

Abstract
Doxorubicin-loaded PEGylated liposomes (commercially available as DOXIL® or Lipodox®) were surface functionalized with a cell-penetrating peptide, octa-arginine (R8). For this purpose, R8-peptide was conjugated to the polyethylene glycol–dioleoyl phosphatidylethanolamine (PEG–DOPE) amphiphilic co-polymer. The resultant R8–PEG–PE conjugate was introduced into the lipid bilayer of liposomes at 2 mol% of total lipid amount via spontaneous micelle-transfer technique. The liposomal modification did not alter the particle size distribution, as measured by Particle Size Analyzer and transmission electron microscopy (TEM). However, surface-associated cationic peptide increased zeta potential of the modified liposomes. R8-functionalized liposomes (R8-Dox-L) markedly increased the intracellular and intratumoral delivery of doxorubicin as measured by flow cytometry and visualizing by confocal laser scanning microscopy (CLSM) compared to unmodified Doxorubicin-loaded PEGylated liposomes (Dox-L). R8-Dox-L delivered loaded Doxorubicin to the nucleus, being released from the endosomes at higher efficiency compared to unmodified liposomes, which had marked entrapment in the endosomes at tested time point of 1 h. The significantly higher accumulation of loaded drug to its site of action for R8-Dox-L resulted in improved cytotoxic activity in vitro (cell viability of 58.5 ± 7% for R8-Dox-L compared to 90.6 ± 2% for Dox-L at Dox dose of 50 μg/mL for 4 h followed by 24 h incubation) and enhanced suppression of tumor growth (348 ± 53 mm3 for R8-Dox-L, compared to 504 ± 54 mm3 for Dox-L treatment) in vivo compared to Dox-L. R8-modification has the potential for broadening the therapeutic window of pegylated liposomal doxorubicin treatment, which could lead to lower non-specific toxicity.
Keywords: Doxorubicin, Liposomes, Octa-arginine, Drug delivery, Apoptosis
1. Introduction
Liposomes, nanosized phospholipid vesicles, have found wide application in drug delivery as pharmaceutical carriers [1]. The advantages of using liposomes as a drug delivery vehicle include the biocompatibility, biodegradability, monodispersity, non-immunogenicity, and high capacity for entrapment of both water-soluble and insoluble drugs. Liposomes and other nanoparticles can preferentially deliver chemotherapeutic drugs to a tumor site due to their small size and long systemic circulation. Nanoparticles take advantage of a tumor’s leaky blood vessels associated with enlarged gaps between endothelial cells that range from 100 to 400 nm that allow increased extravasation of nanosized pharmaceutical drug carriers into a tumor site [2–4]. In spite of the various advantages of liposomal nanocarriers including a size (~100 nm) which allows preferential accumulation of liposomes in a tumor and reduced toxic effects of chemotherapy on normal tissues, the drawbacks including rapid renal clearance and recognition by the reticulo-endothelial system have prompted liposomal system modification to obtain higher therapeutic efficacy. To address the rapid renal clearance issue, the liposomes can be coated with polyethylene glycol (PEG) to make them long-circulating, which allows them to accumulate in the areas with a compromised, leaky vasculature [5–8]. Pegylated liposomal doxorubicin (Dox), commercially available as Doxil®/Caelyx or Lipodox®, is an example of this long-circulating liposome formulation. Dox-loaded pegylated liposomes have been indispensible for application in metastatic, breast, and ovarian cancer [9]. This formulation decreases the toxic effects associated with free Dox administration. However, PEG-modified liposomes have poorer cellular internalization [10]. The therapeutic efficacy of pegylated liposomal Dox was not dramatically increased due mainly to poor cell penetration and slower drug release from the liposomes [9,11]. Therefore, it is obvious that improved intracellular delivery would lead to the increased cytosolic drug concentration required for enhanced drug action.
The emerging concept for improvement of drug action involves efficient intracellular or specific organelle-targeted drug delivery [12–15]. Due to the inability of the nanocarriers to passively cross the cell membrane barrier as do small molecule drugs (which require energy-dependant endocytosis for internalization), cell penetration enhancers have been utilized that accelerate cytoplasmic delivery [16,17]. In this regard, many peptide sequences have been identified that promote translocation of a variety of cargos across the cell membrane [18]. The most commonly used cell-penetrating peptide (CPP) is TATp, which is derived from the 86-mer trans-activating transcriptional activator (TAT protein), encoded by the human immunodeficiency virus type-1 (HIV-1) [16,19–21]. The poly-arginine CPP also has translocation activity very similar to TATp [22]. In particular, synthetic oligoarginine peptides mimic the HIV-1 TAT protein (Tat-(48–60)). The optimum chain length of poly-arginine for promotion of efficient translocation is 8 arginine units (R8) [22].
R8 has been used as a penetration enhancer in some of the previously reported studies. R8-conjugated Smac peptide suppressed the inhibitor of apoptosis (IAP) proteins [23]. Octaarginine and pH-sensitive fusogenic peptide-modified nanoparticles were utilized for liver gene delivery [24]. The Harashima group studied the intracellular fate of R8-modified liposomes [25,26]. Khalil et al. demonstrated that liposomes modified with a high density of R8 were taken up mainly by macropinocytosis [27]. A possible internalization pathway for the R8-modified liposomes with a fusogenic composition was investigated in our recent study [28].
To enhance the therapeutic efficacy by addressing the drawback of poor penetration of cell membranes, we modified a pegylated liposomal doxorubicin, Lipodox®. In this study, for the first time, the possibility of enhanced anticancer activity of a relatively short CPP, R8-modified pegylated liposomal Dox, was investigated.
2. Materials and methods
2.1. Materials
2.1.1. Chemicals
Pegylated Liposomal Doxorubicin (Lipodox®, 2 mg/mL of Dox) was purchased from Sun Pharmaceutical Ind. Ltd. (Gujarat, India). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000](ammonium salt) (PEG2K–DOPE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was from Avanti Polar Lipids (AL, USA). Octa-arginine peptide (RRRRRRRR, M.W. 1267.46 Da) was synthesized by the Tufts University Core Facility (Boston, MA). NPC–PEG2K–NPC was obtained from Laysan Bio (AL, USA). Thiazoyl blue tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St. Louis, MO). Annexin V-Alexa Fluor® 488 conjugate, Annexin-binding buffer 5× concentrate, Hoechst 33342, and Transferrin-Alexa Fluor 488 were purchased from Molecular Probes (Eugene, OR). Para-formaldehyde was from Electron Microscopy Sciences (Hatfield, PA). The Trypan blue solution was purchased from Hyclone (Logan, UT). Fluoromount-G was from Southern Biotech (Birmingham, AL).
2.1.2. Cell lines
The murine mammary carcinoma cell line, 4T1, and normal mouse fibroblast cells, NIH-3T3, were purchased from the American Type Culture Collection (Mansas, VA). Dulbecco’s modified Eagle’s media (DMEM) and heat-inactivated fetal bovine serum (FBS) was obtained from Gibco (Carlsbad, CA). Concentrated penicillin/streptomycin stock solution was from CellGro® (Herndon, VA). All other chemical and solvents were of analytical grade, purchased from Sigma-Aldrich, and used without further purifications.
2.1.3. Animals
Female BALB/c mice (6–8 weeks old) were purchased from Charles River Laboratories, MA, USA. All animal procedures were performed according to an animal care protocol approved by Northeastern University Institutional Animal Care and Use Committee. Mice were housed in groups of 5 at 19–23 °C with a 12 h light-dark cycle and allowed free access to food and water.
2.2. Methods
2.2.1. Synthesis of R8–PEG2K–DOPE
pNP–PEG2K–DOPE, the starting material to synthesize R8–PEG2K–DOPE, was synthesized and purified according to an established procedure with modification [15,29]. Briefly, into the solution of NPC–PEG2K–NPC (1 g, 0.5 mmol) in chloroform, a DOPE (37.2 mg, 0.05 mmol) solution in chloroform and 20 μL of triethylamine were added drop wise. The reaction mixture was stirred overnight at room temperature. On the following day, the reaction mixture was evaporated at reduced pressure using a rotary evaporator and freeze-dried. The dry, crude reaction mixture was dissolved in 0.01 M HCl solution and purified by gel filtration chromatography using a Cl4B column with the 0.01 M HCl as eluent. The purified fractions were freeze-dried, weighed, and dissolved in chloroform to obtain a 10 mg/mL solution for storage at −80 °C. For the synthesis of R8–PEG2K–DOPE, R8 (7.4 mg, 5.86 μmol) and triethylamine (10 μL) dissolved in DMF (200 μL) were added to a mixture of pNP–PEG2K–DOPE (10 mg, 3.9 μmol) in 1 mL chloroform. The reaction mixture was stirred at room temperature overnight. On the following day, the chloroform was evaporated on a rotary evaporator at reduced pressure and freeze-dried. This mixture was dissolved in PBS, pH 8.4, stirred at room temperature for 4 h, and dialyzed against water overnight using a cellulose ester membrane (MWCO. 2000 Da). The dialysate was freeze-dried to obtain a solid white fluffy product which was dissolved in methanol at 5 mg/mL and stored at −80 °C.
2.2.2. Modification of liposomes with PEG2K–DSPE or R8–PEG2K–DOPE
Pegylated Dox-loaded liposomes (Lipodox®) were modified with R8–PEG2K–DOPE by the post-insertion method [30–32]. Lipodox was provided as a sterile, translucent, liposomal dispersion of 10 mL vial. The Doxorubicin HCl was at concentration 2 mg/mL at a pH of 6.5. The liposomes were composed of N-(carbonyl-methoxy-polyethylene glycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt (mPEG–DSPE), 3.19 mg/mL; fully hydrogenated soy phosphatidylcholine (HSPC), 9.58 mg/mL; and cholesterol, 3.19 mg/mL. The inserted co-polymer was 2 mol% of the total lipids in the liposomes. Briefly, Lipodox®) solution (1000 μL) was added to the dry lipid film of PEG2K–DSPE (0.90 mg, 0.45 μmol) or R8–PEG2K–DOPE (1.64 mg, 0.45 μmol). The modified liposomal solutions, Dox-L (modified with PEG2K–DSPE) and R8-Dox-L (modified with R8–PEG2K–DOPE), were vortexed for 5 min and stirred overnight at 4 °C for complete hydration of the lipid film.
Empty liposomes were prepared with the following the lipid composition: fully hydrogenated soy phosphatidylcholine (9.58 mg/mL), cholesterol (3.19 mg/mL), and mPEG–DSPE (3.19 mg/mL). The liposome suspension was stirred overnight at 4 °C with R8–PEG2K–DOPE (2 mol% of total lipids).
2.2.3. Liposomal characterization
The liposomal size and size distribution were measured by dynamic light scattering (DLS) using a Coulter® N4-Plus Submicron Particle Sizer (Coulter Corporation, Miami, FL). Size distribution was confirmed by using transmission electron microscopy (TEM) (Jeol, JEM-1010, Tokyo, Japan). Liposome surface charge was measured with a Zeta Phase Analysis Light Scattering (PALS) Ultrasensitive Zeta Potential Analyzer (Brookhaven Instruments, Holtsville, NY).
2.2.4. Cell culture
Cancer cells (4T1) and normal mouse fibroblasts (NIH-3T3) were grown in DMEM with 2 mM L-glutamine, supplemented with 10% (v/v) heat-inactivated FBS, 100 units/mL penicillin G, and 100 μg/mL streptomycin. Cultures were maintained in a humidified atmosphere at 37 °C with 5% CO2.
2.2.5. Cell association with liposomes by FACS analysis
The cell association with liposomes was assessed by FACS analysis. After the initial passage in 75 cm2 tissue culture flasks (Corning Inc., NY), 4T1 cells (4 × 105/well) were seeded in 6-well tissue culture plates. The following day, the cells were incubated with Dox-L and R8-Dox-L at a Dox concentration of 6 μg/mL in 2 mL of serum-free media for 1 h and 4 h incubation periods. The media were removed; the cells were washed several times, trypsinized, suspended in 1 mL PBS, and then centrifuged at 1000 RPM for 5 min. The cell pellet was suspended in PBS, pH 7.4, before analysis for Dox fluorescence using a BD FACS Caliber flow cytometer. The cells were gated using forward (FSC-H)-versus side-scatter (SSC-H) to exclude debris and dead cells before analysis of 10,000 cell counts.
2.2.6. Cellular internalization by confocal microscopy
Cellular uptake of liposomes was visualized with confocal microscopy. After the initial passage in tissue culture flasks, 4T1 cells (4 × 104 cells) were grown in complete media on circular cover glasses placed in 12-well tissue culture plates in complete media. The following day, cells were incubated with Dox-L or R8-Dox-L at a Dox concentration of 6 μg/mL for 1.5 h in serum-free media. After the incubation period, the cells on the cover-slips were washed with PBS (four times) and Transferrin-Alexa Fluor 488 was added at 10 μg/mL for 20 min, followed by Hoechst 33342 at 5 μg/mL for 5 min, washed thoroughly with PBS, and fixed with 4% para-formaldehyde for 10 min at room temperature. The cover-slips were washed thoroughly with PBS and mounted cell-side down on superfrost microscope slides with fluorescence-free glycerol-based mounting medium (Fluoromount-G; Southern Biotechnology Associates) and viewed with a Zeiss confocal laser scanning microscope (Zeiss LSM 700) equipped with UV (Ex/Em. 385/470 nm), FITC (Ex/Em. 548/595 nm), and a rhodamine filter (Ex/Em. 548/719 nm) for imaging. The LSM picture files were analyzed using Image J software.
2.2.7. Early apoptotic marker determination, Annexin V assay
The procedure for Annexin V labeling was carried out according to the manufacturer’s protocol. Briefly, 4T1 cells were seeded in 12-well plates at 8 × 104/well. After incubation for 4 h with Dox-L and R8-Dox-L at Dox concentration of 15 μg/mL, the cells were incubated for an additional 18 h, trypsinized, washed with cold binding buffer, and re-suspended in binding buffer (200 μL) with or without Annexin V-Alexa Fluor 488 conjugate (15 μL) for 15 min in dark. The Dox-L and R8-Dox-L treated cells without the Annexin V-Alexa Fluor 488 conjugate treatment were used for compensating the interference of Dox fluorescence in FACS study. The cells were diluted with binding buffer to a total volume of 400μL and analyzed immediately by flow cytometry.
2.2.8. MTT assay
The 4T1 cells were seeded in 96 well microplates in phenol red-free DMEM media at a density of 5 × 103 and 3 × 103 cells/well for 24 and 48 h, respectively. On the following day, the cells were incubated with Dox-L or R8-Dox-L at Dox concentrations up to 100 μg/mL for 4 h, the media were removed, and the cells were incubated for an additional 24 and 48 h in fresh complete media. After incubation, the old media were removed and the cells were treated with MTT solution (5 mg/mL) in serum/phenol red-free DMEM for 4 h. At the end of the incubation, cell viability was estimated by the ability of the cells to reduce the yellow dye, MTT, to a purple formazan product. The media were removed and replaced with 100 μL of SDS solution (20%) in 0.01 N HCl for 4 h to dissolve the formazan crystals. The absorbance was read at 570 nm using a microplate reader (Synergy HT multimode microplate reader, Biotek Instrument, Winooski, VT). Blank readings obtained from the treatment well with no cells were subtracted from each reading. For the evaluation of empty liposome cytotoxicity, 4T1 and NIH-3T3 cells were seeded in 96 well plates at 5 × 103 cells/well. On the following day, the cells were treated with empty or R8-modified liposomes at a lipid concentration range of 0–125 μg/mL for 24 h. The cell viability was determined by MTT assay following the above outlined protocol.
2.2.9. In vivo tumor xenograft
A subcutaneous tumor was established in the left flank of the BALB/c mice by inoculating 2 × 106 4T1 cells (suspended in 100 μL PBS). The time for detectable appearance of the tumor was usually 15–20 days. The length and width of the tumor were measured with calipers at 3 days intervals, and the tumor volume was calculated using the formula (width2 × length)/2.
2.2.10. Doxorubicin localization in tumor
The 4T1 tumor-bearing mice with an average tumor volume of 200 mm3 were administered Dox-L or R8-Dox-L i.v at a Dox dose of 10 mg/kg or PBS. The animals were sacrificed with CO2 at 4, 24 h or 72 h after injection and the tumors isolated. The isolated tumors were washed quickly with PBS, pH 7.4, and frozen immediately by immersion in tissue freezing media and stored at −80 °C. Tumor slices (8 μm) of the frozen tumors were cryo-sectioned using a Cryotome Cryostat, mounted on superfrost plus slides, treated with Hoechst 33342 at 5 μg/mL for 5 min, washed and fixed in 4% para-formaldehyde for 10 min at RT, and visualized using fluorescence microscopy.
2.2.11. Tumor volume reduction study
Intravenous treatment with PBS, Dox-L, or R8-Dox-L was started after the tumor volume reached 50–100 mm3 at Dox doses of 0.5 mg/kg. Mouse groups were as follows: (i) PBS (controls); (ii) Dox-L; and (iii) R8-Dox-L (n = 6 per group). Injections via tail vein were once every 3 days. The tumor volume and body weight were recorded at 3 days interval for all tumor-bearing mice for 16 days until the tumor size of control group animals reached 1000 mm3.
2.2.12. TUNEL assay
A TUNEL assay was performed on the frozen tumor sections to measure the pro-apoptotic effect at 24 and 72 h following Dox-L or R8-Dox-L treatment at a Dox dose of 100 μg/mL. Tumor slices (8 μm) were cryo-sectioned, mounted on superfrost plus slides, fixed in 4% para-formaldehyde for 10 min at RT and permeabilized with proteinase K (20 μg/mL) for 15 min at RT. A TUNEL assay was performed on tissue sections using the FragEl™ DNA fragmentation detection Kit following manufacturer’s instructions for frozen sections. The TUNEL positive cells were examined with fluorescence microscope equipped with a FITC-filter. Four random images obtained from two different tumors for each treatment group were analyzed using Spot Advanced software.
2.2.13. Statistical analysis
The data were assessed for statistical significance using Student’s paired t-test and p values calculated with the Graph Pad Prism 5 software (GraphPad Software, Inc; San Diego, CA). All numerical data are expressed as mean ± SD, n = 3–4, from three different experiments. P values ≤0.05 were considered statistically significant. *, **, *** in figures indicated p values ≤0.05, 0.01, and 0.001, respectively.
3. Results
3.1. Physicochemical characterization of liposomes
Average particle sizes of Dox-L and R8-Dox-L at day +1 after synthesis were 90.9 ± 17.4 and 86.7 ± 23.9 nm and at day +15 were 84.4 ± 24.2 and 86.7 ± 23.9 nm, respectively (Supplementary Fig. 1 (S-1)). The result was confirmed using transmission electron microscopy for Dox-L and R8-Dox-L (Fig. 1). The average zeta potential values of Dox-L and R8-Dox-L at day +1 were −35.68 ± 4.31 and −14.24 ± 1.16 nm and at day +15 were −33.0 ± 8.43 and −11.66 ± 4.31 nm, respectively (Fig. S-1).
Fig. 1.

Schematic representation of (A) the synthesis of R8-conjugated PEG–PE and (B) the preparation of the unmodified and R8-modified Dox-loaded liposomes by the micelle-transfer technique. (C) TEM image of control and modified liposomes (left, Dox-L, and right R8-Dox-L). Scale bar. 100 nm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Cellular association of liposomes
A time-dependent (1 h and 4 h) cell association study was performed using flow cytometry and confocal laser scanning microscopy (Fig. 2). The geometric means of Dox fluorescence for Dox-L and R8-Dox-L were 8.77 ± 0.12 and 14.11 ± 0.53 for 1 h and 18.77 ± 0.98 and 37.29 ± 0.90 for 4 h, respectively. The cell association of Dox fluorescence was 2.14-fold and 2.64-fold higher for R8-Dox-L compared to Dox-L at 1 and 4 h exposures, respectively. Confocal microscopy confirmed the FACS analysis result that the cells treated with R8-Dox-L demonstrated higher Dox fluorescence compared to Dox-L at both the time points.
Fig. 2.

Cellular uptake of Dox-L and R8-Dox-L in vitro and in vivo by 4T1 monolayers and tumor xenografts. (A) Representative dot plot of 4T1 cells and comparison of fluorescence due to Dox-labeling by FACS analysis, upon incubation with Dox-L and R8-Dox-L, equivalent to 6 μg/mL of Dox for 1 h and 4 h. (B) Images of 4T1 cells, incubated with Dox-L and R8-Dox-L (Dox dose, 6 μg/mL), visualized by confocal laser scanning microscopy (left). Fluorescence microscopy images of the sections of a 4T1 tumor xenograft, showing the localization of Dox (right) upon the intravenous administration of liposomal Dox at 10 mg/kg. The Dox signal was tracked in the red channel (Ex/Em. 548/595 nm). Nuclei were stained with Hoechst 33342 and tracked in UV (Ex/Em. 385/470 nm). Objective, 40×. Scale bar, 25 μm. The significance of difference between the mean was analyzed by Student’s t-test, ***Indicates p < 0.001.
3.3. Doxorubicin localization in tumor
Dox fluorescence in tumor sections treated with R8-Dox-L was higher at all the time points (4, 24, and 72 h) compared to Dox-L (Fig. 2). The Dox accumulation in tumor was highest after 24 h for both the treatment, which eventually reduced by 72 h as indicated by eventual decreased fluorescence of the Dox signal.
3.4. Assessment of intracellular co-localization
To assess the intracellular localization of Dox delivered by Dox-L or R8-Dox-L, we performed a co-localization analysis using nuclei staining and an endosomal marker. The yellow and purple spots in the merged pictures indicate the overlap of green with red and blue with red fluorescence, respectively. The confocal fluorescence micrographs of Dox-L or R8-Dox-L dosed cells stained for nuclei and endosome visualization are represented in Fig. 3A. R8-Dox-L treatment led to strong accumulation of Dox in the nuclei as indicated by strong purple spots in the merged pictures of column 1 and 2 resulting from the co-localization of red and blue signals, whereas treatment with Dox-L had relatively poor Dox internalization. Merged pictures of column 2 and 3 indicate co-localization of Dox with the endosomes, represented in column 5. Dox-L-treated cells demonstrated higher intensity of yellow signal indicative of endosomal co-localization in the merged picture compared to R8-Dox-L treatment (column 5). A orthogonal section of the merged picture of Dox-L or R8-Dox-L treated cells in a XY plane was used to detail localization behavior (Fig. 3B). Pearson’s coefficient obtained after analyzing the merged pictures by Image J software were plotted to compare co-localization. Pearson’s coefficients for the co-localization of Dox signal with nuclei and endosomal stain were 0.28 ± 0.03/0.96 ± 0.002 and 0.81 ± 0.01/0.50 ± 0.08 for Dox-L/R8-Dox-L treatment, respectively.
Fig. 3.

Intracellular fate of Dox delivered by Dox-L and R8-Dox-L. (A) Confocal microscopy images of 4T1 cells, treated with Dox-L and R8-Dox-L at Dox dose of 6 μg/mL for 1 h: (1) nuclei stained by Hoechst 33342 at 1 μg/mL for 5 min; (2) Dox-signal; (3) endosomes stained by Transferrin-Alexafluor 488 at 10 μg/mL for 20 min; (4) merged picture of 1 and 2, representing fluorescence localization of Dox-signal in nuclei; (5) merged picture of 2 and 3, representing co-localization of Dox in endosomes; (6) merged picture of all the fluorescence. (B) Representative view of the orthogonal sections of the merged image of the cells in the XY plane, obtained by Zeiss LSM image browser software (left). Yellow and purple signals in the merged images indicate the co-localization of the red and green, red, and blue fluorescence, respectively. Analysis of fluorescence intensity-co-localization (Pearson’s coefficient), obtained from the merged pictures (n = 6) from Dox-L and R8-Dox-L-treated cells, by Image J software (Right).
3.5. Assessment of early apoptosis marker
Binding of fluorescently labeled Annexin V to the cell surface bound early apoptotic marker, phosphatidyl serine, was used to detect early apoptosis. The representative histogram and quadrant plot obtained by FACS analysis are shown in Fig. 4A. The histogram showed higher shift in the fluorescence intensity of R8-Dox-L compared to Dox-L. Quadrant statistics demonstrated that 79.8% of the total cell population treated with R8-Dox-L shifted to the lower right quadrant compared to 8.2% with Dox-L treatment due to the higher Annexin V labeling.
Fig. 4.

Effect of Dox-loaded liposomes on cell death and initiation of apoptosis in vitro and in vivo. (A) Representative FACS histogram plot showing the effect of Dox-formulations on initiation of apoptotic activity in vitro (left). The apoptotic marker phosphatidyl serine, expressed on the cell surface due to initiation of apoptosis, was labeled with its ligand, FITC-labeled Annexin V, and the fluorescence of the ligand was tracked to detect the apoptotic cell population by flow cytometry. (B) Assessment of cell viability of 4T1 cells treated with Dox-loaded liposomes at Dox concentration of 0–100 μg/mL for 12, 24, and 48 h. The significance of difference between the mean was analyzed by Student’s t-test, ***, ** indicates p < 0.001 and 0.01, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.6. Assessment of cytotoxicity
The cytotoxic effect of Dox delivered to the cells by Dox-L or R8-Dox-L was evaluated by MTT assay. R8-Dox-L demonstrated higher cytotoxicity compared to Dox-L. No cytotoxicity was observed 12 h after the treatment. However, R8-Dox-L demonstrated 63.6 ± 8.3% and 46.5 ± 5.3% cell viability compared to 85.2 ± 3.4% and 70.4 ± 1.1% for Dox-L at the highest tested Dox dose of 100 μg/mL at 24 and 48 h, respectively. To prove that the cytotoxic effect of R8-modified liposomes is because of the toxicity of Dox, we tested the Dox-free liposomes for their effect on cell viability (Fig. 5). The unmodified empty liposomes and R8-modified liposomes demonstrated no apparent cytotoxicity up to 125 μg/mL for 24 h.
Fig. 5.
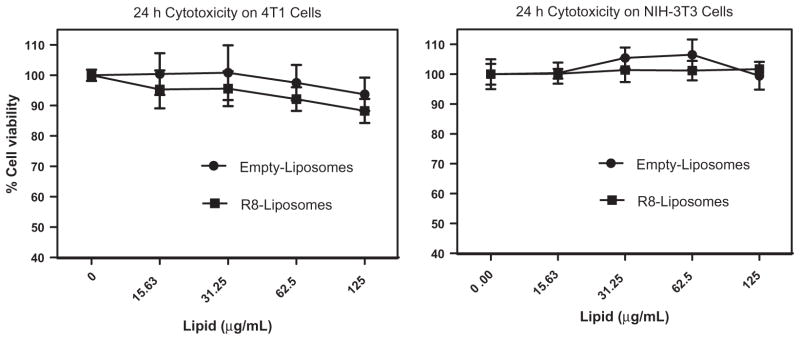
Effect of empty (Dox-free) plain and R8-modified liposomes on cell viability. Cells were incubated with the formulations for 24 h at a Lipid dose range 0–125 μg/mL. The percentage cell viability was determined by considering untreated cells as 100% viable.
3.7. Assessment of in vivo therapeutic efficacy
3.7.1. Tumor volume reduction study
The result from the tumor volumes recorded over the course of tumor growth reduction study demonstrated that the R8-Dox-L treatment suppressed the tumor growth significantly more than the Dox-L treatment (Fig. 6). Over the 16-day period, the tumor volume increased from 64.6 ± 4.3 to 348.0 ± 53.4 mm3 for R8-Dox-L, whereas for Dox-L, the volume increased from 66.6 ± 9.7 to 504.2 ± 54.9 mm3. The body weight was uniform throughout the study indicating no apparent general toxicity (data not shown).
Fig. 6.

(A) Comparison of the efficiency of the formulations in inhibiting tumor growth. The 4T1-tumor-bearing mice were injected i.v with the formulations at a Dox dose of 0.5 mg/kg at alternate days starting from day 1 after tumor volume reached 50–100 mm3 (total eight injections). The tumor volume was measured periodically and plotted as a function of time. Data are expressed as mean ± SEM, n = 6, and *p < 0.05, Student’s t-test. (B). Detection of apoptotic cells in frozen tumor sections treated with Dox-L and R8-Dox-L, determined by TUNEL assay and visualized by fluorescence microscopy. The left panel shows the sections stained with Hoechst 33342 and the right panel shows the TUNEL staining. Magnification ×20 objective.
3.7.2. TUNEL assay
Nuclear DNA fragmentation, a marker of apoptosis, was followed by TUNEL assay of the cryostat sections of the frozen tumors isolated from the mice of PBS, Dox-L, or R8-Dox-L treatment groups at 24 and 72 h time points. The fluorescence microscopy image demonstrated significantly higher apoptotic cell death in tumors treated with R8-Dox-L compared to Dox-L at 24 and 72 h as indicated by green dots (TUNEL positive cells) attributed to FITC-labeled TdT. No green dots indicative of TUNEL positive cells were observed in the tumor sections from the Dox-L treatment group.
4. Discussion
Dox is considered one of the most effective “first line” anticancer chemotherapeutic drugs, since it has been effective against many types of cancer including leukemia, lymphomas, breast, uterine, ovarian, and lung cancers [33,34]. Dox prevents the replication and transcription in rapidly growing cancer cells by intercalating between base pairs of the DNA strands. A positively charged mannose amine in Dox allows efficient binding to the negatively charged nucleic acid phosphate diester groups and the anthraquinone planar ring structure enables Dox to intercalate into the double stranded DNA. In addition, Dox inhibits the enzyme topoisomerase II and prevents the relaxation of super-coiled DNA, another way of blocking transcription and replication. Apart from this, a major biological effect of Dox is that it forms iron-mediated oxygen free radicals that cause oxidative damage to DNA, proteins, and cell membrane lipids. Dox has a high affinity for negatively charged cardiolipin, a mitochondrial membrane lipid. Therefore, this free radical production and the affinity for mitochondrial membrane protein associated with Dox treatment can lead to mitochondria-rich myocardial tissue damage and severe cardiotoxicity.
The unique advantages of using liposomal Dox are the decreased presence of free Dox in the circulation and its preferential accumulation in the tumor area which reduce cardiotoxicity significantly. However, conventional liposomes are attacked by plasma opsonins leading to membrane destabilization. Modified liposomes prepared by “steric stabilization” (pegylation) reduce non-specific interactions with cellular proteins. However, pegylated liposomal system reduces liposomal uptake by the target cells. Suppression of intracellular free Dox concentration is considered a barrier to achievement of the optimum therapeutic efficacy of this otherwise highly potent anticancer drug. To address this issue, various cell penetration enhancers such as certain peptides have been used to increase the cytosolic translocation of the drug loaded nanocarriers. The synthetic, relatively short peptide, octa-arginine (R8) which resembles the peptide sequences of HIV-1TAT peptide, has found application as efficient cell penetrating peptide [23,26,27]. Previous report indicated that the cellular uptake mechanism of high density arginine-rich peptide-modified liposomes was macropinocytosis, whereas low-density R8-modification led to clathrin-mediated endocytosis [27]. The uptake mechanism influenced their intracellular trafficking, resulting in marked difference in the efficacy of loaded cargo. The highly charged basic octa-arginine moiety has demonstrated well established cell transport properties. While investigating intracellular trafficking of D, and L-enantiomer of octa-arginine, it was reported that D-octa-arginine (D-R8) was additionally bound to the nucleolus, whereas both of them translocated in the cytoplasm and labeled nucleus [35]. However, the role of octa-arginine as a promoter for nuclear delivery has to be investigated in detail.
In our study, to improve the cell penetration and therapeutic efficacy of doxorubicin-loaded pegylated liposomes (Doxil®, Lipodox®) (Dox-L), we anchored R8 to the surface by inserting a R8-conjugated PEG–DOPE co-polymer. R8-Dox-L had a significantly higher cellular association compared to Dox-L at both the tested time points of 1 and 4 h. To assess the intracellular localization of Dox delivered by Dox-L and R8-Dox-L, a co-localization analysis using a nuclear stain and an endosomal marker was performed. The treatment with R8-Dox-L generated a marked accumulation of Dox in nuclei as indicated by the bright purple color of the nucleus resulting from the co-localization of blue and red signals. Dox-L failed to deliver Dox to the nuclei at +1 h. However, with Dox-L, a strong green fluorescence was observed in the merged picture of Dox and endosomes indicating that the higher Dox accumulation was confined to the endosomes. Endosomal entrapment of Dox was also observed in R8-Dox-L treatment. However, the extent of the endosomal localization was much lower than observed with Dox-L treatment. With Dox-L treatment, the top, right hand picture of slices through different planes in the orthogonal sections of the merged images indicated that the yellow signal is from inside the cells, whereas cells treated with R8-Dox-L lacked yellow signal. Pearson’s coefficients obtained by analysis of the merged images of the cells indicated that R8-Dox-L treated cells had very high co-localization with nuclei and less endosomal entrapment compared to Dox-L. Such a dramatic improvement in the efficacy of the modified nanocarrier represents a significant step for modification of pegylated liposomal Dox.
The clear improvement in the intracellular delivery efficiency was expected to lead to a more efficient anti-tumor drug action. The ability of the formulations to induce apoptosis by detection of cell surface-associated phosphatidyl serine was evaluated. The mechanism underlying the initiation of apoptosis involves the translocation of phosphatidylserine receptor to the outer cell surface, which binds to its ligand, annexin V. Apoptosis is detected and quantified by the degree of labeling of the cells with the Annexin V, Alexa Fluor® 488 conjugate. The treatment with R8-Dox-L induced significantly higher apoptosis in vitro and in vivo, under specified conditions than with Dox-L treatment. The cytotoxic ability of Dox-L due to the effect of R8-modification was also investigated. The data suggest that the treatment with R8-Dox-L dramatically increases the Dox concentration in the cell, which gives rise to higher cytotoxicity than Dox-L. The cytotoxic effect of R8-modified liposomes was not because of the toxicity of the liposomal carriers. The formulations were non-toxic at a lipid dose range of 0–125 μg/mL for 24 h. In vitro tumor growth inhibition study indicated higher retardation of tumor growth rate by R8-Dox-L treatment than by Dox-L suggesting that the modification on the nanocarrier surface allowed R8-Dox-L to deliver more Dox in the tumor cells resulting in higher therapeutic efficacy compared to Dox-L. The TUNEL assay proved that the R8-Dox-L treatment induced higher apoptosis than Dox-L treatment at specified time points.
5. Conclusion
We surface-modified PEGylated Dox-loaded liposomes, Lipodox®, with a relatively short synthetic cell-penetrating peptide, octa-arginine (R8), using R8–PEG–DOPE. The modification resulted in significant increase in Dox delivery to the cells compared to unmodified Dox liposomes. The R8-modified liposomes delivered more Dox to the cytoplasm which eventually gets accumulated in the nuclei, the site of Dox action, with less Dox entrapped in the endosomes. The higher intracellular Dox delivery led to more pronounced in vitro cancer cell apoptosis, greater cytotoxicity, and the suppression of tumor growth in mice. Further modifications could be performed to shield cell penetrating peptides in the systemic circulation to achieve a higher in vivo anti-tumor efficacy [32]. However, this simple surface modification of liposomal Dox addresses the issue of poor penetration of PEGylated liposomes and provides a method that can significantly improve its therapeutic efficacy. Such a modified liposomal Doxorubicin formulation could have a significant impact in cancer chemotherapy.
Supplementary Material
Acknowledgments
This work was supported by the NIH Grants RO1 CA121838 and RO1 CA 128486 to Vladimir P. Torchilin. We also thank Dr. William Hartner for his helpful advice in editing the manuscript.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ejpb.2012.12.021.
References
- 1.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 2.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42:419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 3.Gabizon AA. Selective tumor localization and improved therapeutic index of anthracyclines encapsulated in long-circulating liposomes. Cancer Res. 1992;52:891–896. [PubMed] [Google Scholar]
- 4.Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta. 1991;1066:29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- 5.Torchilin VP, Klibanov AL, Huang L, O’Donnell S, Nossiff ND, Khaw BA. Targeted accumulation of polyethylene glycol-coated immunoliposomes in infarcted rabbit myocardium. FASEB J. 1992;6:2716–2719. doi: 10.1096/fasebj.6.9.1612296. [DOI] [PubMed] [Google Scholar]
- 6.Torchilin VP, Narula J, Halpern E, Khaw BA. Poly(ethylene glycol)-coated anti-cardiac myosin immunoliposomes: factors influencing targeted accumulation in the infarcted myocardium. Biochim Biophys Acta. 1996;1279:75–83. doi: 10.1016/0005-2736(95)00248-0. [DOI] [PubMed] [Google Scholar]
- 7.Torchilin VP, Omelyanenko VG, Papisov MI, Bogdanov AA, Jr, Trubetskoy VS, Herron JN, Gentry CA. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta. 1994;1195:11–20. doi: 10.1016/0005-2736(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 8.Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010:3–53. doi: 10.1007/978-3-642-00477-3_1. [DOI] [PubMed] [Google Scholar]
- 9.Judson I, Radford JA, Harris M, Blay JY, van Hoesel Q, le Cesne A, van Oosterom AT, Clemons MJ, Kamby C, Hermans C, Whittaker J, Donato di Paola E, Verweij J, Nielsen S. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2001;37:870–877. doi: 10.1016/s0959-8049(01)00050-8. [DOI] [PubMed] [Google Scholar]
- 10.Duggan ST, Keating GM. Pegylated liposomal doxorubicin: a review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi’s sarcoma. Drugs. 2011;71:2531–2558. doi: 10.2165/11207510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 12.Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 13.Biswas S, Dodwadkar NS, Deshpande PP, Torchilin VP. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium–PEG–PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J Control Release. 2012;159:393–402. doi: 10.1016/j.jconrel.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswas S, Dodwadkar NS, Piroyan A, Torchilin VP. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials. 2012;33:4773–4782. doi: 10.1016/j.biomaterials.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biswas S, Dodwadkar NS, Sawant RR, Koshkaryev A, Torchilin VP. Surface modification of liposomes with rhodamine-123-conjugated polymer results in enhanced mitochondrial targeting. J Drug Target. 2011;19:552–561. doi: 10.3109/1061186X.2010.536983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–48. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- 17.Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57:637–651. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4:1449–1452. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 19.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 20.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 21.Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GG. Cell transfection in vitro and in vivo with nontoxic TAT peptide–liposome–DNA complexes. Proc Natl Acad Sci USA. 2003;100:1972–1977. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Mashima T, Sato S, Mochizuki M, Sakamoto H, Yamori T, Oh-Hara T, Tsuruo T. Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide. Cancer Res. 2003;63:831–837. [PubMed] [Google Scholar]
- 24.Khalil IA, Hayashi Y, Mizuno R, Harashima H. Octaarginine- and pH sensitive fusogenic peptide-modified nanoparticles for liver gene delivery. J Control Release. 2011;156:374–380. doi: 10.1016/j.jconrel.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 25.El-Sayed A, Khalil IA, Kogure K, Futaki S, Harashima H. Octaarginine- and octalysine-modified nanoparticles have different modes of endosomal escape. J Biol Chem. 2008;283:23450–23461. doi: 10.1074/jbc.M709387200. [DOI] [PubMed] [Google Scholar]
- 26.Khalil IA, Kogure K, Futaki S, Harashima H. Octaarginine-modified liposomes: enhanced cellular uptake and controlled intracellular trafficking. Int J Pharm. 2008;354:39–48. doi: 10.1016/j.ijpharm.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Khalil IA, Kogure K, Futaki S, Harashima H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 28.Koshkaryev A, Piroyan A, Torchilin VP. Bleomycin in octaarginine-modified fusogenic liposomes results in improved tumor growth inhibition. Cancer Lett. doi: 10.1016/j.canlet.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torchilin VP, Levchenko TS, Lukyanov AN, Khaw BA, Klibanov AL, Rammohan R, Samokhin GP, Whiteman KR. P-Nitrophenylcarbonyl–PEG–PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochim Biophys Acta. 2001;1511:397–411. doi: 10.1016/s0005-2728(01)00165-7. [DOI] [PubMed] [Google Scholar]
- 30.Allen TM, Sapra P, Moase E. Use of the post-insertion method for the formation of ligand-coupled liposomes. Cell Mol Biol Lett. 2002;7:889–894. [PubMed] [Google Scholar]
- 31.Ishida T, Iden DL, Allen TM. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett. 1999;460:129–133. doi: 10.1016/s0014-5793(99)01320-4. [DOI] [PubMed] [Google Scholar]
- 32.Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264–273. doi: 10.1016/j.jconrel.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19:670–686. [PubMed] [Google Scholar]
- 34.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 35.Fretz MM, Penning NA, Al-Taei S, Futaki S, Takeuchi T, Nakase I, Storm G, Jones AT. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem J. 2007;403:335–342. doi: 10.1042/BJ20061808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.