

Abstract
Myeloid sarcoma (MS) is a presentation of acute myeloid leukemia (AML) as a tumor mass outside of the bone marrow. Viable cells from MS are frequently unavailable for cytogenetic studies. We therefore investigated whether chromosomal microarray analysis (CMA) using formalin-fixed paraffin-embedded (FFPE) tissues can detect clinically important genetic abnormalities in MS. CMA successfully identified genomic aberrations in six cases of MS, and in two cases it revealed multiple abnormalities equivalent to a complex karyotype, thus predicting a poor outcome. CMA using FFPE material is therefore a feasible and clinically applicable approach for detection of prognostically significant genomic abnormalities in MS.
Keywords: Chromosomal microarrays, myeloid sarcoma, cytogenetics
1. Introduction
Myeloid sarcoma (MS) is a tumor of malignant myeloid precursor cells, occurring as a mass lesion in extramedullary sites [1]. MS occurs in 2–9% of patients with acute myeloid leukemia (AML), developing concurrently, before or after the diagnosis of bone marrow disease. In rare cases MS manifests as an isolated tumor mass without evidence of bone marrow infiltration [2]; it may also be the first manifestation of relapse after treatment of AML by chemotherapy or hematopoietic stem cell transplantation [3, 4].
Cytogenetic and molecular analyses are essential for the accurate classification of AML. However, fresh tissue from MS is often unavailable for these studies, since the solid-tumor morphology of MS frequently leads to late recognition of its hematopoietic nature. The lack of information regarding associated genetic abnormalities hampers both clinical management, as well as investigations of molecular mechanisms underlying MS.
The literature on genomic alterations in MS mostly includes individual case reports and studies of chromosome abnormalities in corresponding bone marrow leukemia. The largest case series reported to date was by Pileri and colleagues, who analyzed cytogenetic abnormalities in 74 cases of MS [5]. However, even in this study, karyotyping was performed on bone marrow samples, while the MS tissues were only investigated for targeted abnormalities by fluorescent in situ hybridization (FISH).
When MS is followed by more generalized AML, cytogenetic results eventually become available by analysis of the bone marrow samples. Cases where MS remains the only manifestation of acute leukemia are exceedingly rare, and their possibly unique biology and genetics are poorly understood. Information about chromosomal and molecular mutations is particularly sparse for MS without bone marrow involvement.
Only one study used array Comparative Genomic Hybridization (aCGH) to evaluate a series of MS samples [6]. This study included seven cases, and was performed using bacterial-artificial-chromosome (BAC) arrays which have low resolution and cannot identify all abnormality types detectable by high-density array platforms. Furthermore, DNA was isolated from fresh-frozen MS tissue, which is typically not available in the clinical setting. Array analysis has also been successfully used to characterize genomic aberrations in individual reported cases of MS [7], but has never been investigated as a potential tool for routine clinical testing of these tumors.
The goal of our study was to determine if CMA using FFPE samples allows detection of prognostically important genomic abnormalities in MS, in cases for which cytogenetic results could not be obtained from fresh tissue at the time of presentation.
2. Materials and Methods
2.1 Patients and DNA Samples
After approval by the University of Chicago and the University Hospital Carl Gustav Carus Institutional Review Boards, six cases of MS without bone marrow involvement were identified by searching available clinical and research databases at both institutions. The patients’ electronic medical records were reviewed, and relevant clinical data were abstracted, including age, sex, tumor site and results of laboratory studies (where available). The FFPE specimens were obtained from the Pathology archives; upon DNA isolation, both the samples and the corresponding datasets were assigned study numbers and de-identified.
2.2 Conventional Cytogenetics and Fluorescence In Situ Hybridization (FISH)
For cases with available fresh tissue (2/6 cases), conventional cytogenetic analysis was performed following standard procedures. Twenty G-banded metaphase cells were examined, and the chromosomal abnormalities were described according to the International System for Human Cytogenetic Nomenclature (ISCN). FISH testing was performed using commercially available probes (Vysis/Abbott Molecular, Abbott Park, IL) according to the manufacturer’s instructions.
2.3 DNA extraction and CMA analysis
Genomic DNA was extracted with Qiagen DNeasy Blood & Tissue Kit (Qiagen Inc Valencia, CA), according to the manufacturer’s instructions. SNP array testing was performed using the Affymetrix CytoScan™ HD arrays (Affymetrix Inc., Santa Clara, CA), and following the manufacturer’s recommendations, The data were analyzed with the Chromosome Analysis Suite (ChAS) software from Affymetrix (Affymetrix Inc., Santa Clara, CA).
3. Results
Six cases of MS without bone marrow or blood disease and with available FFPE material were identified, and available demographic, clinical and pathology data are summarized in Table 1. Conventional cytogenetic testing was successfully performed at diagnosis only for cases 1 and 2; for the remaining 4 cases, fresh samples were either not made available for karyotyping, or were inadequate for a complete chromosome analysis (Table 2). CMA was initially performed on the two cases with available karyotypes, to determine the concordance of the results between CMA and conventional cytogenetics; we subsequently proceeded with the analysis of the remaining four cases, for which karyotype information was unavailable.
Table 1.
Demographic, hematologic, and clinical data of cases of MS without blood or bone marrow involvement.
| Cases | Age/ Sex |
Initial Site | Histopathology of the Mass | Flow cytometry and immunohistochemistry |
Primary Therapy |
Outcome |
|---|---|---|---|---|---|---|
| 1 | 44/F | small bowel | Extensive infiltration of small bowel by intermediate to large sized blast cells with variably prominent nucleoli, extending from the mucosal to the serosal surface. Focally, tingible body macrophages imparted a ‘starry sky’ appearance. Many neoplastic cells revealed fine granules, and eosinophilic myelocytes were noted throughout the biopsies, consistent with MS, FAB M4eo. | Positive for CD45 (leukocyte common antigen), CD34 and the myeloid marker CD33. Strong expression of myeloperoxidase (MPO), CD43 and CD117. Scattered TdT positive cells, no expression of B- or T-cell markers. | Cytarabine, Daunorubicin, Etoposide | Sustained remission for over 5 years. |
| 2 | 83/M | scrotum | Diffuse proliferation of medium sized cells with a high mitotic rate and blast-like appearance with fine chromatin; findings consistent with MS with expression of monocytic markers. | Blasts with a monocytic profile: CD15+, CD11b+, CD13+, CD33+, HLA-DR+, CD38+, CD56+; 20% of cells coexpressed CD15 and CD34 by flow cytometry. The cells revealed immunoreactivity for CD43 and vimentin, lysozyme, CD14 (weak) and CD68, and were negative for B- and T-cell markers, CD117 and MPO. | Gemtuzumab ozogamicin | Relapsed and died of infection. |
| 3 | 62/F | left breast | Sheets of blasts with high nuclear-cytoplasmic ratio, vesicular chromatin, and prominent nucleoli in a background of geographic necrosis and numerous tingible body macrophages, imparting a "starry sky" appearance. Consistent with MS with monocytic/myelomonocytic differentiation. | CD45 (dim), CD34 (partial), CD13, CD33+, CD117+, CD15+, CD7+, and HLA-DR+. Strong immunoreactivity for CD68 and lysozyme with focal positivity for CD117 and CD34. Scattered MPO positivity. CD20 and CD3 negative. | Daunorubicin Cytarabine + Sorafenib | Relapsed and died of progressive disease. |
| 4 | 42/F | clavicle | Sheets of blasts with vesicular chromatin. | CD45+, CD33+, CD43+, MPO+, CD68+, CD34-, CD56-, CD14-. Negative for cytokeratin, AE1/AE3, CAM 5.2, EMA, S100, TLE1, PAX5, CD20, CD3 and CD5. | HiDAC+ Mitoxantrone | Early local relapse; in remission after HSCT. |
| 5 | 25/M | mediastinal mass | Consistent with MS. | Strong aberrant CD56 and CD7 expression. | Cytarabine, Daunorubicin, Etoposide /HSCT | Early local relapse; died from HSCT complications. |
| 6 | 76/M | testis | Infiltration by blastoid cells of the testis, epididymis, rete testis and proximal spermatic cord into the tunica albuginea, with a negative tumor margin in the spermatic cord. | Positive for CD33 and CD68 and negative for CD20 and CD79a. | Patient refused therapy | The patient died from progressive disease. |
HSCT- hematopoietic stem cell transplantation
Table 2.
Conventional cytogenetics, FISH, and SNP-array findings in cases of MS without blood or bone marrow involvement.
| Case | CC-Mass | CC-BM | FISH | Molecular Studies | CMA results |
|---|---|---|---|---|---|
| 1 | 46,XX,inv(16)(p13.1q2 2)[7]/47,idem,+22[7]/4 6,XX[7] | ND | ND | ND | CN-LOH 6p25.3-p21.32 Gain of chr22 |
| 2 | 47,XX,+8[20] | ND | +8 | FLT3 D835 mutation positive | Gain of chr8 CN-LOH chr13 |
| 3 | Inadequate | 46,XX[20] | Negative for t(8;21)-RUNX1/RUNX1T1 and inv(16)-CBFB | NPM1, FLT3-ITD mutation positive | CN-LOH chr13 |
| 4 | ND | ND | Negative for t(8;21)-RUNX1/RUNX1T1 and inv(16)-CBFB | Negative for NPM1, FLT3 BCR/ABL1 | CN-LOH 11pter-p11.2 |
| 5 | ND | 46,XY[20] | Negative for t(8;21)-RUNX1/RUNX1T1, 11(q23)-MLL, inv(16)-CBFB; positive for +19 | Negative for NPM1, FLT3 mutations | Gain of chr7, chr19 Loss 1p36.33-p36.22, 6q27, 13q12.3-q31.1 |
| 6 | ND | ND | ND | Negative for NPM1, FLT3 mutations | Loss 1p21.1-p12, 1q23.1-q23.3, 15q11.2-q14 Gain 1q21.1-q23.1, 1q23.3-q44, 2q12.1-q36.3, 16pter-p11.1, 19p13.3-p11 Gain chr12, Loss chr13 CN-LOH 8q21.11-q24.11 |
ND-not determined
Case 1
The patient was a 40-year-old Caucasian woman who presented with small bowel obstruction due to a tumor mass. An exploratory laparotomy and small bowel resection were performed. Histopathology, flow cytometry and immunohistochemistry were diagnostic of MS (Figure 1, Table 1). Cytogenetic evaluation revealed the following karyotype: 46,XX,inv(16)(p13.1q22)[7]/47, idem,+22[7]/46,XX[7]. The patient received induction chemotherapy with cytarabine, daunorubicin and etoposide, following the dose and schedule of the Cancer and Leukemia Group B (CALGB) 10503 clinical trial. She subsequently completed three courses of consolidation chemotherapy with high-dose cytarabine (HiDAC) following the CALGB schedule. She remains in first remission for over 5 years. CMA detected gain of chromosome 22 which was also observed in a subpopulation of cells by karyotype analysis. The inv(16) could not be identified by CMA since this was a completely balanced rearrangement. However, CMA detected an additional abnormality (CN-LOH 6p25.3-p21.32), the significance of which is unknown.
Figure 1.

Biopsy of the small bowel tumor mass from the patient with AML with inv(16) (Case 1). The panel on the left is showing infiltration of mucosa and serosa by MS. The upper panel on the right is a higher power showing sheets of intermediate to large sized blast cells with variably prominent nucleoli. Some neoplastic cells revealed fine granules, and eosinophilic myelocytes were noted throughout the biopsy. The lower right panel is showing positive CD34 staining.
Case 2
The patient was a 73-year-old Caucasian male who presented to an outside hospital with a right scrotal mass. Biopsy of the scrotal mass was diagnostic of MS. The patient was initially treated with imatinib mesylate because chronic myeloid leukemia was suspected, but the treatment was soon switched to gemtuzumab ozogamicin, resulting in a complete remission (CR). Approximately a year after initial presentation, the patient relapsed with skin lesions. Cytogenetic evaluation of a skin lesion sample revealed trisomy 8: 47,XY,+8[20]. For this relapse the patient was treated with cytarabine and gemtuzumab ozogamicin, and had another complete response. However, the skin lesions subsequently recurred and continued to progress, and an autologous stem cell transplant was planned. The patient was treated with cytarabine and etoposide for stem cell mobilization, but died from infection. CMA confirmed gain of chromosome 8 detected by conventional cytogenetics, and also identified CN-LOH for the entire chromosome 13. This prompted molecular testing of the FLT3 gene, which maps to chromosome 13. The FLT3 analysis was negative for an internal tandem duplication (ITD) mutation, but showed the presence of a D835 tyrosine kinase domain (TKD) mutation (Supplementary material, Figure 2).
Case 3
The patient was a 61-year-old previously healthy Caucasian woman who presented with a palpable superficial lump under the left breast of one month duration. A mammogram revealed 2 to 3 cm masses in the right breast, left axilla, left breast and subcutaneously below the left breast. All 4 lesions were strongly positron emission tomography (PET) avid. Histopathology, flow cytometry and immunohistochemical analyses, performed on a needle biopsy of a subcutaneous lesion, established the diagnosis of MS. Molecular studies revealed the presence of both NPM1 and FLT3-ITD mutations. The patient received induction with daunorubicin and cytarabine plus sorafenib, followed by two courses of intermediate dose cytarabine plus sorafenib, on a clinical trial for AML with FLT3-ITD. She achieved a CR and her subcutaneous masses resolved. She completed one year of maintenance with sorafenib at 400 mg BID, but relapsed several months later and died from progressive disease. CMA showed no copy number gains or losses, but detected CN-LOH for the entire chromosome 13.
Case 4
The patient was a 40-year-old, previously healthy Caucasian woman who presented with right-sided neck pain. Magnetic resonance imaging (MRI) examination of the neck revealed an abnormal signal centered on her right sternoclavicular joint with marrow changes in the medial clavicular head. Pathology studies of an open biopsy sample were diagnostic of MS. The patient received induction chemotherapy with HiDAC and Mitoxantrone. Repeat PET scan revealed interval metabolic improvement of the fluorodeoxyglucose (FDG) avid tumor in the right clavicle. She received HiDAC consolidation. Subsequently the patient suffered local relapse without bone marrow involvement, and received local irradiation followed by reinduction chemotherapy and allogeneic stem cell transplantation in second remission. She remains alive with chronic GVHD of the skin two years post- transplant without evidence of recurrent AML. CMA was negative for copy number abnormalities, but showed CN-LOH at 11p, affecting a 46.8 Mb region between 11pter-p11.2.
Case 5
The patient was a 25-year-old man with a history of primary mediastinal MS treated at an outside institution. He had originally presented with left cervical lymphadenopathy but a biopsy revealed only fibro-adipose tissue. Upon further workup he was found to have a mediastinal mass that was biopsied and diagnosed as MS (strongly PET avid), reportedly with strong expression of CD56 and CD7. He underwent induction therapy with daunorubicin, etoposide and cytarabine without complications. Followup imaging showed only minimal reduction in the size of the mediastinal mass but resolution of the adenopathy. He received one cycle of HiDAC consolidation. After several months he developed new left-sided cervical adenopathy and was referred for evaluation and treatment. Biopsy was consistent with recurrent MS. An allogeneic transplant was performed using umbilical cord stem cells and a partially matched family member donor. The patient achieved complete remission (PET-negative) post-transplant but expired of transplant associated complications nine months later. CMA detected gains of chromosomes 7 and 19 as well as segmental copy number loss (deletions) at 1p36.33-p36.22, 6q27 and 13q12.3-q31.1 (Table 2, Figure 2). The presence of multiple genomic abnormalities by CMA is equivalent to a complex karyotype, which is associated with a poor prognosis in AML.
Figure 2.
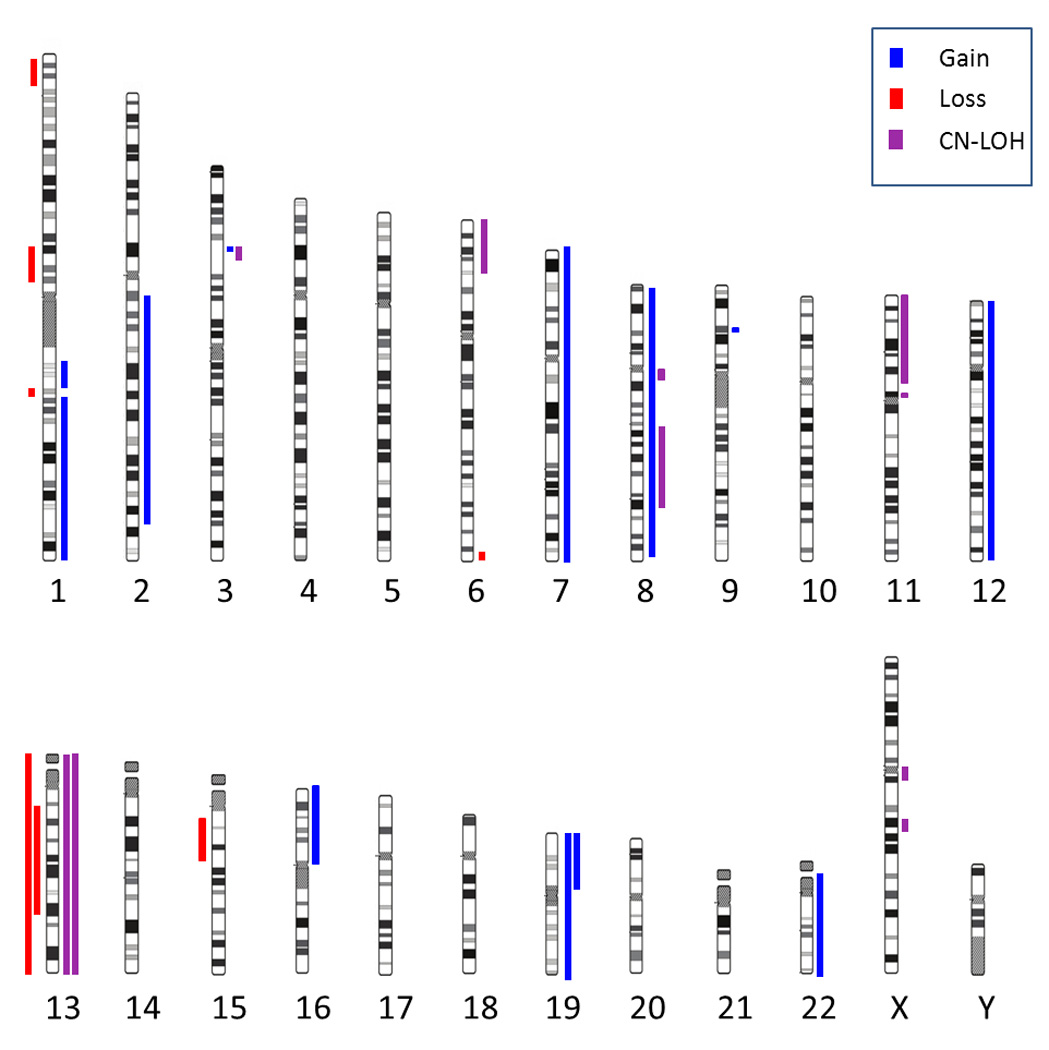
Summary of copy number alterations and CN-LOH events observed by CMA in six cases of MS without bone marrow disease. Abnormalities detected in each individual case are listed in Table 2 and Supplementary table 1.
Case 6
The patient was a 76-year old male referred to the urology department for a palpable mass in his right testicle. A biopsy showed a tumor suspected to be a non-Hodgkin Lymphoma, and the right testicle was removed. Histomorphology and immunohistochemistry revealed MS, with high proliferative activity as measured by the fraction of Ki-67-positive tumor cells (80%). At the time of the removal of the right testicle, a biopsy of the left testicle was normal. Imaging studies at the time revealed a 3 cm presacral soft tissue mass of unclear etiology. The patient refused chemotherapy and only received supportive care. A few months later he was admitted to the hospital due to pain radiating to his right leg. The PET/CT scan showed an enlarging presacral mass (Supplementary figure 1); a biopsy showed the suspected MS. The patient still refused chemotherapy and missed his appointment for radiation treatment. He subsequently returned with a mass in his left (remaining) testicle; the tumor was removed and the pathology confirmed MS. Shortly after surgery, the patient developed paraparesis of both lower extremities and a conus-cauda syndrome; he received radiation of the spine (T4-T7), but developed significant lymphadenopathy and died shortly thereafter due to progressive disease. CMA was performed on multiple samples obtained during the course of the disease, including the original tumor from the right testicle, the biopsy of the presacral mass and the tumor mass from the left testicle. Identical complex genomic abnormalities were detected in all samples, and are listed in Table 2 and in Supplementary Table 1.
Detailed CMA results for all 6 cases are presented in Table 2, Figure 2, and Supplementary Table 1.
4. Discussion
Cytogenetic evaluation is important for prognostic and diagnostic classification of leukemia [8, 9]. However, conventional cytogenetic analysis can only be performed from cells that are capable of actively dividing in tissue culture. Without a prior history of AML, MS is often a diagnostic challenge [10, 11]; by the time it is recognized as a hematologic tumor, fresh tissue is often not available for karyotype analysis. Considering the logistic challenges of conventional cytogenetic analysis for MS, we demonstrated that CMA may be a useful platform for detecting prognostically important chromosomal abnormalities in MS samples in diagnostic laboratories.
CMA results were obtained successfully for all six available FFPE MS samples, although the age and methods of processing varied widely between the samples, with the oldest case being more than 9 years old. One important consideration is that FFPE tissues may occasionally yield insufficient amounts of DNA, or the DNA may be too degraded for array testing [12]. Laboratories offering CMA on FFPE tissues will have to anticipate that DNA from a subset of samples will be of poor quality, and that acceptable results will not be routinely obtainable with the array platforms used for fresh samples. However, a specialized array technology has recently been developed for FFPE samples (OncoScan™, Affymetrix, Santa Clara, CA) [13]. Further study will be required to determine its utility in the evaluation of MS samples.
A comparison of the detected abnormalities between CMA and conventional cytogenetics in cases 1 and 2 showed that CMA easily identified unbalanced chromosomal abnormalities (trisomy 22 in case 1 and trisomy 8 in case 2), and in both cases it also revealed CN-LOH abnormalities which were undetectable by karyotyping. However, CMA failed to identify the inv(16) in case 1, since this technology cannot detect completely balanced chromosomal rearrangements. Conventional cytogenetic analysis therefore remains the test of choice when fresh material is available, since in addition to gains and losses of chromosomal material it also detects balanced rearrangements which play an important role in pathogenesis of AML.
The ability of CMA to reveal previously undetected abnormalities of clinical significance in MS is best illustrated by cases 3 and 6 in this study. Multiple copy number gains, losses and CN-LOH regions detected in these cases are analogous to a complex karyotype, which is a well-established predictor of aggressive disease and poor outcome in AML; in these two cases CMA therefore revealed information that would have been useful in clinical management. However, even in the cases where CMA did not detect abnormalities of direct prognostic importance, it revealed previously unknown chromosomal aberrations (Table 2), thus allowing better genetic characterization and more complete understanding of the molecular pathogenesis for each studied case.
The spectrum of mutations detected in this study is consistent with the reports of the most common that have previously been described in association with MS include inv(16) in Case 1 [15, 16], trisomy 8 in Case 2 [17, 18, 19], FLT3 mutations in cases 2 and 3 [20] and NPM1 mutation in case 3 [21].
Complex chromosomal rearrangements, revealed by CMA in two cases in this study, have not been specifically investigated in MS, but have been described in multiple case reports and case series [22]. CMA is particularly well suited for detecting cases with multiple unbalanced genomic rearrangements, and considering the prognostic importance of complex karyotypes in AML, CMA should be used for timely identification of these high risk patients.
The prognostic relevance of specific cytogenetic and molecular mutations in MS has not been formally investigated, but likely parallels the prognostic implications of the same mutations in the bone marrow disease. This study included a small number of cases which were not uniformly treated; however, the patient with a genetic marker of good prognosis [inv(16)] has done well, while the patients with adverse genetic markers (complex genomic abnormalities, FLT3 mutation) had recurrent and progressive disease. Since MS is typically treated with systemic chemotherapy and hematopoietic stem cell transplantation in the same fashion as bone marrow AML, it is important to identify specific genetic lesions that aid in determination of the most effective treatment regimen [23].
Cases 2 and 3 illustrate how correlation of cytogenetic, CMA and molecular studies allows better insight into molecular pathogenesis of leukemia. Both cases showed acquired CN-LOH for chromosome 13 by CMA and were shown by molecular testing to carry FLT3 mutations. CN-LOH at the FLT3 locus on 13q has been described as a frequent abnormality in FLT3 mutation positive AML [24], and some studies even suggest that the resulting increase in the mutated allele burden may be associated with more aggressive disease [24, 25]. CN-LOH is a common mechanism in tumor cells to increase allelic burden of oncogene mutations or lose the wild-type copies of tumor-suppressor genes; in addition to FLT3co-occurrence with CN-LOH has been described for mutated RUNX1, EZH2 and other genes implicated in AML [26]. CN-LOH detection by CMA can therefore inform further molecular testing by revealing chromosomal locations of likely mutated oncogenes and tumor suppressor genes.
Case 6 had multiple samples available for CMA testing, which were obtained from different extramedullary sites over the disease course. The presence of the same genomic alterations in samples collected many months apart from separate anatomic locations showed the persistence and spreading of the original malignant clone which initially presented in the right testicle. This case illustrates how CMA can be used to study clonal evolution in MS. For example, there is currently little information about concordance of genetic abnormalities between extramedullary sites and bone marrow disease in cases of generalized AML; it has not been determined how frequently the extamedullary disease has distinct genetic or cytogenetic abnormalities from the disease in the bone marrow. An extamedullary tumor that develops concurrently with bone marrow involvement may either represent the original population of tumor cells or a clonal evolution from the disease that initiated in the bone marrow. CMA alone or in combination with next-generation sequencing may be a valuable tool to address research questions related to the original sites and cells of origin of MS, and to investigate clonal evolution in extramedullary AML. In summary, this study confirmed the feasibility and clinical utility of CMA testing for MS. We propose that CMA using FFPE tissue, as well as molecular testing for FLT3NPM1 and possibly other prognostically relevant molecular mutations should be performed on every MS sample, especially if results of conventional cytogenetic analysis are unavailable or inconclusive. Implementation of novel array platforms that allow successful analysis on small amounts of partially degraded DNA from FFPE tissue should facilitate routine implementation of CMA in advancing both research and clinical management of MS.
Supplementary Material
Acknowledgements
This work was funded by the American Cancer Society Institutional Research Grant (#IRG-58-004) and the University of Chicago Comprehensive Cancer Center Support Grant (#P30 CA14599).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
KMM, MS and PR performed the experiments. FS, RL, WS, GE, FK and KZ contributed patient samples and clinical data. FS, KO and GR analyzed and interpreted the data. KMM, MS, FS, KO and GR wrote the paper. GR designed and coordinated the research study.
References
- 1.Byrd JC, Edenfield WJ, Shields DJ, Dawson NA. Extramedullary myeloid cell tumors in acute nonlymphocytic leukemia: a clinical review. J Clin Oncol. 1995;13(7):1800–1816. doi: 10.1200/JCO.1995.13.7.1800. [DOI] [PubMed] [Google Scholar]
- 2.Neiman RS, Barcos M, Berard C, Bonner H, Mann R, Rydell RE, et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer. 1981;48(6):1426–1437. doi: 10.1002/1097-0142(19810915)48:6<1426::aid-cncr2820480626>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 3.Clark WB, Strickland SA, Barrett AJ, Savani BN. Extramedullary relapses after allogeneic stem cell transplantation for acute myeloid leukemia and myelodysplastic syndrome. Haematologica. 2010;95(6):860–863. doi: 10.3324/haematol.2010.025890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimizu H, Saitoh T, Hatsumi N, Takada S, Handa H, Jimbo T, et al. Prevalence of extramedullary relapses is higher after allogeneic stem cell transplantation than after chemotherapy in adult patients with acute myeloid leukemia. Leuk Res. 2013;37(11):1477–1481. doi: 10.1016/j.leukres.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 5.Pileri SA, Ascani S, Cox MC, Campidelli C, Bacci F, Piccioli M, et al. Myeloid sarcoma: clinicopathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia. 2007;21(2):340–350. doi: 10.1038/sj.leu.2404491. [DOI] [PubMed] [Google Scholar]
- 6.Deeb G, Baer MR, Gaile DP, Sait SN, Barcos M, Wetzler M, et al. Genomic profiling of myeloid sarcoma by array comparative genomic hybridization. Genes Chromosomes Cancer. 2005;44(4):373–383. doi: 10.1002/gcc.20239. [DOI] [PubMed] [Google Scholar]
- 7.Stolzel F, Hackmann K, Kuithan F, Mohr B, Fussel M, Oelschlagel U, et al. Clonal evolution including partial loss of human leukocyte antigen genes favoring extramedullary acute myeloid leukemia relapse after matched related allogeneic hematopoietic stem cell transplantation. Transplantation. 2012;93(7):744–749. doi: 10.1097/TP.0b013e3182481113. [DOI] [PubMed] [Google Scholar]
- 8.Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–365. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- 9.Rollig C, Bornhauser M, Thiede C, Taube F, Kramer M, Mohr B, et al. Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: evaluation of the proposed reporting system. J Clin Oncol. 2011;29(20):2758–2765. doi: 10.1200/JCO.2010.32.8500. [DOI] [PubMed] [Google Scholar]
- 10.Traweek ST, Arber DA, Rappaport H, Brynes RK. Extramedullary myeloid cell tumors. An immunohistochemical and morphologic study of 28 cases. Am J Surg Pathol. 1993;17(10):1011–1019. doi: 10.1097/00000478-199310000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Menasce LP, Banerjee SS, Beckett E, Harris M. Extra-medullary myeloid tumour (granulocytic sarcoma) is often misdiagnosed: a study of 26 cases. Histopathology. 1999;34(5):391–398. doi: 10.1046/j.1365-2559.1999.00651.x. [DOI] [PubMed] [Google Scholar]
- 12.Tuefferd M, de Bondt A, Van den Wyngaert I, Talloen W, Gohlmann H. Microarray profiling of DNA extracted from FFPE tissues using SNP 6.0 Affymetrix platform. Methods Mol Biol. 2011;724:147–160. doi: 10.1007/978-1-61779-055-3_10. [DOI] [PubMed] [Google Scholar]
- 13.Chandler WM, Rowe LR, Florell SR, Jahromi MS, Schiffman JD, South ST. Differentiation of malignant melanoma from benign nevus using a novel genomic microarray with low specimen requirements. Arch Pathol Lab Med. 2012;136(8):947–955. doi: 10.5858/arpa.2011-0330-OA. [DOI] [PubMed] [Google Scholar]
- 14.Al-Khateeb H, Badheeb A, Haddad H, Marei L, Abbasi S. Myeloid sarcoma: clinicopathologic, cytogenetic, and outcome analysis of 21 adult patients. Leuk Res Treatment. 2011;2011:523168. doi: 10.4061/2011/523168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xavier SG, Fagundes EM, Hassan R, Bacchi C, Conchon M, Tabak DG, et al. Granulocytic sarcoma of the small intestine with CBFbeta/MYH11 fusion gene: report of an aleukaemic case and review of the literature. Leuk Res. 2003;27(11):1063–1066. doi: 10.1016/s0145-2126(03)00070-5. [DOI] [PubMed] [Google Scholar]
- 16.McKenna M, Arnold C, Catherwood MA, Humphreys MW, Cuthbert RJ, Bueso-Ramos C, et al. Myeloid sarcoma of the small bowel associated with a CBFbeta/MYH11 fusion and inv(16)(p13q22): a case report. J Clin Pathol. 2009;62(8):757–759. doi: 10.1136/jcp.2008.063669. [DOI] [PubMed] [Google Scholar]
- 17.Schaich M, Schlenk RF, Al-Ali HK, Dohner H, Ganser A, Heil G, et al. Prognosis of acute myeloid leukemia patients up to 60 years of age exhibiting trisomy 8 within a non-complex karyotype: individual patient data-based meta-analysis of the German Acute Myeloid Leukemia Intergroup. Haematologica. 2007;92(6):763–770. doi: 10.3324/haematol.11100. [DOI] [PubMed] [Google Scholar]
- 18.Sen F, Zhang XX, Prieto VG, Shea CR, Qumsiyeh MB. Increased incidence of trisomy 8 in acute myeloid leukemia with skin infiltration (leukemia cutis) Diagn Mol Pathol. 2000;9(4):190–194. doi: 10.1097/00019606-200012000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Agis H, Weltermann A, Fonatsch C, Haas O, Mitterbauer G, Mullauer L, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol. 2002;81(2):90–95. doi: 10.1007/s00277-001-0412-9. [DOI] [PubMed] [Google Scholar]
- 20.Ansari-Lari MA, Yang CF, Tinawi-Aljundi R, Cooper L, Long P, Allan RH, et al. FLT3 mutations in myeloid sarcoma. Br J Haematol. 2004;126(6):785–791. doi: 10.1111/j.1365-2141.2004.05124.x. [DOI] [PubMed] [Google Scholar]
- 21.Falini B, Lenze D, Hasserjian R, Coupland S, Jaehne D, Soupir C, et al. Cytoplasmic mutated nucleophosmin (NPM) defines the molecular status of a significant fraction of myeloid sarcomas. Leukemia. 2007;21(7):1566–1570. doi: 10.1038/sj.leu.2404699. [DOI] [PubMed] [Google Scholar]
- 22.Stolzel F, Rollig C, Radke J, Mohr B, Platzbecker U, Bornhauser M, et al. (1)(8)F-FDG-PET/CT for detection of extramedullary acute myeloid leukemia. Haematologica. 2011;96(10):1552–1556. doi: 10.3324/haematol.2011.045047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bakst RL, Tallman MS, Douer D, Yahalom J. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118(14):3785–3793. doi: 10.1182/blood-2011-04-347229. [DOI] [PubMed] [Google Scholar]
- 24.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res. 2001;61(19):7233–7239. [PubMed] [Google Scholar]
- 25.Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3- activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgibbon J, Smith LL, Raghavan M, Smith ML, Debernardi S, Skoulakis S, et al. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005;65(20):9152–9154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.