT, Suhagia BN, Pathik SB. HDL elevation and lipid lowering therapy: current scenario and future perspectives. Frontiers in Cardiovascular Drug Discovery. 2010;1:32–60. doi: 10.2174/157489007782418973. [DOI] [PubMed] [Google Scholar]
- 31.Probst MC, Thumann H, Aslanidis C, et al. Screening for functional sequence variations and mutations in ABCA1. Atherosclerosis. 2004;175(2):269–279. doi: 10.1016/j.atherosclerosis.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 32.Albrecht C, Baynes K, Sardini A, et al. Two novel missense mutations in ABCA1 result in altered trafficking and cause severe autosomal recessive HDL deficiency. Biochimica et Biophysica Acta. 2004;1689(1):47–57. doi: 10.1016/j.bbadis.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Daimon M, Kido T, Baba M, et al. Association of the ABCA1 gene polymorphisms with type 2 DM in a Japanese population. Biochemical and Biophysical Research Communications. 2005;329(1):205–210. doi: 10.1016/j.bbrc.2005.01.119. [DOI] [PubMed] [Google Scholar]
- 34.Lawn RM, Wade DP, Garvin MR, et al. The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway. Journal of Clinical Investigation. 1999;104(8):R25–R31. doi: 10.1172/JCI8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Eckardstein A, Langer C, Engel T, et al. ATP binding cassette transporter ABCA1 modulates the secretion of apolipoprotein E from human monocyte-derived macrophages. The FASEB Journal. 2001;15(9):1555–1561. doi: 10.1096/fj.00-0798com. [DOI] [PubMed] [Google Scholar]
- 36.Hayden MR, Clee SM, Brooks-Wilson A, Genest J, Jr., Attie A, Kastelein JJP. Cholesterol efflux regulatory protein, Tangier disease and familial high density lipoprotein deficiency. Current Opinion in Lipidology. 2000;11(2):117–122. doi: 10.1097/00041433-200004000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Marcil M, Brooks-Wilson A, Clee SM, et al. Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. The Lancet. 1999;354(9187):1341–1346. doi: 10.1016/s0140-6736(99)07026-9. [DOI] [PubMed] [Google Scholar]
- 38.Hobbs HH, Rader DJ. ABC1: connecting yellow tonsils, neuropathy, and very low HDL. The Journal of Clinical Investigation. 1999;104(8):1015–1017. doi: 10.1172/JCI8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frikke-Schmidt R, Nordestgaard BG, Jensen GB, Tybjærg-Hansen A. Genetic variation in ABC transporter A1 contributes to HDL cholesterol in the general population. The Journal of Clinical Investigation. 2004;114(9):1343–1353. doi: 10.1172/JCI20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcil M, Yu L, Krimbou L, et al. Cellular cholesterol transport and efflux in fibroblasts are abnormal in subjects with familial HDL deficiency. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(1):159–169. doi: 10.1161/01.atv.19.1.159. [DOI] [PubMed] [Google Scholar]
- 41.Brousseau ME, Eberhart GP, Dupuis J, et al. Cellular cholesterol efflux in heterozygotes for Tangier disease is markedly reduced and correlates with high density lipoprotein cholesterol concentration and particle size. Journal of Lipid Research. 2000;41(7):1125–1135. [PubMed] [Google Scholar]
- 42.Clee SM, Kastelein JJP, van Dam M, et al. Age and residual cholesterol efflux affect HDL cholesterol levels and coronary artery disease in ABCA1 heterozygotes. The Journal of Clinical Investigation. 2000;106(10):1263–1270. doi: 10.1172/JCI10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitz NF, Cronican AA, Saleem M, et al. Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. Journal of Neuroscience. 2012;32(38):13125–13136. doi: 10.1523/JNEUROSCI.1937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaefer EJ, Zech LA, Schwartz DE, Brewer HB., Jr. Coronary heart disease prevalence and other clinical features in familial high-density lipoprotein deficiency (Tangier disease) Annals of Internal Medicine. 1980;93(2):261–266. doi: 10.7326/0003-4819-93-2-261. [DOI] [PubMed] [Google Scholar]
- 45.Li B, Krishnan VG, Mort ME, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25(21):2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly. 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 49.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Research. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Research. 2009;19(9):1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305(5685):869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 52.Corbo RM, Scacchi R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a “thrifty” allele? Annals of Human Genetics. 1999;63, part 4:301–310. doi: 10.1046/j.1469-1809.1999.6340301.x. [DOI] [PubMed] [Google Scholar]
- 53.Weissglas-Volkov D, Pajukanta P. Genetic causes of high and low serum HDL-cholesterol. Journal of Lipid Research. 2010;51(8):2032–2057. doi: 10.1194/jlr.R004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singaraja RR, Visscher H, James ER, et al. Specific mutations in ABCA1 have discrete effects on ABCA1 function and lipid phenotypes both in vivo and in vitro. Circulation Research. 2006;99(4):389–397. doi: 10.1161/01.RES.0000237920.70451.ad. [DOI] [PubMed] [Google Scholar]
- 55.Nagao K, Tomioka M, Ueda K. Function and regulation of ABCA1—membrane meso-domain organization and reorganization. The FEBS Journal. 2011;278(18):3190–3203. doi: 10.1111/j.1742-4658.2011.08170.x. [DOI] [PubMed] [Google Scholar]
- 56.Fitzgerald ML, Morris AL, Rhee JS, Andersson LP, Mendez AJ, Freeman MW. Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. The Journal of Biological Chemistry. 2002;277(36):33178–33187. doi: 10.1074/jbc.M204996200. [DOI] [PubMed] [Google Scholar]
- 57.Vaughan AM, Tang C, Oram JF. ABCA1 mutants reveal an interdependency between lipid export function, apoA-I binding activity, and Janus kinase 2 activation. Journal of Lipid Research. 2009;50(2):285–292. doi: 10.1194/jlr.M800366-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frikke-Schmidt R, Sing CF, Nordestgaard BG, Tybjærg-Hansen A. Gender- and age-specific contributions of additional DNA sequence variation in the 5′ regulatory region of the APOE gene to prediction of measures of lipid metabolism. Human Genetics. 2004;115(4):331–345. doi: 10.1007/s00439-004-1165-z. [DOI] [PubMed] [Google Scholar]
- 59.Frikke-Schmidt R, Nordestgaard BG, Schnohr P, Tybjærg-Hansen A. Single nucleotide polymorphism in the low-density lipoprotein receptor is associated with a threefold risk of stroke: a case-control and prospective study. European Heart Journal. 2004;25(11):943–951. doi: 10.1016/j.ehj.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 60.Clee SM, Zwinderman AH, Engert JC, et al. Common genetic variation in ABCA1 is associated with altered lipoprotein levels and a modified risk for coronary artery disease. Circulation. 2001;103(9):1198–1205. doi: 10.1161/01.cir.103.9.1198. [DOI] [PubMed] [Google Scholar]
- 61.Reddy MV, Iatan I, Weissglas-Volkov D, et al. Exome sequencing identifies 2 rare variants for low high-density lipoprotein cholesterol in an extended family. Circulation: Cardiovascular Genetics. 2012;5(5):538–546. doi: 10.1161/CIRCGENETICS.112.963264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wellington CL, Yang Y, Zhou S, et al. Truncation mutations in ABCA1 suppress normal upregulation of full-length ABCA1 by 9-cis-retinoic acid and 22-R-hydroxycholesterol. Journal of Lipid Research. 2002;43(11):1939–1949. doi: 10.1194/jlr.m200277-jlr200. [DOI] [PubMed] [Google Scholar]
- 63.Wang S, Gulshan K, Brubaker G, Hazen SL, Smith JD. ABCA1 mediates unfolding of apolipoprotein AI N terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(6):1197–1205. doi: 10.1161/ATVBAHA.112.301195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Denis M, Haidar B, Marcil M, Bouvier M, Krimbou L, Genest J. Characterization of oligomeric human ATP binding cassette transporter A1: potential implications for determining the structure of nascent high density lipoprotein particles. The Journal of Biological Chemistry. 2004;279(40):41529–41536. doi: 10.1074/jbc.M406881200. [DOI] [PubMed] [Google Scholar]
- 65.Bertolini S, Pisciotta L, Seri M, et al. A point mutation in ABC1 gene in a patient with severe premature coronary heart disease and mild clinical phenotype of Tangier disease. Atherosclerosis. 2001;154(3):599–605. doi: 10.1016/s0021-9150(00)00587-6. [DOI] [PubMed] [Google Scholar]
- 66.Kim WS, Hill AF, Fitzgerald ML, Freeman MW, Evin G, Garner B. Wild type and tangier disease ABCA1 mutants modulate cellular amyloid-β production independent of cholesterol efflux activity. Journal of Alzheimer's Disease. 2011;27(2):441–452. doi: 10.3233/JAD-2011-110521. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Burnett JR, Near S, et al. Common and rare ABCA1 variants affecting plasma HDL cholesterol. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(8):1983–1989. doi: 10.1161/01.atv.20.8.1983. [DOI] [PubMed] [Google Scholar]
- 68.Nishida Y, Hirano K, Tsukamoto K, et al. Expression and functional analyses of novel mutations of ATP-binding cassette transporter-1 in Japanese patients with high-density lipoprotein deficiency. Biochemical and Biophysical Research Communications. 2002;290(2):713–721. doi: 10.1006/bbrc.2001.6219. [DOI] [PubMed] [Google Scholar]
- 69.Wang N, Lan D, Gerbod-Giannone M, et al. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. The Journal of Biological Chemistry. 2003;278(44):42906–42912. doi: 10.1074/jbc.M307831200. [DOI] [PubMed] [Google Scholar]
- 70.Ho Hong S, Rhyne J, Zeller K, Miller M. Novel ABCA1 compound variant associated with HDL cholesterol deficiency. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2002;1587(1):60–64. doi: 10.1016/s0925-4439(02)00066-2. [DOI] [PubMed] [Google Scholar]
- 71.Walter M, Kerber S, Fechtrup C, Seedorf U, Breithardt G, Assmann G. Characterization of atherosclerosis in a patient with familial high-density lipoprotein deficiency. Atherosclerosis. 1994;110(2):203–208. doi: 10.1016/0021-9150(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 72.Koldamova RP, Lefterov IM, Staufenbiel M, et al. The liver X receptor ligaun T0901317 decreases amyloid β production in vitro and in a mouse model of Alzheimer's disease. The Journal of Biological Chemistry. 2005;280(6):4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- 73.Slatter TL, Williams MJA, Frikke-Schmidt R, Tybjærg-Hansen A, Morison IM, McCormick SPA. Promoter haplotype of a new ABCA1 mutant influences expression of familial hypoalphalipoproteinemia. Atherosclerosis. 2006;187(2):393–400. doi: 10.1016/j.atherosclerosis.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 74.Suetani RJ, Sorrenson B, Tyndall JDA, Williams MJA, McCormick SPA. Homology modeling and functional testing of an ABCA1 mutation causing Tangier disease. Atherosclerosis. 2011;218(2):404–410. doi: 10.1016/j.atherosclerosis.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 75.Kelly L, Karchin R, Sali A. Protein interactions and disease phenotypes in the ABC transporter superfamily. Proceedings of the Pacific Symposium on Biocomputing; 2007; pp. 51–63. [PubMed] [Google Scholar]
- 76.Pisciotta L, Bocchi L, Candini C, et al. Severe HDL deficiency due to novel defects in the ABCA1 transporter. Journal of Internal Medicine. 2009;265(3):359–372. doi: 10.1111/j.1365-2796.2008.02019.x. [DOI] [PubMed] [Google Scholar]
- 77.Fasano T, Bocchi L, Pisciotta L, Bertolini S, Calandra S. Denaturing high-performance liquid chromatography in the detection of ABCA1 gene mutations in familial HDL deficiency. Journal of Lipid Research. 2005;46(4):817–822. doi: 10.1194/jlr.D400038-JLR200. [DOI] [PubMed] [Google Scholar]
- 78.Brousseau ME, Schaefer EJ, Dupuis J, et al. Novel mutations in the gene encoding ATP-binding cassette 1 in four tangier disease kindreds. Journal of Lipid Research. 2000;41(3):433–441. [PubMed] [Google Scholar]
- 79.Huang W, Moriyama K, Koga T, et al. Novel mutations in ABCA1 gene in Japanese patients with Tangier disease and familial high density lipoprotein deficiency with coronary heart disease. Biochimica et Biophysica Acta. 2001;1537(1):71–78. doi: 10.1016/s0925-4439(01)00058-8. [DOI] [PubMed] [Google Scholar]
- 80.Quazi F, Molday RS. Differential phospholipid substrates and directional transport by ATP binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. The Journal of Biological Chemistry. 2013;288(48):34414–34426. doi: 10.1074/jbc.M113.508812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kolovou V, Kolovou G, Marvaki A, et al. ATP-binding cassette transporter A1 gene polymorphisms and serum lipid levels in young Greek nurses. Lipids in Health and Disease. 2011;10, article 56 doi: 10.1186/1476-511X-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frikke-Schmidt R, Nordestgaard BG, Jensen GB, Steffensen R, Tybjærg-Hansen A. Genetic variation in ABCA1 predicts ischemic heart disease in the general population. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(1):180–186. doi: 10.1161/ATVBAHA.107.153858. [DOI] [PubMed] [Google Scholar]
- 83.Kitjaroentham A, Hananantachai H, Tungtrongchitr A, Pooudong S, Tungtrongchitr R. R219K polymorphism of ATP binding cassette transporter A1 related with low HDL in overweight/obese Thai males. Archives of Medical Research. 2007;38(8):834–838. doi: 10.1016/j.arcmed.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 84.Cenarro A, Artieda M, Castillo S, et al. A common variant in the ABCA1 gene is associated with a lower risk for premature coronary heart disease in familial hypercholesterolaemia. Journal of Medical Genetics. 2003;40(3):163–168. doi: 10.1136/jmg.40.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tregouet D, Ricard S, Nicaud V, et al. In-depth haplotype analysis of ABCA1 gene polymorphisms in relation to plasma ApoA1 levels and myocardial infarction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(4):775–781. doi: 10.1161/01.ATV.0000121573.29550.1a. [DOI] [PubMed] [Google Scholar]
- 86.Morgan TM, Krumholz HM, Lifton RP, Spertus JA. Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. Journal of the American Medical Association. 2007;297(14):1551–1561. doi: 10.1001/jama.297.14.1551. [DOI] [PubMed] [Google Scholar]
- 87.Li Y, Tang K, Zhou K, et al. Quantitative assessment of the effect of ABCA1 R219K polymorphism on the risk of coronary heart disease. Molecular Biology Reports. 2012;39(2):1809–1813. doi: 10.1007/s11033-011-0922-z. [DOI] [PubMed] [Google Scholar]
- 88.Kolovou V, Marvaki A, Karakosta A, et al. Association of gender, ABCA1 gene polymorphisms and lipid profile in Greek young nurses. Lipids in Health and Disease. 2012;11, article 62 doi: 10.1186/1476-511X-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xiao Z, Wang J, Chen W, Wang P, Zeng H. Association studies of several cholesterol-related genes (ABCA1, CETP and LIPC) with serum lipids and risk of Alzheimer's disease. Lipids in Health and Disease. 2012;11, article 163 doi: 10.1186/1476-511X-11-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wavrant-De Vrièze F, Compton D, Womick M, et al. ABCA1 polymorphisms and Alzheimer's disease. Neuroscience Letters. 2007;416(2):180–183. doi: 10.1016/j.neulet.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neufeld EB, Stonik JA, Demosky SJ, Jr., et al. The ABCA1 transporter modulates late endocytic trafficking: insights from the correction of the genetic defect in Tangier disease. The Journal of Biological Chemistry. 2004;279(15):15571–15578. doi: 10.1074/jbc.M314160200. [DOI] [PubMed] [Google Scholar]
- 92.Schaefer EJ, Blum CB, Levy RI, et al. Metabolism of high density lipoprotein apolipoproteins in Tangier disease. The New England Journal of Medicine. 1978;299(17):905–910. doi: 10.1056/NEJM197810262991701. [DOI] [PubMed] [Google Scholar]
- 93.Huang J, Huang W, Li H, et al. Relationship between the I883M polymorphism of ATP-binding cassette transporter 1 gene and cardiovascular disease. Shandong Medical Journal. 2009;9 [Google Scholar]
- 94.Sandhofer A, Iglseder B, Kaser S, Morè E, Paulweber B, Patsch JR. The influence of two variants in the adenosine triphosphate-binding cassette transporter 1 gene on plasma lipids and carotid atherosclerosis. Metabolism. 2008;57(10):1398–1404. doi: 10.1016/j.metabol.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 95.Hodoglugil U, Williamson DW, Huang Y, Mahley RW. Common polymorphisms of ATP binding cassette transporter A1, including a functional promoter polymorphism, associated with plasma high density lipoprotein cholesterol levels in Turks. Atherosclerosis. 2005;183(2):199–212. doi: 10.1016/j.atherosclerosis.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 96.Jensen MK, Pai JK, Mukamal KJ, Overvad K, Rimm EB. Common genetic variation in the ATP-binding cassette transporter A1, plasma lipids, and risk of coronary heart disease. Atherosclerosis. 2007;195(1):e172–e180. doi: 10.1016/j.atherosclerosis.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 97.Porchay-Baldérelli I, Péan F, Emery N, et al. Relationships between common polymorphisms of adenosine triphosphate-binding cassette transporter A1 and high-density lipoprotein cholesterol and coronary heart disease in a population with type 2 diabetes mellitus. Metabolism: Clinical and Experimental. 2009;58(1):74–79. doi: 10.1016/j.metabol.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 98.Brousseau ME, Bodzioch M, Schaefer EJ, et al. Common variants in the gene encoding ATP-binding cassette transporter 1 in men with low HDL cholesterol levels and coronary heart disease. Atherosclerosis. 2001;154(3):607–611. doi: 10.1016/s0021-9150(00)00722-x. [DOI] [PubMed] [Google Scholar]
- 99.Harada T, Imai Y, Nojiri T, et al. A common Ile 823 Met variant of ATP-binding cassette transporter A1 gene (ABCA1) alters high density lipoprotein cholesterol level in Japanese population. Atherosclerosis. 2003;169(1):105–112. doi: 10.1016/s0021-9150(03)00135-7. [DOI] [PubMed] [Google Scholar]
- 100.Schaefer EJ, Anderson DW, Zech LA, et al. Metabolism of high density lipoprotein subfractions and constituents in Tangier disease following the infusion of high density lipoproteins. Journal of Lipid Research. 1981;22(2):217–228. [PubMed] [Google Scholar]
- 101.Villarreal-Molina MT, Aguilar-Salinas CA, Rodríguez-Cruz M, et al. The ATP-binding cassette transporter A1 R230C variant affects HDL cholesterol levels and BMI in the Mexican population: association with obesity and obesity-related comorbidities. Diabetes. 2007;56(7):1881–1887. doi: 10.2337/db06-0905. [DOI] [PubMed] [Google Scholar]
- 102.Aguilar-Salinas CA, Canizales-Quinteros S, Rojas-Martínez R, et al. The non-synonymous Arg230Cys variant (R230C) of the ATP-binding cassette transporter A1 is associated with low HDL cholesterol concentrations in Mexican adults: a population based nation wide study. Atherosclerosis. 2011;216(1):146–150. doi: 10.1016/j.atherosclerosis.2010.10.049. [DOI] [PubMed] [Google Scholar]
- 103.Sorrenson B, Suetani RJ, Williams MJA, et al. Functional rescue of mutant ABCA1 proteins by sodium 4-phenylbutyrate. Journal of Lipid Research. 2013;54(1):55–62. doi: 10.1194/jlr.M027193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hong SH, Rhyne J, Zeller K, Miller M. ABCA1Alabama: a novel variant associated with HDL deficiency and premature coronary artery disease. Atherosclerosis. 2002;164(2):245–250. doi: 10.1016/s0021-9150(02)00106-5. [DOI] [PubMed] [Google Scholar]
- 105.Bungert S, Molday LL, Molday RS. Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. The Journal of Biological Chemistry. 2001;276(26):23539–23546. doi: 10.1074/jbc.M101902200. [DOI] [PubMed] [Google Scholar]