

Abstract
Charcot-Marie-Tooth disease (CMT) is the most common inherited neuropathy characterized by clinical and genetic heterogeneity. Although more than 30 loci harboring CMT-causing mutations have been identified, many other genes still remain to be discovered for many affected individuals. For two consanguineous families with CMT (axonal and mixed phenotypes), a parametric linkage analysis using genome-wide SNP chip identified a 4.3 Mb region on 12q24 showing a maximum multipoint LOD score of 4.23. Subsequent whole-genome sequencing study in one of the probands, followed by mutation screening in the two families, revealed a disease-specific 5 bp deletion (c.247−10_247−6delCACTC) in a splicing element (pyrimidine tract) of intron 2 adjacent to the third exon of cytochrome c oxidase subunit VIa polypeptide 1 (COX6A1), which is a component of mitochondrial respiratory complex IV (cytochrome c oxidase [COX]), within the autozygous linkage region. Functional analysis showed that expression of COX6A1 in peripheral white blood cells from the affected individuals and COX activity in their EB-virus-transformed lymphoblastoid cell lines were significantly reduced. In addition, Cox6a1-null mice showed significantly reduced COX activity and neurogenic muscular atrophy leading to a difficulty in walking. Those data indicated that COX6A1 mutation causes the autosomal-recessive axonal or mixed CMT.
Main Text
Charcot-Marie-Tooth disease (CMT) is the most common inherited neuromuscular disease characterized by clinical and genetic heterogeneity, which has been traditionally subdivided into two major subgroups, demyelinating and axonal forms. Initially, genes encoding major myelin proteins including peripheral myelin protein 22 (PMP22 [MIM 601097])1 and myelin protein zero (MPZ [MIM 159440])2 were identified as genes involved in demyelinating CMT as well as mitofusin 2 (MFN2 [MIM 608507])3 in axonal CMT. So far, mutations causing CMT have been identified in more than 30 loci. To identify the genetic background of Japanese CMT, we analyzed the disease-causing mutation in about 350 affected individuals; however, we could not identify the causative mutations in about 50% of demyelinating CMT and 80% of axonal CMT.
We studied two families with affected members from consanguineous marriages at different sites in Japan (Figure 1A). The affected siblings of family 1 initially had slightly reduced median motor nerve conduction velocities (NCVs) and onion bulb formation in the sural nerve at young ages, but they had motor NCVs below 38 m/s when they were aged 30–39 years, indicating mixed CMT (Tables S1 and S2 available online). The affected member of family 2 had slightly reduced median motor NCVs at age 39, indicating axonal phenotype (Tables S3 and S4). Although the two families have no record of affinal connections each other, their affected members share similar disease phenotypes and a deduced mode of inheritance (i.e., recessive). Informed consent was obtained from all subjects, and all procedures were approved by the Research Ethics Committees of Yamagata University Faculty of Medicine and Niigata University School of Medicine. For these families, family 1 with two affected members from the marriage between second cousins and family 2 with one from that between first cousins once removed (Figure 1A), we carried out a parametric linkage study using genome-wide 6.5K SNP chip. A genome-wide SNP genotyping was performed for ten members from the two families with the HumanLinkage V Panel (Illumina). A multipoint linkage analysis was performed with Allegro v.2 and a human genetic map based on NCBI dbSNP Build 123 and identified a significant linkage in a 4.3 Mb region on 12q24 covered by nine SNPs showing multipoint LOD scores of 3.85–4.23 at θ = 0.00 (Figures 1B, 1C, and S1; Table 1).
Figure 1.
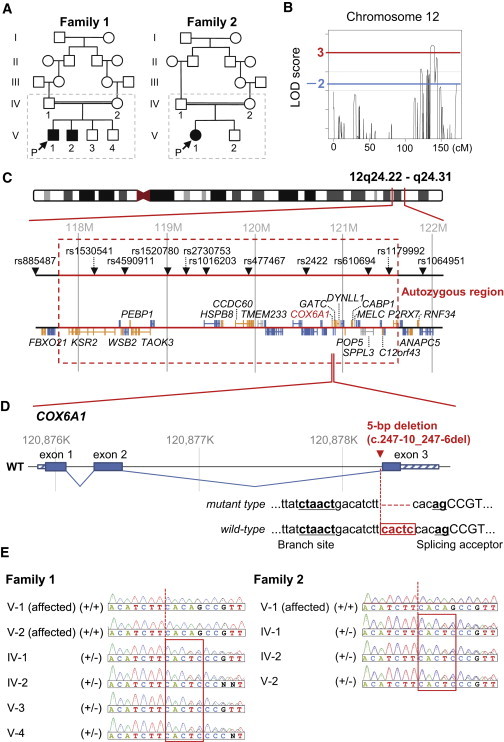
Genomic Analyses in the CMT Families
(A) Pedigrees of two consanguineous CMT families, from which a total of ten members in broken line boxes were sampled.
(B) Multipoint LOD scores on chromosome 12 using these ten members from two families with the HumanLinkage V SNP chip. We extracted genomic DNA from peripheral blood using the QIAamp DNA spin columns (QIAGEN) and quantified them using the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies). Genotyping was performed with the GoldenGate Genotyping Universal-32 kit on a BeadArray Reader System (Illumina) according to the manufacturer’s assay guide. Genotype calls were made using the Genotyping module of the GenomeStudio v2009.1 software (Illumina). A multipoint linkage analysis was performed with Allegro v.24 and a human genetic map based on NCBI dbSNP Build 123 (see also Figure S1).
(C) Gene map in the significant linkage region on 12q24. These physical coordinates are taken from UCSC Genome Browser build hg19, RefSeq, and dbSNP 138 (UCSC Genome Browser database: 2014 update).
(D) A disease-specific 5 bp deletion (c.247−10_247−6delCACTC) in the pyrimidine tract near to the splicing acceptor near to the third exon of COX6A1.
(E) Validation and distribution of the 5 bp deletion among the CMT family members by Sanger-based PCR direct sequencing (see also Table S10).
Table 1.
Multipoint LOD Scores on 12q24 and Estimated Haplotypes in the Significant Linkage Region
| Markers | Positionsa | LOD Score |
Family 1 |
Family 2 |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IV-1 (Father) | IV-2 (Mother) | V-1 (Affected) | V-2 (Affected) | V-3 (Brother) | V-4 (Brother) | IV-1 (Father) | IV-2 (Mother) | V-1 (Affected) | V-2 (Brother) | |||||||||||||
| rs885487 | 117539934 | −infinity | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 1 | 1 | 1 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2 |
| rs1530541 | 118229731 | 4.0597 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2∗ | 2∗ | 2 | 2∗ | 2 | 2∗ | 1 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 1 |
| rs4590911 | 118562757 | 4.2433 | 1∗ | 2 | 1∗ | 2 | 1∗ | 1∗ | 1∗ | 1∗ | 1∗ | 2 | 1∗ | 2 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2 |
| rs1520780 | 119039725 | 4.2338 | 2∗ | 1 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2∗ | 2∗ | 1 | 2∗ | 1 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2 |
| rs2730753 | 119171974 | 4.2317 | 2∗ | 1 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2∗ | 2∗ | 1 | 2∗ | 1 | 2∗ | 2 | 2∗ | 1 | 2∗ | 2∗ | 2∗ | 2 |
| rs1016203 | 119423542 | 4.2249 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2∗ | 2∗ | 2 | 2∗ | 2 | 2∗ | 2 | 2∗ | 1 | 2∗ | 2∗ | 2∗ | 2 |
| rs477467 | 119927992 | 4.1386 | 2∗ | 2 | 2∗ | 1 | 2∗ | 2∗ | 2∗ | 2∗ | 2∗ | 2 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2 |
| rs2422 | 120565188 | 4.0416 | 1∗ | 2 | 1∗ | 1 | 1∗ | 1∗ | 1∗ | 1∗ | 1∗ | 2 | 1∗ | 2 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2 |
| 5 bp del | 120878247 | +∗ | − | +∗ | − | +∗ | +∗ | +∗ | +∗ | +∗ | − | +∗ | − | +∗ | − | +∗ | − | +∗ | +∗ | +∗ | − | |
| rs610694 | 121304826 | 3.8859 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2∗ | 2∗ | 2 | 2∗ | 2 | 1∗ | 1 | 1∗ | 2 | 1∗ | 1∗ | 1∗ | 1 |
| rs1179992 | 121495432 | 3.8466 | 1∗ | 2 | 1∗ | 2 | 1∗ | 1∗ | 1∗ | 1∗ | 2∗ | 2 | 1∗ | 2 | 2∗ | 2 | 2∗ | 1 | 2∗ | 2∗ | 2∗ | 2 |
| rs1064951 | 121878659 | −1.7247 | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2∗ | 2 | 2∗ | 2 | 2∗ | 2∗ | 2∗ | 2 |
Haplotypes were reconstructed using Allegro v.2 with “HAPLOTYPE” option from each family data including 5 bp deletion (c.247−10_247−6delCACTC). Asterisk indicates the putative disease-associated haplotypes in each family.
Physical coordinate is taken from dbSNP 138 (UCSC Genome Browser, 2014 update).
Subsequent whole-genome sequencing for the proband from family 1 was performed on a Genome Analyzer IIx system (Illumina), followed by annotation, filtering, and assessing potential effects of variants (Tables S5 and S6). It yielded enough data (a mean depth of 49.6×) to detect sequence variants by comparison with a reference sequence (Table S5). Of a total of 3,716,455 variants (Table S7), after filtering out known or heterozygous variants, six were suspected to be deleterious by a majority rule approach using five prediction methods: PolyPhen-2, Grantham, PROVEAN, SIFT, and Mutation Taster (Table 2; see also Table S8 for a relaxed rule). Among them, only a 5 bp deletion (c.247−10_247−6delCACTC; RefSeq accession number NM_004373.3) was located just in a long run of homozygosity spanning 3.7 Mb, between rs10774925 and rs503720, in the significant linkage region on 12q24 (Figures 1C and 1D; Tables 1 and S9; see also Figure S2 and Table S10). In the two families, the 5 bp deletion was cosegregated perfectly with the disease state (Figures 1E and S3). Mutation screening by PCR assay detected the 5 bp deletion in none of 1,452 control chromosomes from diverse ethnic groups (Table S11).
Table 2.
List of Homozygous and Potentially Deleterious Variants Nominated by at Least Three of Five Prediction Methods
| Chr | Start |
Alleles |
Gene (MIM ID) | Types | Accession |
Changes |
||
|---|---|---|---|---|---|---|---|---|
| Ref | Variant | Nucleotide | Protein | |||||
| 12 | 120878247 | CACTC | – | COX6A1 | splicing | NM_004373.3 | c.247−10_247−6delCACTC | NA |
| 2 | 114257705 | C | – | FOXD4L1 (MIM 611084) | frame-shift | NM_012184.4 | c.876del | p.Gly293Alafs∗151 |
| 14 | 24470691 | – | A | DHRS4L2 (MIM 615196) | frame-shift | NM_001193637.1 | c.204_205insA | p.His69Thrfs∗69 |
| 19 | 20807178 | – | A | ZNF626 (NA) | frame-shift | NM_001076675.2 | c.1505dup | p.Ile503Hisfs∗95 |
| 2 | 114257443 | A | C | FOXD4L1 (MIM 611084) | nonsynonymous | NM_012184.4 | c.610A>C | p.Lys204Gln |
| 8 | 126443464 | G | T | TRIB1 (MIM 609461) | nonsynonymous | NM_025195.3 | c.320G>T | p.Arg107Leu |
To assess potential variant effects for nonsynonymous variants, we used PolyPhen-2 v.2.2.2 (HumVar; score > 0.85),5 Grantham Score from SeattleSeq Annotation v.137 (score > 151), PROVEAN v.1.1.3 (score < −2.5),6 SIFT (score < 0.05),7 and Mutation Taster (predicted as “disease causing”)8 and listed potentially deleterious variants voted by at least two prediction methods. Furthermore, we also referred gene expression in human whole brain on BioGPS (GeneAtlas U133A, gcrma data set)9,10 (see also Table S8).
As expected from the fact that this disease-specific 5 bp deletion disrupted an intronic splicing element (pyrimidine tract11–13) adjacent to the boundary of the second intron and the third exon of cytochrome c oxidase subunit VIa polypeptide 1 (COX6A1 [MIM 602072]) whose expression is almost ubiquitous excepting skeletal muscles (see Su et al.10 for human and Figure S4 for mouse), quantitative RT-PCR assay detected the significantly reduced (<1/5 of controls; p < 0.001) expression of the normally spliced form of COX6A1 in peripheral white blood cells from two affected members of family 1 (Figure 2A, see also Table S12). Consistent with these findings, their cultured cell lines showed statistically significant reductions in the COX6A1 expression (p < 0.001; Figure 2B, see also Table S12), cytochrome c oxidase (COX) activity (Figure 2C), and total ATP contents (Figure 2D) in these affected individuals relative to controls. Detailed descriptions are found in the legends of figures and tables for all experiments including expression analysis of COX6A1 mRNA (see also Figure S5 and Table S12), determination of COX activity, and ATP amount. We performed two additional experiments: (1) the qRT-PCR using three primers specific to the mutant transcript showed its low level (Figure S6 and Table S12); and (2) the immunoblot showed no band at around 11.0 kDa for the mutant protein (Figure S7). These results suggest that the deletion leads to alternative splicing events triggering a potential nonsense-mediated RNA decay.
Figure 2.
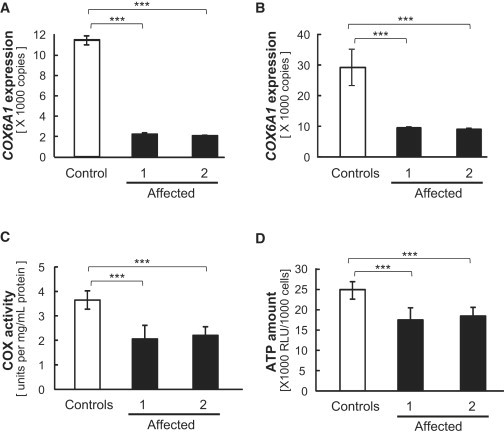
COX6A1 Expression and COX Activity Levels
(A and B) COX6A1 expression in fresh whole-blood from a control and the two affected individuals from family 1 (A) and in EBV-transformed B cell lines from four controls and the two affected individuals (B). Lymphoblastoid cells from two affected members in the family 1 were immortalized by infection with the Epstein-Barr virus (VR-1492; American Type Culture Collection). Immortalized cells from the four healthy Japanese individuals (HEV0031, HEV0032, HEV0038, and HEV0041) were provided by RIKEN BioResource Center. Total RNA was extracted using QIAamp RNA Blood Mini Kit or AllPrep DNA/RNA Kit (QIAGEN) according to the manufacturer’s instructions with on-column DNase I treatment. After determining RNA concentrations using Quant-iT RiboGreen RNA Assay Kit (Life Technologies), 400 ng of total RNA per 40 μl reaction was used to synthesize cDNA using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies) with random primers. Absolute quantification for COX6A1 was performed using a custom TaqMan assay (Table S12). PCR products were ligated into pGEM-T easy vector (Promega) and isolated plasmid DNA was then linearized by EcoRI digestion. Before use, plasmid concentration was determined by Quant-iT PicoGreen dsDNA Assay Kit and serial dilutions were performed to generate standard curve. Real-time PCR was conducted using TaqMan Universal Master Mix II (Life Technologies) with a 7500 Fast real-time PCR system (Life Technologies). Each reaction was run in triplicate and contained 2 μl of cDNA template in a final reaction volume of 20 μl and data were analyzed with 7500 Software v.2.0.2.
(C and D) COX activity (C) and ATP amount (D) in mitochondrial fractions from the same four controls and two affected individuals’ cell lines. For the determination of COX activity, we used a Cytochrome c Oxidase Assay kit (Sigma-Aldrich). Mitochondrial fractions were obtained from cells by homogenization in homogenization buffer (20 mM HEPES-KOH [pH 7.4], 220 mM mannitol, 70 mM sucrose, 1 mM EDTA, 1× protease inhibitor). The determination of COX activity was based on a colorimetric assay that quantifies the oxidation of ferrocytochrome c to ferricytochrome c via cytochrome c oxidase, a reaction that results in a decrease in absorbance at 550 nm. After measurement of absorbance, COX activity was calculated according to manufacturer’s instructions. The amount of ATP was measured using a Lumino assay detecting cellular-ATP kit (CA100; Wako Pure Chemical Industries). The collected cells (1,000 cells per well) were used according to manufacturer’s instructions. All experiments were triplicated per sample and tested using t test for the difference between controls mean and each affected individual mean. ∗∗∗p < 0.001. The error bars represent the standard deviation.
Finally, we examined commercially provided Cox6a1-null mice (Figure S8 and Table S13). All experimental procedures were performed according to the animal welfare regulations of Yamagata University Faculty of Medicine and Shiga University of Medical Science, and the study protocol was approved by the Animal Subjects Committee of Yamagata University Faculty of Medicine and Shiga University of Medical Science. The null mice showed lack of COX6A1 products (Figure 3A) and remarkable behavioral phenotypes including difficulty in walking (Figure 3B; Movie S1). Histological examinations revealed that the null mice had thinned sciatic nerves (Figure 3C) and neurogenic muscular changes including small angular fiber and small group atrophy (Figure 3D), although no remarkable neurodegeneration was found. In addition, consistent with the findings in the affected individuals, the null mice showed statistically significant reductions in COX activity and ATP contents in liver cells (p < 0.001; Figures 4A and 4B) and delayed motor NCV (p < 0.01; Figure 4C) relative to control animals.
Figure 3.
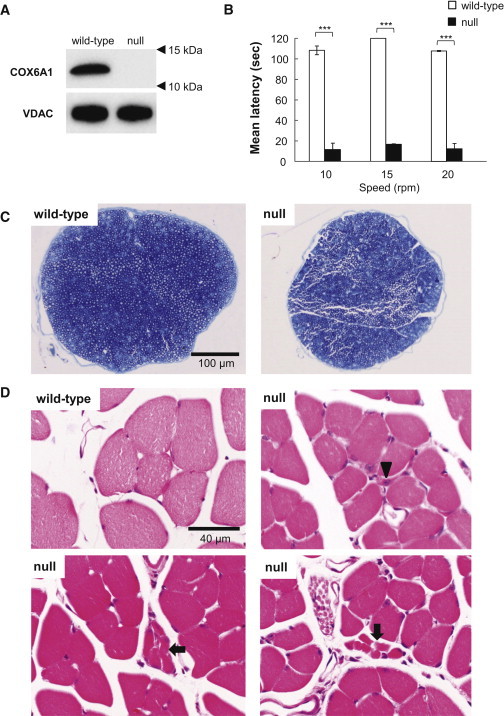
Characterization of Cox6a1 Knockout Mice
(A) Immunoblot of COX6A1 and voltage-dependent anion channel (VDAC) as control in a wild-type and Cox6a1 knockout null mice. Mice aged 7–8 weeks, three (one male and two female) knockout and three (two male and one female) wild-type were anesthetized and perfused with 10 mM PBS. Mitochondria fractions were obtained from liver tissue and applied to immunoblot. 20 μg of protein was loaded into a SDS-PAGE and transferred to the polyvinylidene difluoride membrane. The primary antibody for COX6A1 (mouse monoclonal, ab110265; abcam) is diluted 1:1,000 and secondary antibody for anti-COX6A1 (anti-mouse IgG, HRP-conjugated, 315-035-003; Jackson ImmunoResearch) is diluted 1:10,000. All blotting is carried out in 5% skim-milk/TBS solutions at room temperature for 1 hr.
(B) Motor coordination and balance was assessed as the latency to fall in the rota-rod. Mice aged 7–8 weeks, four (two male and two female) knockout and four (two male and two female) wild-type were used. Each mouse underwent the same 4 day procedure on a rota-rod (MK-660A; Muromachi Kikai). The first 3 days were used to train the mice (four sessions of 60 s each, walking at 20 rpm). The test sessions were run on the last day. The mice performed two series of three trials (10, 15, and 20 rpm) at each speed, with a 10 min rest period between trials. The latency to fall was recorded with a cut-off at 120 s. The difference between the wild-type and knockout null mice means were tested using t test. ∗∗∗p < 0.001. The error bars represent the standard deviation.
(C and D) Histological examinations by toluidine blue staining sections of mice sciatic nerves at lower magnification (C) and hematoxylin-eosin staining sections of mice lower limb muscles (D). Arrow indicates a smaller number of fibers are involved in small group atrophy and arrowhead indicates small angular fibers despite the limited numbers. Mice aged 7–8 weeks, four (two male and two female) knockout and four (two male and two female) wild-type were anesthetized and perfused with 10 mM PBS, followed by a fixative of 4% paraformaldehyde (for leg muscle) or 2.5% glutaraldehyde (for sciatic nerve) in 0.1 M phosphate buffer. Sciatic nerve specimens were fixed in 2.5% glutaraldehyde in phosphate buffer for 2 hr at room temperature. After postfixation with 1% OsO4, the tissues were embedded in epoxy resin. Tissue blocks were sectioned at 1 mm thickness and stained with toluidine blue for light microscopic examination. For the histological analysis of leg muscle, mice tissues were postfixed in 4% paraformaldehyde for 48 hr at 4°C. The muscle tissues were dissected out and then incubated overnight in 10% sucrose in phosphate buffer. After snap freezing with CO2 gas, tissue blocks were sectioned at 20 μm thickness in a cryostat and stained with hematoxylin and eosin.
Figure 4.
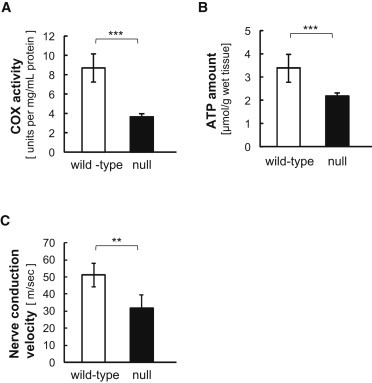
COX Activity and Electrophysiological Analysis in Mice
(A and B) COX activity in mitochondrial fractions from livers (A) and ATP amount in liver homogenates (B) from three wild-type and knockout null mice, respectively. Experiments for COX activity and ATP amount were triplicated per sample and all experiments were tested using t test for the difference between the wild-type and knockout mice means.
(C) Nerve conduction velocity of sciatic nerve from mice. Studies were demonstrated at 7–10 weeks of age, six (three male and three female) wild-type and seven (five male and two female) null mice with Sapphire (Medelec) under anesthesia of pentobarbital sodium (5 mg/kg i.p.). At the right dorsal femoral, sciatic nerve was exposed by opening up overlying skin and electrically stimulated using needle electrodes at the sciatic notch and at the knee joint level under 37°C ± 0.5°C. The compound muscle action potential (CMAP) evoked by the two parts of stimuli were recorded at gastrocnemius. Motor nerve conduction velocity (NCV) was calculated with dividing the distance between the sciatic notch and knee joint level by the delta latency between the two CMAP curves. Asterisks indicate the level of statistical significance (∗∗p < 0.01; ∗∗∗p < 0.001). The error bars represent the standard deviation.
Haplotype estimation around the linkage region reveals that the 5 bp deletion is harbored in background haplotypes that differ between family 1 and 2 (Tables 1 and S14; Figures S9 and S10), suggesting that this mutation occurred independently on each the haplotype origin. The two independent mutations in human CMT families and the null allele in mice consistently indicate that the 5 bp deletion can cause the disease phenotype through deficiency of COX6A1 leading to the reduced COX activity in affected individuals’ peripheral nerves as well. The COX6A1 is expressed in the sciatic nerve as well as other tissues including lung, kidney, liver, and brain. Despite its expression in multiple tissues, the affected individuals carrying COX6A1 mutation show no signs and symptoms other than that of the peripheral nervous system. It is interesting to note that the surfeit 1 (SURF1 [MIM 185620]) encodes one of assembly factors of COX whose mutations cause demyelinating CMT accompanying with axonal loss14 leading to multisystem involvement with nystagmus, hearing loss, kyphoscoliosis, and brain MRI abnormalities. In contrast, COX6A1 is small polypeptide required for the stability of holoenzyme. It is likely that the focal symptoms of our affected individuals are due to such narrow function, hypomorphic nature of the mutant allele, and/or the possibility for the residual amount of COX6A1 to warrant sufficient COX activity in other tissues as to maintain bioenergetic compensation. As another possibility, COX6A1 deficiency might be compensated by heart/muscle isoform, cytochrome c oxidase subunit VIa polypeptide 2 (COX6A2) in the other tissues or interfere with forming a stable COX assembly especially in the mitochondria of peripheral nervous system. Such mitochondrial respiratory chain deficiency might increase the vulnerability of nerve cells.15–18 Finally, the tissue specificity might reflect the differential vulnerability to COX6A1 deficiency among them, as shown in many known examples such as TAF1 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 250 kDa (TAF1),19 huntintin (HTT),20 and superoxide dismutase 1, soluble (SOD1).21 However, a molecular mechanism between potential dysfunctions of the respiratory chain and the disease phenotype is still unclear. Our findings warrant further mechanistic analysis of structural analysis of COX holoenzyme, COX activity, and assembly in different tissues, including brain, heart, and, as a “positive control,” skeletal muscle. To disentangle their high-order functional interplay, the results from our genetic study would warrant further mechanistic analyses of the mitochondrial involvements in the peripheral nervous system. On the other hand, the multiple mutation events resulting in the same deletion in the pyrimidine tract imply that this site may be a mutational hotspot in human, so that there is a possibility that this deletion would be found across the world, especially from families with consanguineous loop, under the same scenario as Charcot-Marie-Tooth disease type 2F (CMT2F [MIM 606595])22 or hereditary motor and sensory neuropathy, proximal type (HMSN-P [MIM 604484]).23,24
Acknowledgments
We would like to thank Kaoru Honaga, Michiyuki Kawakami, Akio Kimura, and Keiko Murayama for clinical information. This work was supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan to K.H. (grant number 25461537), A.A. (grant number 25860842), and G.T. (grant number 25129701) and Grant-in-Aid from the Global Center of Excellence program of the Japan Society for the Promotion of Science, “Formation of an International Network for Education and Research of Molecular Epidemiology.”
Contributor Information
Gen Tamiya, Email: gtamiya@genetix-h.com.
Kiyoshi Hayasaka, Email: hayasaka@med.id.yamagata-u.ac.jp.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
BioGPS, http://biogps.org/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
PROVEAN, http://provean.jcvi.org/index.php
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
SNP Genetic Mapping, http://integrin.ucd.ie/cgi-bin/rs2cm.cgi
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 2.Hayasaka K., Himoro M., Sato W., Takada G., Uyemura K., Shimizu N., Bird T.D., Conneally P.M., Chance P.F. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat. Genet. 1993;5:31–34. doi: 10.1038/ng0993-31. [DOI] [PubMed] [Google Scholar]
- 3.Züchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 4.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 5.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 8.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 9.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W., 3rd, Su A.I. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim L.P., Burge C.B. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. USA. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirose Y., Chiba K., Karasugi T., Nakajima M., Kawaguchi Y., Mikami Y., Furuichi T., Mio F., Miyake A., Miyamoto T. A functional polymorphism in THBS2 that affects alternative splicing and MMP binding is associated with lumbar-disc herniation. Am. J. Hum. Genet. 2008;82:1122–1129. doi: 10.1016/j.ajhg.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raben N., Nichols R.C., Martiniuk F., Plotz P.H. A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II) Hum. Mol. Genet. 1996;5:995–1000. doi: 10.1093/hmg/5.7.995. [DOI] [PubMed] [Google Scholar]
- 14.Echaniz-Laguna A., Ghezzi D., Chassagne M., Mayençon M., Padet S., Melchionda L., Rouvet I., Lannes B., Bozon D., Latour P. SURF1 deficiency causes demyelinating Charcot-Marie-Tooth disease. Neurology. 2013;81:1523–1530. doi: 10.1212/WNL.0b013e3182a4a518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 16.Suter U., Scherer S.S. Disease mechanisms in inherited neuropathies. Nat. Rev. Neurosci. 2003;4:714–726. doi: 10.1038/nrn1196. [DOI] [PubMed] [Google Scholar]
- 17.Pitceathly R.D., Murphy S.M., Cottenie E., Chalasani A., Sweeney M.G., Woodward C., Mudanohwo E.E., Hargreaves I., Heales S., Land J. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology. 2012;79:1145–1154. doi: 10.1212/WNL.0b013e3182698d8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rinaldi C., Grunseich C., Sevrioukova I.F., Schindler A., Horkayne-Szakaly I., Lamperti C., Landouré G., Kennerson M.L., Burnett B.G., Bönnemann C. Cowchock syndrome is associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 2012;91:1095–1102. doi: 10.1016/j.ajhg.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makino S., Kaji R., Ando S., Tomizawa M., Yasuno K., Goto S., Matsumoto S., Tabuena M.D., Maranon E., Dantes M. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am. J. Hum. Genet. 2007;80:393–406. doi: 10.1086/512129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawa A., Wiegand G.W., Cooper J., Margolis R.L., Sharp A.H., Lawler J.F., Jr., Greenamyre J.T., Snyder S.H., Ross C.A. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat. Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- 21.Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 22.Evgrafov O.V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C.L., Schagina O., Verpoorten N., Van Impe K., Fedotov V. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 23.Maeda K., Kaji R., Yasuno K., Jambaldorj J., Nodera H., Takashima H., Nakagawa M., Makino S., Tamiya G. Refinement of a locus for autosomal dominant hereditary motor and sensory neuropathy with proximal dominancy (HMSN-P) and genetic heterogeneity. J. Hum. Genet. 2007;52:907–914. doi: 10.1007/s10038-007-0193-7. [DOI] [PubMed] [Google Scholar]
- 24.Lee S.S., Lee H.J., Park J.M., Hong Y.B., Park K.D., Yoo J.H., Koo H., Jung S.C., Park H.S., Lee J.H. Proximal dominant hereditary motor and sensory neuropathy with proximal dominance association with mutation in the TRK-fused gene. JAMA Neurol. 2013;70:607–615. doi: 10.1001/jamaneurol.2013.1250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.