

Abstract
We performed a genome-wide association study on 1,292 individuals with abdominal aortic aneurysms (AAAs) and 30,503 controls from Iceland and The Netherlands, with a follow-up of top markers in up to 3,267 individuals with AAAs and 7,451 controls. The A allele of rs7025486 on 9q33 was found to associate with AAA, with an odds ratio (OR) of 1.21 and P = 4.6 × 10−10. In tests for association with other vascular diseases, we found that rs7025486[A] is associated with early onset myocardial infarction (OR = 1.18, P = 3.1 × 10−5), peripheral arterial disease (OR = 1.14, P = 3.9 × 10−5) and pulmonary embolism (OR = 1.20, P = 0.00030), but not with intracranial aneurysm or ischemic stroke. No association was observed between rs7025486[A] and common risk factors for arterial and venous diseases—that is, smoking, lipid levels, obesity, type 2 diabetes and hypertension. Rs7025486 is located within DAB2IP, which encodes an inhibitor of cell growth and survival.
AAA, defined as an increase in the aortic diameter of ≥50% or an increase in the infrarenal diameter of ≥30 mm (ref. 1), is a common disease with a prevalence of up to 9% in men over 65 years of age2. The main risk factors for the development of AAA include advanced age, male gender, smoking, atherosclerosis and family history1. This disorder is a serious public health problem, accounting for more than 150,000 hospital admissions, 40,000 repair operations3 and 15,000 deaths annually in the United States2. The mainstay of treatment is surveillance and surgery of aneurysms at high risk of rupture, judged primarily by size and growth rate4. Unfortunately, most AAAs are asymptomatic until near-rupture or rupture, a catastrophic event with very high mortality5.
There is a substantial genetic contribution to the risk of AAA. A recent twin study showed an estimated 70% heritability6 and others have shown increased incidence of AAA in first-degree relatives of affected individuals7,8. Several studies have attempted to identify genetic risk variants. Linkage studies have reported two loci that map to chromosomes 19q13 and 4q31 (refs. 9,10); however, predisposing sequence variants in these regions have not been found.
Recently, a common sequence variant on chromosome 9p21 (rs10757278) near CDKN2A and CDKN2B was found to associate with AAA (OR = 1.31, P = 1.2 × 10−12)11, an observation confirmed in independent studies12,13. One AAA genome-wide association study (GWAS) has been published, and this study yielded a locus on 3p12.3, tagged by rs7635818 (OR = 1.33, P = 0.0028)14. However, this finding has yet to be confirmed in independent samples.
We performed a GWAS using 452 Icelandic and 840 Dutch individuals with AAA, and 27,712 Icelandic and 2,791 Dutch controls, genotyped with the Illumina HumanHap370 or HumanHap610 SNP chips. Partial genotype information on an additional 536 Icelanders with AAA not typed with the SNP chips, but closely related to genotyped individuals, was also used in the analysis (Online Methods). We identified 293,677 SNPs that passed quality criteria, and we tested these for association with AAA in the Icelandic and Dutch sample sets separately. The results for the Icelandic sample set were adjusted using the method of genomic control, dividing the χ2 statistic by λg = 1.143, whereas no adjustment was needed for the Dutch sample set (λg = 1). The genome-wide association results for the two populations were combined assuming a fixed-effect model15 (Supplementary Fig. 1).
In the combined analysis of the two discovery sample sets, three correlated SNPs achieved genome-wide significance (P < 1.6 × 10−7). These SNPs are located at the previously associated CDKN2A-CDKN2B locus on 9p21. The associated ORs range from 1.25 to 1.27 (P = 1.6 × 10−7 to P = 1.9 × 10−8; Supplementary Table 1). To search for additional sequence variants associated with AAA, we selected 22 SNPs with P < 5.5 × 10−5 (excluding the 9p21 locus) for genotyping in additional AAA sample sets of European ancestry from Belgium, Canada, New Zealand and the UK (follow-up set 1 with 1,665 individuals with AAA and 1,931 controls). Nineteen of the 22 SNPs were successfully genotyped in all sample sets (Supplementary Table 2). After the results for the two discovery sets and follow-up set 1 were combined, one SNP, rs7025486[A] on 9q33, was associated with AAA at a genome-wide significance level (OR = 1.24, P = 1.8 × 10−9; Supplementary Table 3). Additional rs7025486 genotypes were available for 302 individuals with AAA from New Zealand who were included in follow-up set 1. For further validation, we genotyped rs7025486 in four additional sample sets of European ancestry from Denmark, The Netherlands (Nijmegen) and two US populations in Pittsburgh and Danville, Pennsylvania (follow-up set 2), totaling 1,300 people with AAA and 5,520 controls. The observed effect was weaker in set 2 than in set 1, (OR = 1.11 compared to 1.28, Table 1); however, this difference was not significant (P = 0.079). In the combined analysis of the discovery sets and follow-up sets 1 and 2, rs7025486[A] was associated with AAA with OR = 1.21 and P = 4.6 × 10−10 (Table 1). No significant heterogeneity was observed in the effect estimates between the study populations (Phet = 0.37). Analysis of 520 SNPs in a 630-kb region centered on rs7025486[A], with genotypes imputed using the CEU HapMap data16, did not yield additional SNPs that associated with AAA after adjustment for the number of tests done (data not shown). The previously identified14 3p12.3 sequence variant rs7635818 does not associate with AAA in our two discovery sample sets (P = 0.76).
Table 1. Association of rs7025486[A] with abdominal aortic aneurysm.
| Sample seta | nca | naa | rs7025486[A] | Phetc | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| fcb | fab | OR (95% CI) | P | ||||
| Discovery samples | |||||||
| Icelandd | 27,712 | 452 | 0.298 | 0.347 | 1.25 (1.10–1.42) | 0.00063 | |
| The Netherlands (Utrecht) | 2,791 | 840 | 0.286 | 0.32 | 1.17 (1.04–1.33) | 0.012 | |
| Combined | 30,503 | 1,292 | 1.21 (1.11–1.32) | 2.9 × 10−5 | |||
| Follow-up set 1 | |||||||
| Belgium | 266 | 172 | 0.227 | 0.253 | 1.15 (0.84–1.58) | 0.39 | |
| Canada | 150 | 196 | 0.28 | 0.306 | 1.13 (0.82–1.58) | 0.45 | |
| New Zealand | 848 | 1,144 | 0.226 | 0.273 | 1.28 (1.11–1.49) | 0.00097 | |
| UK | 667 | 455 | 0.216 | 0.278 | 1.40 (1.15–1.70) | 0.0008 | |
| Combined | 1,931 | 1,967 | 1.28 (1.15–1.41) | 3.6 × 10−6 | |||
| Follow-up set 2 | |||||||
| Denmark | 4,380 | 297 | 0.274 | 0.306 | 1.17 (0.98–1.41) | 0.087 | |
| The Netherlands (Nijmegen) | 301 | 147 | 0.287 | 0.248 | 0.82 (0.60–1.12) | 0.22 | |
| US (Denville) | 380 | 758 | 0.253 | 0.278 | 1.14 (0.93–1.39) | 0.2 | |
| US (Pittsburg) | 459 | 98 | 0.245 | 0.281 | 1.20 (0.85–1.70) | 0.3 | |
| Combined | 5,520 | 1,300 | 1.11 (0.98–1.25) | 0.081 | |||
| Follow-up sets 1 and 2 | 7,451 | 3,267 | 1.20 (1.11–1.30) | 3.8 × 10−6 | |||
| All combined | 37,954 | 4,559 | 1.21 (1.14–1.28) | 4.6 × 10−10 | 0.37 | ||
Association of rs7025486[A] with AAA in the two discovery sample sets and in eight replication sample sets
The number of controls nc and affected individuals na
Frequency in controls fc and in affected individuals fa
P value for the test of heterogeneity in the effect estimates.
We included 536 ungenotyped Icelanders with AAA in the analysis and adjusted the P value for relatedness of the Icelandic individuals by dividing the χ2 statistic by the genomic-control factor λg = 1.143.
To evaluate potential epistatic interaction between the new AAA variant rs7025486[A] and the previously established AAA variant rs10757278[G] on 9p21, we compared, for the two loci, the results from a full genotype model including additive and dominant effects to the results when the full model includes epistatic interaction effects (Online Methods). In all the AAA case-control sample sets combined (except for the Danish sample set, for which we did not have rs10757278[G] typed), there was no significant difference between the two models (P = 0.42), indicating the absence of epistatic effects.
rs7025486[A] is within intron 1 of the DAB2IP (encoding the DAB2-interacting protein), also called AIP1 (encoding the ASK1-interacting protein) (Fig. 1). DAB2IP is a member of the RAS-GTPase–activating protein family17. DAB2IP has been shown to suppress cell survival and proliferation through suppression of the PI3K-Act and RAS pathways and to induce apoptosis through activation of ASK1, a member of the JNK and p38 MAPK path-ways18. DAB2IP expression is often found to be downregulated in human cancers19,20. According to gene expression databases in the portal BioGPS, DAB2IP is expressed in many tissues, and our data further demonstrate DAB2IP expression in cardiovascular tissue such as aortic smooth-muscle cells, heart cells and human umbilical vein endothelial cells (HUVECs), with the highest expression by far in HUVECs (Supplementary Fig. 2). The sequence variant rs7025486[A] showed nominally significant correlation with the level of DAB2IP expression in adipose tissue (P = 0.018), mammary artery (P = 0.016) and ascending aorta (P = 0.026); however, the observed correlation was weak, and the direction of the effect was not the same in all tissues (Supplementary Table 4). Many studies have underscored the pivotal role of the PI3K-Akt signaling pathway in the vascular endothelium, which affects endothelial cell proliferation and survival, endothelial cell migration and nitric oxide production21,22. Furthermore, the JNK pathway has been implicated in the pathogenesis of AAA both in mice and in humans23. Lastly, a recent study has also identified DAB2IP as an endogenous inhibitor of VEGFR2-mediated signaling24, an important regulator of angiogenesis control.
Figure 1.
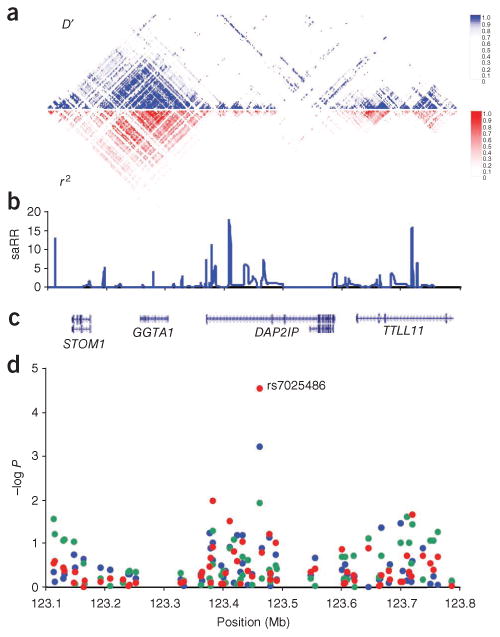
The 9q33 locus. (a) The pairwise correlation structure in a 700-kb interval (123.1–123.8 Mb, NCBI B36) on chromosome 9. The upper plot shows pairwise D' for 490 common SNPs (with minor allele frequency > 5%) from the HapMap (version 22) CEU data set. The lower plot shows the corresponding r2 values. (b) Estimated sex-averaged recombination rates (saRR) in centimorgans per Mb from the HapMap Phase II data30. (c) Location of known genes in the region. (d) Schematic view of the association with AAA in the discovery sample sets from Iceland (blue dots) and The Netherlands (green dots), and in the two sample sets combined (red dots), for all 66 markers tested in the GWAS in the region. All panels use the same horizontal scale shown in d.
The previously discovered AAA variant rs10757278[G] at the CDKN2A-CDKN2B locus on chromosome 9p21 was originally identified as a risk variant for myocardial infarction and coronary artery disease (CAD) in two GWAS25,26. It has since been shown to affect the risk of two distinct aneurysmal diseases, AAA and intracranial aneurysm, with comparable effects. The same variant also affects the risk of peripheral arterial disease (PAD) and large artery and cardiogenic stroke, but with less effect11. In light of the broad vascular effect of the 9p21 AAA risk variant, we tested for association between rs7025486[A] at the DAB2IP locus and other vascular diseases. We tested for association with myocardial infarction in seven sample sets of European ancestry (6,096 individuals with myocardial infarction and 10,757 controls; Table 2), with PAD in seven European sample sets (3,690 individuals with PAD and 12,271 controls; Table 2), with ischemic stroke in Icelandic individuals (2,360 individuals with stroke and 5,863 controls) and with intracranial aneurysm in three European sample sets (1,045 individuals with intracranial aneurysm and 6,418 controls; Supplementary Table 5). We also included four sample sets of venous thromboembolism (VTE) (1,908 individuals with VTE, of which 811 had pulmonary embolism, and 7,055 controls; Table 3), as recent studies have pointed to a link between VTE and cardiovascular diseases27,28. For the Icelandic sample sets we used 5,863 Icelandic population controls without history of vascular diseases who were not included in the control set of 27,712 individuals used for the AAA GWAS. The frequency of the risk variant in this set is 0.292, which is not significantly different from the frequency of 0.298 in the control set used in the genome-wide analysis of AAA (P = 0.29).
Table 2. Association of rs7025486[A] with myocardial infarction and peripheral arterial disease.
| Sample set | nca | naa | rs7025486[A] | Phetc | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| fcb | fab | OR (95% CI) | P | ||||
| Myocardial Infarction | |||||||
| Icelandd | 5,864 | 2,631 | 0.292 | 0.300 | 1.04 (0.97–1.12) | 0.27 | |
| Italy | 383 | 637 | 0.208 | 0.240 | 1.21 (0.97–1.50) | 0.088 | |
| New Zealand | 848 | 529 | 0.226 | 0.249 | 1.13 (0.94–1.36) | 0.18 | |
| US (Atlanta) | 933 | 386 | 0.258 | 0.286 | 1.15 (0.95–1.39) | 0.14 | |
| US (Baltimore) | 1,564 | 183 | 0.267 | 0.290 | 1.12 (0.88–1.43) | 0.35 | |
| US (Durham) | 705 | 1,191 | 0.239 | 0.268 | 1.16 (1.00–1.36) | 0.049 | |
| US (Philadelphia) | 461 | 540 | 0.245 | 0.269 | 1.13 (0.92–1.38) | 0.23 | |
| Combined | 10,758 | 6,097 | 1.09 (1.04–1.15) | 0.0012 | 0.73 | ||
| Early onset myocardial infarction | |||||||
| Icelande | 5,863 | 723 | 0.292 | 0.328 | 1.17 (1.04–1.31) | 0.0071 | |
| Italy | 383 | 194 | 0.208 | 0.245 | 1.24 (0.92–1.66) | 0.15 | |
| New Zealand | 848 | 73 | 0.226 | 0.178 | 0.74 (0.48–1.13) | 0.17 | |
| US (Atlanta) | 933 | 223 | 0.258 | 0.318 | 1.34 (1.07–1.68) | 0.011 | |
| US (Baltimore) | 1,564 | 132 | 0.267 | 0.288 | 1.11 (0.84–1.47) | 0.46 | |
| US (Durham) | 705 | 595 | 0.239 | 0.273 | 1.20 (1.00–1.43) | 0.047 | |
| US (Philadelphia) | 461 | 199 | 0.245 | 0.274 | 1.16 (0.89–1.52) | 0.27 | |
| Combined | 10,757 | 2,139 | 1.18 (1.09–1.27) | 3.1 × 10−5 | 0.44 | ||
| Peripheral artery disease | |||||||
| Icelandf | 5,863 | 1,575 | 0.292 | 0.327 | 1.18 (1.03–1.25) | 0.00022 | |
| Austria | 423 | 458 | 0.249 | 0.266 | 1.09 (0.88–1.35) | 0.42 | |
| Denmark | 4,380 | 455 | 0.274 | 0.297 | 1.12 (0.96–1.30) | 0.14 | |
| Italy | 234 | 168 | 0.216 | 0.193 | 0.87 (0.62–1.23) | 0.44 | |
| New Zealand | 848 | 434 | 0.226 | 0.250 | 1.14 (0.92–1.41) | 0.22 | |
| Sweden | 143 | 204 | 0.252 | 0.277 | 1.14 (0.81–1.60) | 0.46 | |
| US (Denville) | 380 | 396 | 0.253 | 0.274 | 1.12 (0.89–1.40) | 0.34 | |
| Combined | 12,271 | 3,690 | 1.14 (1.07–1.21) | 3.9 × 10−5 | 0.79 | ||
Association of rs7025486[A] with myocardial infarction, early onset myocardial infarction and PAD in several sample sets of European descent.
The number of controls nc and affected individuals na
Frequency in controls fc and in affected individuals fa
P value for the test of heterogeneity in the effect estimates.
We included 4,387 ungenotyped Icelanders with myocardial infarction, 667 ungenotyped Icelanders with early onset myocardial infarction and 1,035 ungenotyped Icelanders with PAD in the analysis, yielding na,eff = 4,077, 925 and 1,933, respectively, and adjusted the P values for relatedness of the Icelandic individuals by dividing the χ2 statistic by the corresponding genomic-control factors λg = 1.426, 1.196 and 1.225.
Table 3. Association of rs7025486[A] with venous thromboembolism and pulmonary embolism.
| Sample set | nca | naa | rs7025486[A] | Phetc | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| fcb | fab | OR (95% CI) | P | ||||
| Venous thromboembolism | |||||||
| Icelandd | 5,863 | 1,019 | 0.298 | 0.321 | 1.14 (1.03–1.25) | 0.011 | |
| Canada 1 | 226 | 187 | 0.257 | 0.246 | 0.95 (0.68–1.30) | 0.73 | |
| Canada 2 | 78 | 27 | 0.263 | 0.278 | 1.08 (0.54–2.16) | 0.83 | |
| Spain | 888 | 675 | 0.177 | 0.196 | 1.13 (0.94–1.36) | 0.18 | |
| Combined | 7,055 | 1,908 | 1.12 (1.07–1.22) | 0.0079 | 0.71 | ||
| Pulmonary embolism | |||||||
| Icelande | 5,863 | 479 | 0.298 | 0.336 | 1.21 (1.07–1.37) | 0.0026 | |
| Canada | |||||||
| ACE | 226 | 72 | 0.257 | 0.271 | 1.08 (0.70–1.65) | 0.74 | |
| PEDS | 78 | 26 | 0.263 | 0.288 | 1.14 (0.56–2.29) | 0.72 | |
| Spain | 888 | 234 | 0.176 | 0.218 | 1.30 (1.01–1.67) | 0.045 | |
| Combined | 7,055 | 811 | 1.20 (1.09–1.32) | 0.00030 | 0.97 | ||
Association of rs7025486[A] with VTE and pulmonary embolism in several sample sets of European descent
The number of controls nc and affected individuals na
Frequency in controls fc and in affected individuals fa
P value for the test of heterogeneity in the effect estimates.
We included 1,626 ungenotyped Icelanders with VTE and 901 ungenotyped Icelanders with pulmonary embolism in the analysis, with na,eff = 1,554 and 775, respectively, and adjusted the P values for relatedness of the Icelandic individuals by dividing the χ2 statistic by the corresponding genomic-control factors λg = 1.319 and 1.207.
Although rs7025486[A] has only a modest effect on all myocardial infarction (OR = 1.09, P = 0.0012), when we restricted the analysis to individuals with early onset myocardial infarction, defined as an event before age 50 years for men and 60 years for women, the risk increased substantially (OR = 1.18, P = 3.1 × 10−5), even after adjustment for the number of vascular diseases tested. Regressing the age of onset of myocardial infarction on the number of copies of rs7025486[A] an individual carries showed that each copy decreases the age of onset by approximately 0.48 years (P = 0.034). This is about half the effect of the 9p21 variant rs10757278[G], for which each copy corresponds to a decrease of about 1 year25.
In addition to early onset myocardial infarction, rs7025486[A] associates with increased risk of PAD (OR = 1.14, P = 3.9 × 10−5) and pulmonary embolism (OR = 1.20, P = 0.00030), whereas the effect was weaker for VTE (OR = 1.12, P = 0.0079). When we repeated the association analysis after excluding individuals with known cases of AAA, CAD or PAD from the group with VTE or pulmonary embolism, the observed effect did not change (Supplementary Table 6), indicating that the association with VTE and pulmonary embolism is not simply the consequence of the association between rs7025486[A] and the other cardiovascular diseases. The association of rs7025486[A] with VTE and pulmonary embolism prompted us to test the association of the 9p21 variant (rs10757278 [G]) with VTE and pulmonary embolism. For the four VTE and pulmonary embolism sample sets tested, no association was observed (VTE, OR = 1.05, P = 0.17; pulmonary embolism, OR = 1.04, P = 0.40).
In contrast to the 9p21 variant, rs7025486[A] does not associate with increased risk of intracranial aneurysm (OR = 1.02, P = 0.77) nor with ischemic stroke or its large artery and cardiogenic stroke subtypes (OR = 1.07, 1.05 and 1.09, and P = 0.097, 0.63 and 0.26, respectively; Supplementary Table 5). It should be noted, however, that the sample sets for these phenotypes were small and thus lacked the power to detect variants with weak effects.
To determine whether the observed association with rs7025486[A] is mediated through known risk factors for arterial or venous diseases, some of which are shared, we tested for association between rs7025486[A] and obesity, type 2 diabetes, hypertension, smoking, smoking quantity and serum lipid levels in up to 38,373 Icelandic individuals typed for the variant. None of the tested risk factors associated with rs7025486[A] after adjustment for the number of risk factors tested (Supplementary Table 7).
Our findings show an association between one sequence variant and several arterial diseases as well as the venous disease pulmonary embolism, and a more modest effect was observed with VTE as a whole. Arterial and venous disorders have traditionally been considered two distinct entities; this belief, however, is challenged by a growing body of evidence27–29. The previous discovery that the 9p21 myocardial infarction and CAD variant is associated with intracranial aneurysm, a non-atherosclerotic disease, was unexpected and provided insight into an involved pathophysiological pathway, shifting attention from atherosclerosis and toward vascular remodeling. The current discovery again connects diseases of the vascular system that have traditionally not been considered strongly related. It will be important to validate this observation in independent studies.
In summary, we have discovered, through GWAS, a common sequence variant within DAB2IP that associates with risk of AAA in populations of European ancestry. This is the second risk variant found to date that consistently associates with AAA in many populations. Our data also show that the same variant affects the risk of other related atherosclerotic diseases, myocardial infarction and PAD. This association is independent of the traditional atherosclerosis risk factors. More notably, the variant also associates with the venous disease pulmonary embolism, suggesting that there is a pathophysiological relationship between these arterial and venous diseases and that the risk variant may affect biochemical pathways common to these conditions, such as thrombosis, inflammation, or vascular remodeling and repair.
Online Methods
Subjects
Detailed information on all case-control sample sets is found in the Supplementary Note. All studies were approved by the relevant institutional review boards or ethics committees, and all participants provided written informed consent.
Association analysis
For case-control association analysis, we used a standard likelihood ratio statistic, implemented in the NEMO software31, to calculate two-sided P values and ORs for each individual allele, assuming a multiplicative model for risk32. We tested the correlation between the risk variant and quantitative traits such as age of onset of myocardial infarction, body-mass index, lipid level or smoking quantity (Supplementary Table 5) by regressing the trait on the number of copies of the risk allele an individual carries. We tested for epistatic interaction between rs7025486[A] and rs10757278[G] by correlating the number of risk alleles an individual carries in multiple regression, adjusting for different sample sets by including corresponding indicator variables. This was done separately for the individuals with AAA and the controls, excluding the Danish sample set, for which rs10757278[G] was not genotyped. In all tables, allelic frequencies are presented for the markers and all reported P values are two-sided.
Familial imputation
For the Icelandic data set, we extended the classical case-control association analysis to include in silico genotypes of affected individuals who were not genotyped but who had genotyped relatives33 among the 40,000 Icelanders (about 13% of all living Icelanders) genotyped with the Illumina SNP chips at deCODE Genetics. For every ungenotyped affected individual, we calculated the probability distribution of the genotypes of his or her relatives, given his or her four possible phased genotypes. In practice, we included only genotypes of the affected individual's parents, children, siblings, half-siblings (and the half-sibling's parents), grandparents, grandchildren (and the grandchildren's parents) and spouses. The contribution of the ungenotyped affected individuals through this familial imputation to the effective sample size of the affected individuals, na,eff, was estimated using the Fisher information.
Genomic control
Some of the individuals in the Icelandic case-control groups are related to each other, causing the χ2 test statistic to have a mean >1 and median >0.455. We estimated the genome-wide inflation factor λg as the average of the 293,677 χ2 statistics to adjust for both relatedness and potential population stratification34. This was done both for the primary AAA genome-wide analysis and for other traits tested in the Icelandic data set, for which a genome-wide analysis was carried out to estimate the inflation factor. The P values presented for the Icelandic case-control groups in Tables 1-3 and in Supplementary Tables 1, 3 and 4 are adjusted using these inflation factors.
Analysis of the New Zealand sample set
Only a portion of the individuals with AAA and controls from New Zealand (594 affected individuals and 527 controls) were directly genotyped for the 19 variants included in Supplementary Table 2 with single SNP genotyping. The rest, and about 54% of individuals typed with single SNP genotyping, were genotyped with the Affymetrix SNP 6.0 array, and either direct or imputed genotypes were available for all the 19 variants. These two data sets were analyzed together in the case-control analysis using the NEMO software. Genotypes not directly genotyped with single SNP genotyping or with the Affymetrix chip were treated as missing, but the imputed genotypes were included in the analysis and used to provide partial information on the missing genotypes. To handle uncertainty with phase and missing genotypes, the maximum-likelihood estimates, likelihood ratios and P values were computed directly for the observed data, and hence the loss of information owing to uncertainty in phase and missing genotypes was automatically captured by the likelihood ratios31.
Meta-analysis
Results from multiple case-control groups, both when we combined the Icelandic and Dutch genome-wide analysis and when we combined the follow-up sets, were combined using a Mantel-Haenszel model15 in which the groups were allowed to have different population frequencies for alleles and genotypes but were assumed to have common relative risks (a fixed-effect model). Heterogeneity in the effect estimate was tested assuming that the estimated ORs for different groups follow a log-normal distribution and using a likelihood ratio χ2 test with degrees of freedom equal to the number of groups compared minus 1.
SNP imputation
Additional SNPs at the 9q33 loci, not genotyped on the Illumina SNP bead-chips, were imputed with the IMPUTE software35 using the HapMap CEU data set (version 22)36 as training set. In all, 520 SNPs in a 630-kb interval that includes DAB2IP and 200 kb upstream and downstream of the gene were imputed for both the Icelandic and the Dutch sample sets. For the Icelandic data set, the analysis was restricted to directly genotyped individuals to avoid complication in combining imputation of SNPs and imputation of ungenotyped individuals.
Epistatic interaction
To test for epistatic interaction between two loci, we considered a general genotype model: log(p/(1 – p) = μ + a1x2 + d1z1 + a2x2 + d2z2 + iaax1x2 + iadx1z2 + idaz1x2 + iddz1z2, where zi = −0.5 and xi = 1 or –1 for the two homozygote genotypes, respectively, and zi = 0.5 and xi = 0 for the heterozygote. μ, a1, d1, a2 and d2 are general genetic parameters corresponding the mean, additive and dominant effects at the two loci, and iaa, ias, ida and idd correspond to epistatic interaction effects37. We performed a four-degrees-of-freedom test of epistatic interaction by comparing the full model to the model without epistatic effects (iaa = ias = ida = idd = 0). When multiple case-control sample sets were tested jointly, indicator variables for each sample set were added to the model.
Supplementary Material
Acknowledgments
We thank the individuals who participated in the study and whose contribution made this work possible. This study was funded in part through grants from the US National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health (R01HL089650-02), and from two projects in the European Community's Seventh Framework Programme (FP7/2007-2013): the Fighting Aneurysmal Disease project grant agreement HEALTH-F2-2008-200647, and ENGAGE project grant agreement HEALTH-F4-2007-201413. The gene expression study in aorta and mammary artery was funded by the Swedish Research Council (12660) and a donation by F. Lundberg. The recruitment of AAA sample sets and controls from Belgium, Canada and Pittsburgh was funded in part by grants from the NHLBI (HL064310 to H.K. and HL044682 to R.E.F.). The Finnish IA sample collection was funded in part by the US National Institute of Neurological Disorders and Stroke (NS034395 to G. Tromp), the American Heart Association, Michigan Affiliate (to G. Tromp) and the University of Kuopio (to A.R.). A.F.B. and Y.M.R. were supported by grants from the Dr. E. Dekker program of the Netherlands Heart Foundation (2009T001 and 2005T014 respectively). New Zealand samples were recruited and genotyped through the Vascular Research Consortium of New Zealand with funding via a program grant from the Health Research Council of New Zealand. Sample collection in Danville, Pennsylvania, was funded by a grant from the American Heart Association (to D.J.C.). UK sample collection was funded partially by grants from the UK Medical Research Council and the British Heart Foundation (to J.T.P.). The recruitment of VTE patients and controls from Spain was funded in part by grants from the Instituto Carlos III (RECAVA RD06/0014/0004, RD06/0014/0039 and 06/0014/0016). DNA isolation of Dutch AAA samples was funded from a grant from the Novartis Foundation for Cardiovascular Excellence. The GeneSTAR Research Program was funded in part by the NHLBI (HL087698 to L.C.B. and HL58625 to D.M.B.). The Danish Study of control subjects was funded by research grants from the Danish Research Council, the Danish Diabetes Association, the Danish Centre for Health Technology Assessment, Novo Nordisk, the Research Foundation of Copenhagen County, the Danish Ministry of Internal Affairs and Health, the Danish Heart Foundation, the Danish Pharmaceutical Association, the Augustinus Foundation, the Ib Henriksen Foundation, the Becket Foundation and the Velux foundation. The Inter99 was initiated by T.J. (principal investigator), K. Borch-Johnsen (co-principal investigator), H. Ibsen and T.F. Thomsen. The steering committee comprises the former two and C. Pisinger.
Footnotes
URL. DAB2IP entry in BioGPS, http://biogps.gnf.org/#goto=genereport&id=153090.
Accession numbers. DAB2IP: MIM 609205.
Note: Supplementary information is available on the Nature Genetics website.
Author Contributions: The study was designed and results interpreted by S.G., A.F.B., H.H., G. Thorleifsson, U.T. and K.S. The first draft was written by S.G., A.F.B., H.H., G. Thorleifsson and U.T. Statistical analysis was performed by G. Thorleifsson, A.K., P.S., G. Masson and G.B.W. Expression analysis was designed and implemented by Adalbjorg Jonasdottir and Aslaug Jonasdottir. Biological material collection and handling was supervised by J.S., S.G., K.P.M. and U.T. Genotyping was supervised by S.G., M.M., J.P.K. and U.T. Collection of vessel wall biopsies and gene expression analyses in aorta and mammary artery were performed by L.F., A.F.-C. and P.E. Those responsible for patient and control ascertainment, recruitment and phenotypic information were S.E.M. and S.G. (AAA and PAD Iceland); A.F.B., M.d.H., J.-P.P.M.d.V., S.E.K., C.J.A.M.Z., S.M.v.S., R.H.G., D.E.G. and J.D.B. (AAA Utrecht, The Netherlands); G.T.J. and A.M.v.R., (AAA, PAD and controls, New Zealand); A.P.M.B. (AAA and controls, Nijmegen, The Netherlands); S.U. and J.S.L. (AAA and PAD, Denmark); N.S., R.L., J.-O.D. and H.K. (AAA and controls, Belgium); H.K. (AAA and controls, Canada); H.K. and R.E.F. (AAA, Pittsburgh); J.R.E. and D.J.C. (AAA, PAD and controls, Danville, Pennsylvania); J.T.P. (AAA, UK); T.M., B.D. and M.H. (PAD and controls, Austria); E.G. and R. Pola (PAD and controls, Italy); B.L. and A.G. (PAD and controls, Sweden); K.A. and G. Thorgeirsson (MI, Iceland); L.C.B. and D.M.B. (MI and controls, Baltimore); G.T.J. and M.J.A.W. (MI, New Zealand); S.H.S. and C.B.G. (MI, Durham, North Carolina); V. Vaccarino, R.S.P., A.M.Z. and A.A.Q. (MI, Atlanta); E.T., G. Malerba, N.M., O.O., P.F.P. and D.G. (MI and controls, Italy); M.P.R. and D.J.R. (MI, Philadelphia); I.O., M.K.M., R. Palmason, V.H. and P.T.O. (VTE, Iceland); N.L., M.F., M.R. and P. W. (VTE and controls, Canada); F.E., J.F., V. Vicente and J.C. (VTE and controls, Spain); S.G., S.S. and E.M.V. (stroke, Iceland); S.G., H.B.M. and E.M.V. (IA, Iceland); G. Tromp and A.R. (IA and controls, Finland); C.W. and Y.M.R. (IA, The Netherlands); G.B.W. and G. Thorleifsson (obesity, Iceland); V.S. (type 2 diabetes, Iceland); H.H. (hypertension, Iceland); G. Thorleifsson and H.S. (smoking, Iceland); T.R., K.K.A., J.d.G., S.H. and L.A.K. (controls, Nijmegen, The Netherlands); V.S., T.H., D.R.W., T.J., N.G. and O.P. (controls, Denmark); H.A. and A.I.L. (controls, Atlanta); and H.S. and D.A.C. (controls, UK). All authors contributed to the final version of the paper.
Competing Financial Interests: The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Crawford CM, Hurtgen-Grace K, Talarico E, Marley J. Abdominal aortic aneurysm: an illustrated narrative review. J Manipulative Physiol Ther. 2003;26:184–195. doi: 10.1016/S0161-4754(02)54111-7. [DOI] [PubMed] [Google Scholar]
- 2.Weintraub NL. Understanding abdominal aortic aneurysm. N Engl J Med. 2009;361:1114–1116. doi: 10.1056/NEJMcibr0905244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Annambhotla S, et al. Recent advances in molecular mechanisms of abdominal aortic aneurysm formation. World J Surg. 2008;32:976–986. doi: 10.1007/s00268-007-9456-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grootenboer N, Bosch JL, Hendriks JM, van Sambeek MR. Epidemiology, aetiology, risk of rupture and treatment of abdominal aortic aneurysms: does sex matter? Eur J Vasc Endovasc Surg. 2009;38:278–284. doi: 10.1016/j.ejvs.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Assar AN, Zarins CK. Ruptured abdominal aortic aneurysm: a surgical emergency with many clinical presentations. Postgrad Med J. 2009;85:268–273. doi: 10.1136/pgmj.2008.074666. [DOI] [PubMed] [Google Scholar]
- 6.Wahlgren CM, Larsson E, Magnusson PK, Hultgren R, Swedenborg J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg. 2010;51:3–7. doi: 10.1016/j.jvs.2009.08.036. discussion 7. [DOI] [PubMed] [Google Scholar]
- 7.Ogata T, et al. J Vasc Surg. Vol. 42. Nova Scotia, Canada: 2005. The lifetime prevalence of abdominal aortic aneurysms among siblings of aneurysm patients is eightfold higher than among siblings of spouses: an analysis of 187 aneurysm families in; pp. 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandford RM, Bown MJ, London NJ, Sayers RD. The genetic basis of abdominal aortic aneurysms: a review. Eur J Vasc Endovasc Surg. 2007;33:381–390. doi: 10.1016/j.ejvs.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 9.Shibamura H, et al. Genome scan for familial abdominal aortic aneurysm using sex and family history as covariates suggests genetic heterogeneity and identifies linkage to chromosome 19q13. Circulation. 2004;109:2103–2108. doi: 10.1161/01.CIR.0000127857.77161.A1. [DOI] [PubMed] [Google Scholar]
- 10.Van Vlijmen-Van Keulen CJ, Rauwerda JA, Pals G. Genome-wide linkage in three Dutch families maps a locus for abdominal aortic aneurysms to chromosome 19q13.3. Eur J Vasc Endovasc Surg. 2005;30:29–35. doi: 10.1016/j.ejvs.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 11.Helgadottir A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–224. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 12.Thompson AR, et al. Sequence variant on 9p21 is associated with the presence of abdominal aortic aneurysm disease but does not have an impact on aneurysmal expansion. Eur J Hum Genet. 2009;17:391–394. doi: 10.1038/ejhg.2008.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bown MJ, et al. Association between the coronary artery disease risk locus on chromosome 9p21.3 and abdominal aortic aneurysm. Circ Cardiovasc Genet. 2008;1:39–42. doi: 10.1161/CIRCGENETICS.108.789727. [DOI] [PubMed] [Google Scholar]
- 14.Elmore JR, et al. Identification of a genetic variant associated with abdominal aortic aneurysms on chromosome 3p12.3 by genome wide association. J Vasc Surg. 2009;49:1525–1531. doi: 10.1016/j.jvs.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 15.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
- 16.Evans DM, Frazer IH, Martin NG. Genetic and environmental causes of variation in basal levels of blood cells. Twin Res. 1999;2:250–257. doi: 10.1375/136905299320565735. [DOI] [PubMed] [Google Scholar]
- 17.Iwashita S, Song SY. RasGAPs: a crucial regulator of extracellular stimuli for homeostasis of cellular functions. Mol Biosyst. 2008;4:213–222. doi: 10.1039/b716357f. [DOI] [PubMed] [Google Scholar]
- 18.Xie D, et al. DAB2IP coordinates both PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proc Natl Acad Sci USA. 2009;106:19878–19883. doi: 10.1073/pnas.0908458106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H, Pong RC, Wang Z, Hsieh JT. Differential regulation of the human gene DAB2IP in normal and malignant prostatic epithelia: cloning and characterization. Genomics. 2002;79:573–581. doi: 10.1006/geno.2002.6739. [DOI] [PubMed] [Google Scholar]
- 20.Qiu GH, et al. Differential expression of hDAB2IPA and hDAB2IPB in normal tissues and promoter methylation of hDAB2IPA in hepatocellular carcinoma. J Hepatol. 2007;46:655–663. doi: 10.1016/j.jhep.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 21.Dimmeler S, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 22.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 23.Yoshimura K, et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, et al. AIP1 functions as an endogenous inhibitor of VEGFR2-mediated signaling and inflammatory angiogenesis in mice. J Clin Invest. 2008;118:3904–3916. doi: 10.1172/JCI36168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helgadottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 26.McPherson R, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prandoni P. Links between arterial and venous disease. J Intern Med. 2007;262:341–350. doi: 10.1111/j.1365-2796.2007.01815.x. [DOI] [PubMed] [Google Scholar]
- 28.Sørensen HT, Horvath-Puho E, Pedersen L, Baron JA, Prandoni P. Venous thromboembolism and subsequent hospitalisation due to acute arterial cardiovascular events: a 20-year cohort study. Lancet. 2007;370:1773–1779. doi: 10.1016/S0140-6736(07)61745-0. [DOI] [PubMed] [Google Scholar]
- 29.Braekkan SK, et al. Family history of myocardial infarction is an independent risk factor for venous thromboembolism: the Tromso study. J Thromb Haemost. 2008;6:1851–1857. doi: 10.1111/j.1538-7836.2008.03102.x. [DOI] [PubMed] [Google Scholar]
- 30.Sabeti PC, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gretarsdottir S, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003;35:131–138. doi: 10.1038/ng1245. [DOI] [PubMed] [Google Scholar]
- 32.Rice JA. Generalized likelihood ratio tests. In: Rice JA, editor. Mathematical Statistics and Data Analysis. Vol. 1. International Thomson Publishing; 1995. pp. 308–310. [Google Scholar]
- 33.Rafnar T, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41:221–227. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 35.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 36.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cordell HJ. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.