

Abstract
Neu-Laxova syndrome (NLS) is a rare autosomal-recessive disorder characterized by a recognizable pattern of severe malformations leading to prenatal or early postnatal lethality. Homozygous mutations in PHGDH, a gene involved in the first and limiting step in L-serine biosynthesis, were recently identified as the cause of the disease in three families. By studying a cohort of 12 unrelated families affected by NLS, we provide evidence that NLS is genetically heterogeneous and can be caused by mutations in all three genes encoding enzymes of the L-serine biosynthesis pathway. Consistent with recently reported findings, we could identify PHGDH missense mutations in three unrelated families of our cohort. Furthermore, we mapped an overlapping homozygous chromosome 9 region containing PSAT1 in four consanguineous families. This gene encodes phosphoserine aminotransferase, the enzyme for the second step in L-serine biosynthesis. We identified six families with three different missense and frameshift PSAT1 mutations fully segregating with the disease. In another family, we discovered a homozygous frameshift mutation in PSPH, the gene encoding phosphoserine phosphatase, which catalyzes the last step of L-serine biosynthesis. Interestingly, all three identified genes have been previously implicated in serine-deficiency disorders, characterized by variable neurological manifestations. Our findings expand our understanding of NLS as a disorder of the L-serine biosynthesis pathway and suggest that NLS represents the severe end of serine-deficiency disorders, demonstrating that certain complex syndromes characterized by early lethality could indeed be the extreme end of the phenotypic spectrum of already known disorders.
Main Text
Neu-Laxova syndrome (NLS [MIM 256520]) is a rare disorder that manifests with severe malformations leading to prenatal or early postnatal lethality.1,2 The hallmark clinical features of this syndrome are a characteristic facies with shortened eyelids, proptosis, and a round gaping mouth, as well as microcephaly, intrauterine growth restriction (IUGR), skin abnormalities (ichthyosis and hyperkeratosis), and flexion deformities. Additionally, limb malformations and significant edema of the hands and feet are frequently observed. Other CNS malformations, such as abnormal gyration, hypoplasia of the corpus callosum, or neural-tube defects, can also be present in NLS.3
As a result of previous reports of both consanguinity in parents of affected individuals and recurrence in subsequent pregnancies, NLS was suggested to be inherited in an autosomal-recessive manner. This was recently confirmed by the identification of homozygous mutations in phosphoglycerate dehydrogenase (PHGDH [MIM 606879]) as a cause of NLS in three consanguineous families.4 PHGDH codes for an enzyme involved in the first and limiting step in L-serine biosynthesis, and mutations in this gene have been previously described as the cause of PHGDH deficiency (MIM 601815).5,6 Here, we provide evidence that NLS is genetically heterogeneous and can also be caused by mutations in phosphoserine aminotransferase 1 (PSAT1 [MIM 610936]) and phosphoserine phosphatase (PSPH [MIM 172480]), two other genes involved in de novo L-serine biosynthesis.
We have collected 12 families in which a diagnosis of NLS was ascertained on the basis of clinical features (Table S1, available online). Eleven of these families show the typical clinical presentation of NLS, albeit with substantial variation in the severity of clinical expression (Figure S1). In one of the families (family 12), the phenotype is somewhat atypical with a lack of limb edema, only mild ichthyosis, and longer postnatal survival, as well as some clinical features that are unusual in NLS (dysplastic kidneys and pancreatic insufficiency) (Table S1). Parental consanguinity was reported in nine families (Figure S2), and microarray analysis revealed a probable distant relationship between the parents of another family (family 4, Figure S2). Because NLS causes early lethality, availability of DNA from affected individuals was a limitation in this study; for some families, only low-quality DNA extracted from formalin-fixed, paraffin-embedded material of the affected fetuses was available. In some families in which no material from a fetus with NLS was obtainable (families 5 and 10), only DNAs from parents as obligate heterozygous mutation carriers were available for study. Written informed consent for molecular studies was obtained from the parents, and the study was conducted in accordance with the principles of the Declaration of Helsinki.
In a total of seven families (five with known parental consanguinity), we initially used microarrays to scan for copy-number variants (CNVs) and genome-wide homozygosity. CNV analysis did not reveal any deletions or duplications assumed to be causative. Because of the limitations regarding material from index individuals, potential regions of homozygosity in the affected individuals from three unrelated consanguineous families were inferred by genotyping of healthy carriers only. From this analysis, no obvious candidate regions of overlapping autozygosity were shared by all or a majority of consanguineous NLS-affected families. Exome sequencing was performed in three unrelated affected fetuses with NLS (families 1, 2, and 7) and in two obligate carriers from different consanguineous families (families 3 and 11). Filtering the results for rare and private DNA sequence variants revealed no single gene harboring potentially disease-causing mutations in more than one affected individual. These findings, together with our initial results from the microarray homozygosity scan, clearly suggest genetic heterogeneity in our study cohort.
Specific reanalysis of the mapping data resulted in the identification of a large homozygous region on chromosome 1 in a consanguineous family (Figure 1 and Table 1); this region overlaps the recently published locus of PHGDH, in which mutations can cause NLS.4 Indeed, we identified a missense mutation (c.793G>A [p.Glu265Lys], RefSeq accession number NM_006623.3) in this family (family 8). Additionally, analysis of the exome of the affected fetus from family 7 showed a heterozygous mutation in PHGDH (c.160C>T [p.Arg54Cys]). A second mutation in PHGDH was only identified after an additional high-resolution microarray analysis, which revealed a small intragenic deletion of at least three exons (Figure S3). We screened the remaining families of our cohort for mutations in PHGDH by Sanger sequencing and identified another homozygous missense mutation (c.856G>C [p.Ala286Pro]) in PHGDH in the index individual from family 9. Thus, in total, three PHGDH missense mutations, (c.160C>T [p.Arg54Cys], c.793G>A [p.Glu265Lys], and c.856G>C [p.Ala286Pro]) were observed in our cohort. Modeling and 3D-structure analysis predict all three residues (PHGDH Arg54, Glu265, and Ala286) to be in close proximity to the interacting site of PHGDH with its substrate, 3-phosphoglycerate (Figure 2). Furthermore, the identified substitutions are predicted to cause steric clashes with the side chains of neighboring residues, which could disrupt binding of the substrate to the enzyme. Interestingly, Arg54 is predicted to form a binding site with residue Arg135, in which a missense substitution (p.Arg135Trp) was recently reported in a newborn with NLS.4
Figure 1.
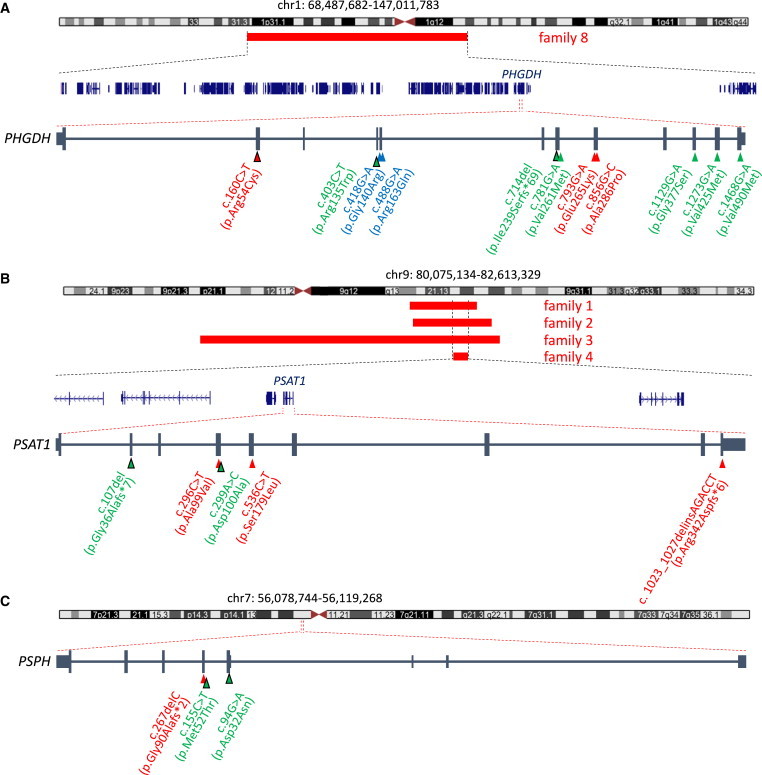
Identification of Mutations in PHGDH, PSAT1, and PSPH in Individuals with NLS by Homozygosity Mapping and Exome and Sanger Sequencing
(A) In one family (family 8), a large homozygous region (68.4–147.0 Mb) on chromosome 1 was identified. This region overlaps the gene PHGDH, in which a mutation has recently been identified to cause NLS. The mutations found in families 7–9 are depicted at the cDNA level (red triangles) with their protein consequences below. PHGDH mutations were identified in a total of three individuals with NLS. These are compared to PHGDH mutations previously identified in individuals with serine-deficiency syndrome (MIM 614023; green triangles)5,6 and NLS (blue triangles).4
(B) Schematic overview of homozygous regions mapped to chromosome 9 in affected individuals from families 1–4. Overlapping homozygous regions (red bars) were identified in four families (family 1 by WES, families 2–4 by microarray analysis); the smallest region of overlap (80.1–82.6 Mb) mapped to chromosome 9. This region contains five protein-coding RefSeq genes (shown in blue), one of which is PSAT1. The mutations found in families 1–6 are depicted at the cDNA level (red triangles) with their protein consequences below. PSAT1 mutations were identified in a total of six individuals with NLS. These are compared to PSAT1 mutations previously identified in an individual with serine-deficiency syndrome (green triangles).7
(C) In one family (family 10), we identified a homozygous PSPH frameshift mutation (56.0–56.1 Mb) located on chromosome 7. The mutations are depicted at the cDNA level (red triangle) with their protein consequences below. A PSPH mutation has been previously reported in a child with serine-deficiency syndrome and Williams-Beuren syndrome (green triangle).8
Triangles outlined in black depict heterozygous mutations, whereas triangles with no outline depict homozygous mutations.
Table 1.
Summary of Genetic Findings in 12 Probands with NLS
| Proband | Gene | cDNA Mutation | Nucleotide Conservation (PhyloP) | EVS Frequency | Protein Substitution | Mode of Mutation Identification | Homozygosity |
|---|---|---|---|---|---|---|---|
| 1a | PSAT1 | c.1023_1027delinsAGACCT | NA | 0 | p.Arg342Aspfs∗6 | WES and CE | chr9: 70,999,315–84,300,694 (rs17081271–rs2244337)b |
| 2a | PSAT1 | c.296C>T | 4.08 | 0 | p.Ala99Val | WES | chr9: 71,704,827–87,455,347 (rs953588–rs4486281)c |
| 3a | PSAT1 | c.536C>T | 6.26 | 0 | p.Ser179Leu | WES of father | chr9: 27,601,332–89,030,832 (rs480866–rs416513)c |
| 4d | PSAT1 | c.296C>T | 4.08 | 0 | p.Ala99Val | CE | chr9: 80,075,134–82,613,329 (rs1547190–rs10780348)c,e |
| 5af | PSAT1 | c.296C>T | 4.08 | 0 | p.Ala99Val | CE | NA |
| 6 | PSAT1 | c.[296C>T];[536C>T] | 4.08; 6.26 | 0; 0 | p.[Ala99Val];[Ser179Leu] | CE | NA |
| 7 | PHGDH | c.160C>T | 1.9 | 0 | p.Arg54Cys; deletione | WES | NA |
| 8a | PHGDH | c.793G>A | 5.53 | 0 | p.Glu265Lys | CE | chr1: 68,487,682–147,011,783 (rs11209200–rs3806218)c |
| 9a | PHGDH | c.856G>C | 3.11 | 0 | p.Ala286Pro | CE | NA |
| 10af | PSPH | c.267delC | NA | 0 | p.Gly90Alafs∗2 | CE | NA |
| 11a | PSPH | c.662G>A | NA | NA | p.Gly221Glug | WES | no overlap with the three genes |
| 12a | none | NA | NA | NA | NA | NA | no overlap with the three genes |
Abbreviations are as follows: CE, capillary electrophoresis and Sanger sequencing; EVS, NHLBI Exome Sequencing Project Exome Variant Server; NA, not available; and WES, whole-exome sequencing.
Consanguineous.
LOH mapped by WES.
LOH mapped by array.
Distant relationship of parents was shown by array data.
Minimally deleted region is chr1: 120,276,386–120,280,721.
No material from the index individual was available for testing.
On the basis of linkage and segregation data, this variant is not considered disease causing.
Figure 2.
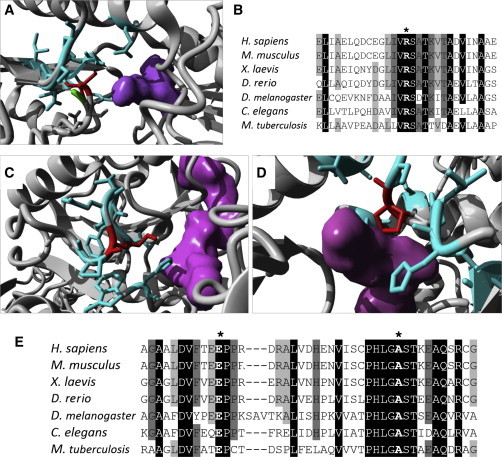
Modeling of PHGDH Substitutions
(A, C, and D) 3D models of human PHGDH show in red the substitutions p.Arg54Cys (A), p.Glu265Lys (C), and p.Ala286Pro (D). All three substitutions lead to clashes with the side chains of neighboring residues (cyan) in close proximity to the binding site of phosphoglycerate and NAD+ (shown in purple).
(B and E) Alignment of PHGDH sequence from multiple organisms shows high conservation of residues Arg54 (B, marked by an asterisk) and Glu265 and Ala286 (E, marked by the left and right asterisks, respectively). Protein sequences for PHGDH orthologs in Homo sapiens (UniProt ID O43175), Mus musculus (UniProt ID Q61753), Xenopus laevis (RefSeq NP_001015929.1), Danio rerio (RefSeq NP_955871.1), Drosophila melanogaster (RefSeq NP_609496.1), Caenorhabditis elegans (RefSeq NP_496868.1), and Mycobacterium tuberculosis (UniProt ID P9WNX3) were obtained from UniProt and Entrez. The 3D structure of wild-type human PHGDH (Protein Data Bank [PDB] ID 2G76) was obtained from the PDB, and substitutions were modeled with the FoldX plugin9 for YASARA.
Exome sequencing data allowed us to map regions of homozygosity in the index individual from family 1, which had not been previously investigated by array-based mapping (Figure S2).10 We identified a large homozygous chromosome 9 region overlapping homozygous intervals in three other families that had previously undergone microarray-based homozygosity analysis (families 2–4). The combined mapping data result in the smallest region of overlap at chr9: 80,075,134–82,613,329 (rs1547190–rs10780348, see Table 1), which contains only five RefSeq genes (Figure 1). Interestingly, one of the genes in this region, PSAT1, encodes an enzyme involved in the L-serine biosynthesis pathway (Figure 3), the same pathway that has already been implicated in the pathogenesis of NLS.4 Moreover, severe craniofacial and brain malformations entailing early lethality have been described in mice with homozygous Psat1 knockout. Indeed, reanalysis of exome sequencing data of the three unrelated fetuses with NLS and two obligate carriers from different families resulted in the identification of mutations in PSAT1 in three of these five families. A homozygous complex indel (c.1023_1027delinsAGACCT [p.Arg342Aspfs∗6], RefSeq NM_058179.3) leading to a frameshift was found in the index individual of family 1. Of note, this complex variant was not correctly called by initial exome sequencing (Figure S4). Furthermore, in the index individual from family 2, we identified a homozygous missense mutation (c.296C>T [p.Ala99Val]), which was also missed in the first analysis. This variant is in direct proximity to a known SNP (rs3739474), which prohibited initial calling of the c.296C>T mutation from color-space exome data (Figure S4). Finally, exome sequencing revealed a heterozygous missense change (c.536C>T [p.Ser179Leu]) in the obligate carrier parent of family 3. We screened the remainder of our cohort for mutations in PSAT1 and identified the same mutation seen in family 2, c.296C>T (p.Ala99Val), in three additional families. Homozygous c.296C>T mutations were found in three of the four families, whereas compound-heterozygous mutations in PSAT1 (c.[296C>T];[536C>T], p.[Ala99Val];[Ser179Leu]) were identified in the remaining one (family 6). All four families with the c.296C>T mutation are from the Middle East and have either Iranian or Turkish ancestry, which suggests a founder mutation. This is supported by the finding that two unrelated families of Turkish origin from our cohort (families 2 and 4) share a 0.74 Mb disease-associated haplotype, including the c.296C>T (p.Ala99Val) mutation.
Figure 3.
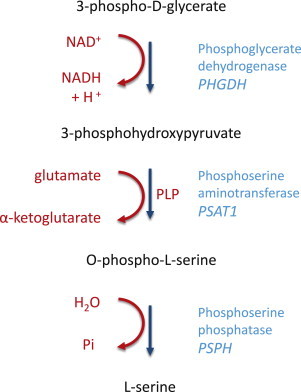
L-Serine Biosynthesis Pathway
De novo biosynthesis of serine is crucial to provide the organism with sufficient levels of this amino acid. The first and limiting step of this pathway is the conversion of 3-phosphoglycerate to 3-phosphohydroxypiruvate by PHGDH (encoded by PHGDH). This is followed by the conversion of 3-phosphohydroxypiruvate to O-phosphoserine by PSAT (encoded by PSAT1). This reaction is accompanied by the transformation of glutamate to α-ketoglutarate and requires the presence of pyridoxal phosphate (PLP). The final step in serine biosynthesis is catalyzed by PSPH (encoded by PSPH), which gives rise to L-serine, the final product of this pathway.
PSAT1 encodes phosphoserine aminotransferase (PSAT), which is the enzyme catalyzing the conversion of 3-phosphohydroxypyruvate into 3-phosphoserine (Figure 3), an essential step in the biosynthesis of serine. To better understand their impact on protein structure and function, we modeled the identified substitutions on the 3D structure of human and Escherichia coli PSAT (Figure 4). The recurrently observed PSAT substitution (p.Ala99Val) affects a highly conserved residue (PhyloP = 4.08) within a beta sheet and is predicted by SIFT and PolyPhen to be deleterious. Furthermore, 3D modeling of PSAT p.Ala99Val predicts that this change leads to protein instability (Figure 4). Interestingly, a mutation resulting in a substitution (c.299A>C [p.Asp100Ala]) has been previously identified in siblings with a nonlethal form of PSAT deficiency (MIM 610992),7 suggesting that this region of the protein is particularly susceptible to functional impairment when altered. The second missense substitution in PSAT, p.Ser179Leu, changes a highly conserved residue (PhyloP = 6.26) and is also predicted to be deleterious by SIFT and PolyPhen. Interestingly, Ser179 is located in a PSAT region close to the binding site of pyridoxal phosphate (PLP), an essential coenzyme in this transamination reaction. 3D modeling of this substitution shows that p.Ser179Leu alters the localization of the side chain of Lys200, which forms part of the binding site for PLP (Figure 4B). This would potentially interfere with the binding of PSAT to its cofactor and therefore hinder the transamination reaction. Finally, the complex indel mutation found in a single family (family 1), PSAT1 c.1023_1027delinsAGACCT (p.Arg342Aspfs∗6), is a frameshift mutation located in the last exon of PSAT1. We observed normal mRNA levels in fibroblasts of a skin biopsy obtained from the stillborn fetus (family 1) at 36 weeks of gestation (Figure S5), supporting that this frameshift mutation does not lead to an unstable RNA and might in fact result in the presence of a protein with an altered C terminus. We analyzed the predicted structural consequences of this substitution, in which the last 30 amino acids of the wild-type protein are replaced by an aberrant sequence of six amino acids. Strikingly, the C-terminal region of PSAT is in close proximity to the area where glutamate, the donor of an amino group in this step, interacts with the enzyme. It is most likely that this complex substitution leads to a radical change in a protein region that is crucial for interaction with glutamate, thus disrupting the function of PSAT (Figure 4).
Figure 4.
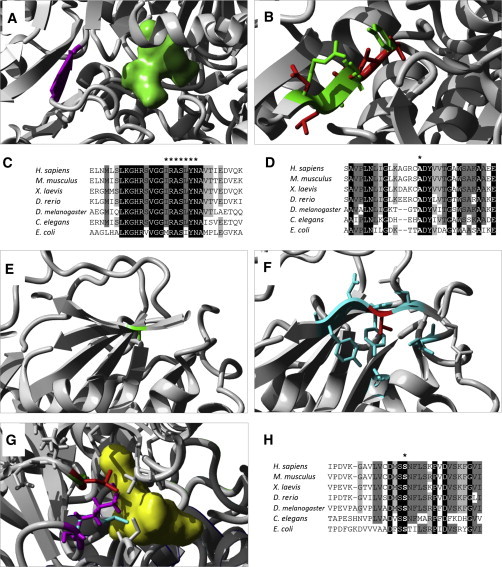
Modeling of PSAT1 Substitutions
(A) A 3D model of PSAT from Escherichia coli shows glutamate (molecule in green) localizing closely to the beta sheet where Arg335 (corresponding to human Arg342 and highlighted in purple) is located.
(B) Modeling of multiple residue substitutions resulting from frameshift p.Arg342fs6∗, in which the side chain of the wild-type residues (in green) overlaps the side chain of the altered residues (in red).
(C) Alignment of PSAT sequence from multiple organisms shows the high conservation of the region surrounding Arg342. The seven altered residues resulting from the frameshift are marked by an asterisk.
(D) Alignment of PSAT sequence shows high conservation of Ala99 (marked by an asterisk).
(E and F) 3D model of human PSAT substitution p.Ala99Val. Residue Ala99 of PSAT is located in a beta sheet (E, in green). Substitution p.Ala99Val (F, in red) is predicted to disrupt the beta sheet by creating clashes between the large side chains of neighboring residues (shown in cyan).
(G) A 3D model of PSAT from E. coli (in white) shows its binding to PLP (molecule in yellow) through the side chain of Lys198 (corresponding to human Lys200, in cyan). When residue Ser177 (corresponding to human Ser179, shown in green) is changed to a leucine (visible in red), the side chain of Lys198 (purple) is displaced and interferes with the binding of PLP.
(H) Alignment of PSAT sequences from multiple organisms shows high conservation of residue S179. Protein sequences for PSAT1 orthologs in Homo sapiens (UniProt ID Q9Y617), Mus musculus (UniProt ID Q99K85), Xenopus laevis (RefSeq NP_001016582.1), Danio rerio (RefSeq NP_956113.1), Drosophila melanogaster (UniProt ID Q9VAN0), Caenorhabditis elegans (UniProt ID P91856), and Escherichia coli (UniProt ID Q8XEA7) were obtained from Uniprot and Entrez. 3D models for human PSAT (PDB ID 3E77) and E. coli serC (PDB ID 1BJO11) were obtained from the PDB.
Given that we found mutations in two genes involved in the serine biosynthesis pathway in the context of NLS, we screened the remainder of our cohort for mutations in PSPH, the gene encoding the third enzyme of this pathway (Figure 3). Using Sanger sequencing, we identified a homozygous frameshift mutation in PSPH (c.267delC [p.Gly90Alafs∗2], RefSeq NM_004577.3) in family 10. This single-base-pair deletion in PSPH is predicted to result in a frameshift leading to a premature stop codon. Because of the mutation’s location, the transcribed RNA is predicted to be unstable and undergo nonsense-mediated decay or to be translated into a significantly truncated, nonfunctional protein. Another PSPH variant, c.662G>A (p.Gly221Glu), was identified in a large family with multiple affected children in two branches of the pedigree (family 11). However, segregation in the expected pattern could not be confirmed (Figure S2), and therefore this variant is not considered disease causing. Furthermore, mapping based on a single disease-associated allele in this multiconsanguineous pedigree was negative for all three loci. In another consanguineous family (family 12), no linkage to any of the three loci was found, and sequencing of the three genes was negative (Figure S2).
All identified sequence variations considered to be disease-causing mutations were validated and shown to segregate with the disease in all families where possible (Figure S2). None of the mutations identified in our cohort have been observed in any of the more than 6,500 individuals reported in the NHLBI Exome Sequencing Project Exome Variant Server. Further supporting the essential role of the serine-biosynthesis-pathway-associated genes PHGDH, PSAT1, and PSPH is the fact that in these genes, truncating variants leading to loss of function are also extremely rare in control individuals; no loss-of-function variants were seen in PSPH and PHGDH, and only two different loss-of-function alleles, observed in just 1 and 3 of 13,006 chromosomes, respectively, have been reported for PSAT1.
We have identified mutations in all three essential genes involved in the L-serine biosynthesis pathway (Figure 3) in 10 out of 12 families affected by NLS. Of these, six families have mutations in PSAT1, one has mutations in PSPH, and only three were found to have mutations in PHGDH, the gene previously described as associated with NLS.4 In addition to the recently published work, our findings show that NLS is a heterogeneous disorder and clearly link this distinct phenotype to the serine de novo biosynthesis pathway.4 Whereas the previous study identified only PHGDH mutations in all investigated NLS-affected families, we found PSAT1 to be the predominant gene in our cohort. It is likely, however, that population selection accounts for these differences. Indeed, we found evidence of a common PSAT1 founder allele in the Turkish-Iranian part of our cohort, and a probable Arab PHGDH founder mutation was identified in the previously published cohort.4 We failed to clearly link the disease to one of the three genes involved in the L-serine biosynthesis pathway in two families, thus leaving the possibility of further genetic heterogeneity of NLS.
Genes involved in the L-serine biosynthesis pathway have been previously described to be associated with a group of congenital metabolism disorders subsumed under the term serine-deficiency disorders.5–8,12 Interestingly, although certain clinical features of these disorders, such as microcephaly and IUGR, overlap with those of NLS, the overall phenotype is significantly less severe and even has adolescent and adult forms. However, previous reports on disorders of serine biosynthesis also include some affected individuals with lethality during infancy.7 This suggests that NLS represents a more severe form within a continuous spectrum of serine-deficiency disorders, and NLS itself might also have variable clinical expression. In this context, it is remarkable that our NLS individuals carrying the PSAT1 mutation c.296C>T (p.Ala99Val) present with a somewhat milder expression of NLS (Figure S1), and a nearby mutation resulting in substitution p.Asp100Ala has been previously identified in a severe form of serine-biosynthesis deficiency.7 It is tempting to hypothesize that the broad phenotypic spectrum observed in disorders of L-serine biosynthesis is probably mainly determined by the level of residual enzyme activity. Indeed, residual enzyme activities measured in persons with nonlethal forms of serine deficiency range between 12% and 25%.4,5 We postulate that the range of enzyme activity in individuals with NLS is even below these levels; however, we had no access to respective samples to test this hypothesis in the current study.
NLS is not the first disorder in which prenatal lethality is caused by mutations in genes previously described as associated with nonlethal diseases; other examples include Smith-Lemli-Opitz syndrome (MIM 270400),13 Cenani-Lenz syndactyly syndrome (MIM 212780),14 and the wide phenotype spectrum associated with mutations in FLNA (MIM 300017).15 Such phenotypic variability might be the direct consequence of the nature of the causative mutation (for instance, hypomorphic versus null-alleles mutations), the zygosity of the condition (for example, a double-allelic hit more severe than a single heterozygous mutation in a gene), and modifier genes or compensatory pathways. The mutation spectrum observed in our cohort includes a majority of missense changes, one frameshift mutation in the last exon of PSAT1, a small intragenic deletion in PHGDH, and only one mutation predicted to represent a functional “null” allele in PSPH. The lack of complete recessive null mutations in PSAT1 and PHGDH in our and the previously published NLS-affected families4 suggests that NLS-associated altered PSAT and PHGDH might still retain some residual function. We thus speculate that NLS is still not the ultimate end of the spectrum and that the most severe mutations leading to a complete defect of PSAT or PHGDH might lead to a phenotype with very early prenatal lethality that has not yet been recognized; this is in line with the finding that mice with homozygous Phgdh knockout show an embryonically lethal phenotype.16 Notably, the only PSPH mutation observed in NLS in one of the families reported here (family 10) is considered to represent a functional null mutation. On the basis of the hypothesis raised above, this could indicate that the last phosphatase step in the L-serine biosynthesis pathway could be bypassed to some extent; low levels of dephosphorylation of O-phosphoserine might occur even in the absence of this enzyme either spontaneously or in response to catalysis by other phosphoserine phosphohydrolases.
Taken together, the human phenotypes of PHGDH, PSAT, and PSPH deficiencies, as well as early lethality in Phgdh- and Psat1-knockout mice, emphasize the critical role of appropriate serine availability in early embryonic and fetal development. Alternatively to de novo synthesis, L-serine can also be derived from three possible other sources: dietary or transplacental intake, degradation of protein and phospholipids, or direct synthesis from glycine by serine hydroxymethyltransferase. Considering the metabolic basis of NLS, it becomes clear that under physiological conditions, these alternative sources cannot compensate for a defect of the L-serine biosynthesis pathway, particularly in the developing fetus. We nevertheless hypothesize that NLS might be a treatable condition when recognized and treated early enough. Hart et al.7 have already shown that treatment with serine and glycine supplementation can be successful in PSAT deficiency when administered, similarly to other serine therapies, immediately after birth.17,18 The main challenge for NLS, however, remains that the first clinical signs might be recognized too late. Shaheen et al. therefore suggested a supplement therapy in subsequent pregnancies for parents who previously had a child affected by NLS.4
Although it remains speculative at this stage, there are some indications on the mechanism through which severe serine deficiency causes significant growth restriction and multiple malformations as seen in NLS. It has been shown at both the cellular19 and the organism levels20,21 that cell proliferation requires high levels of serine to replenish the one-carbon pool required for synthesizing nucleotides and other cellular components.22 Increased replication is sustained by higher serine production from the biosynthesis pathway by increased expression of PHGDH and PSAT1, as seen experimentally in embryonic stem cells,23 or by upregulation or amplification of these genes, as seen in cancer cells.19,20 Taking into account the importance of an appropriate serine supply in proliferating cells, we hypothesize that the impairment of proliferation and nucleotide synthesis in the context of embryonic development represents the major pathophysiological mechanism in NLS. This can certainly cause severe IUGR and microcephaly but might also—in analogy to folate deficiency—explain other malformations that sometimes accompany NLS, such as cleft lip and palate, brain malformations, and spina bifida. NLS represents a metabolic disorder among the heterogeneous group of disorders associated with microcephalic primordial dwarfism (i.e., severe generalized growth impairment).
In conclusion, we provide evidence that the early lethal condition NLS is a genetically heterogeneous disorder that can be caused by mutations in all three genes involved in de novo L-serine biosynthesis: PHGDH, PSAT1, and PSPH. Our findings place NLS at the severe end of the spectrum of serine-deficiency disorders, rare metabolic disorders characterized by neurological manifestations. Thus, this group of disorders represents a paradigmatic example of lethal conditions constituting the most severe end of already known human diseases, and it is therefore of great interest to study lethal human conditions more systematically.24,25 Focusing future studies on disorders causing prenatal lethality is also crucial for identifying genes that are essential for embryonic or fetal development and in which mutations in live-born infants are never seen.
Acknowledgments
The authors are grateful to the Radboud Genomics Technology Center, Saskia Velde-Vissers, and all members of the Genomic Disorders Group and Developmental Genomics Group for technical assistance. A.H. was supported by the Netherlands Organization for Health Research and Development (ZonMW 916-12-095). R.A.-H. was supported by a Radboudumc PhD grant. A.N., G.G., M.N., and D.N were supported through the regional agreement on medical training and clinical research between Stockholm County Council and Karolinska Institutet by grants from Kronprinsessan Lovisa, Frimurare Barnhuset i Stockholm, Karolinska Institutet, and the Swedish Childhood Cancer Foundation. This work was supported in part by the Care4Rare Canada Consortium with funding from Genome Canada and the Canadian Institutes of Health Research.
Contributor Information
Andreas R. Janecke, Email: andreas.janecke@i-med.ac.at.
Alexander Hoischen, Email: alexander.hoischen@radboudumc.nl.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Mouse Genome Informatics, Psat1-knockout mouse phenotype, http://www.informatics.jax.org/allele/MGI:4363603
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Protein Data Bank in Europe, structure of human PHGDH, http://www.ebi.ac.uk/pdbe-srv/view/entry/2g76/summary
Protein Data Bank in Europe, structure of human PSAT, http://www.ebi.ac.uk/pdbe-srv/view/entry/3e77/summary
UniProt, http://www.uniprot.org/
YASARA, http://www.yasara.org
References
- 1.Neu R.L., Kajii T., Gardner L.I., Nagyfy S.F. A lethal syndrome of microcephaly with multiple congenital anomalies in three siblings. Pediatrics. 1971;47:610–612. [PubMed] [Google Scholar]
- 2.Laxova R., Ohara P.T., Timothy J.A. A further example of a lethal autosomal recessive condition in sibs. J. Ment. Defic. Res. 1972;16:139–143. doi: 10.1111/j.1365-2788.1972.tb01585.x. [DOI] [PubMed] [Google Scholar]
- 3.Manning M.A., Cunniff C.M., Colby C.E., El-Sayed Y.Y., Hoyme H.E. Neu-Laxova syndrome: detailed prenatal diagnostic and post-mortem findings and literature review. Am. J. Med. Genet. A. 2004;125A:240–249. doi: 10.1002/ajmg.a.20467. [DOI] [PubMed] [Google Scholar]
- 4.Shaheen R., Rahbeeni Z., Alhashem A., Faqeih E., Zhao Q., Xiong Y., Almoisheer A., Al-Qattan S.M., Almadani H.A., Al-Onazi N. Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. Am. J. Hum. Genet. 2014;94:898–904. doi: 10.1016/j.ajhg.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabatabaie L., de Koning T.J., Geboers A.J., van den Berg I.E., Berger R., Klomp L.W. Novel mutations in 3-phosphoglycerate dehydrogenase (PHGDH) are distributed throughout the protein and result in altered enzyme kinetics. Hum. Mutat. 2009;30:749–756. doi: 10.1002/humu.20934. [DOI] [PubMed] [Google Scholar]
- 6.Klomp L.W., de Koning T.J., Malingré H.E., van Beurden E.A., Brink M., Opdam F.L., Duran M., Jaeken J., Pineda M., Van Maldergem L. Molecular characterization of 3-phosphoglycerate dehydrogenase deficiency—a neurometabolic disorder associated with reduced L-serine biosynthesis. Am. J. Hum. Genet. 2000;67:1389–1399. doi: 10.1086/316886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart C.E., Race V., Achouri Y., Wiame E., Sharrard M., Olpin S.E., Watkinson J., Bonham J.R., Jaeken J., Matthijs G., Van Schaftingen E. Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am. J. Hum. Genet. 2007;80:931–937. doi: 10.1086/517888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veiga-da-Cunha M., Collet J.-F., Prieur B., Jaeken J., Peeraer Y., Rabbijns A., Van Schaftingen E. Mutations responsible for 3-phosphoserine phosphatase deficiency. Eur. J. Hum. Genet. 2004;12:163–166. doi: 10.1038/sj.ejhg.5201083. [DOI] [PubMed] [Google Scholar]
- 9.Van Durme J., Delgado J., Stricher F., Serrano L., Schymkowitz J., Rousseau F. A graphical interface for the FoldX forcefield. Bioinformatics. 2011;27:1711–1712. doi: 10.1093/bioinformatics/btr254. [DOI] [PubMed] [Google Scholar]
- 10.Stránecký V., Hoischen A., Hartmannová H., Zaki M.S., Chaudhary A., Zudaire E., Nosková L., Barešová V., Přistoupilová A., Hodaňová K. Mutations in ANTXR1 cause GAPO syndrome. Am. J. Hum. Genet. 2013;92:792–799. doi: 10.1016/j.ajhg.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hester G., Stark W., Moser M., Kallen J., Marković-Housley Z., Jansonius J.N. Crystal structure of phosphoserine aminotransferase from Escherichia coli at 2.3 A resolution: comparison of the unligated enzyme and a complex with α-methyl-l-glutamate. J. Mol. Biol. 1999;286:829–850. doi: 10.1006/jmbi.1998.2506. [DOI] [PubMed] [Google Scholar]
- 12.van der Crabben S.N., Verhoeven-Duif N.M., Brilstra E.H., Van Maldergem L., Coskun T., Rubio-Gozalbo E., Berger R., de Koning T.J. An update on serine deficiency disorders. J. Inherit. Metab. Dis. 2013;36:613–619. doi: 10.1007/s10545-013-9592-4. [DOI] [PubMed] [Google Scholar]
- 13.Quélin C., Loget P., Verloes A., Bazin A., Bessières B., Laquerrière A., Patrier S., Grigorescu R., Encha-Razavi F., Delahaye S. Phenotypic spectrum of fetal Smith-Lemli-Opitz syndrome. Eur. J. Med. Genet. 2012;55:81–90. doi: 10.1016/j.ejmg.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Lindy A.S., Bupp C.P., McGee S.J., Steed E., Stevenson R.E., Basehore M.J., Friez M.J. Truncating mutations in LRP4 lead to a prenatal lethal form of Cenani-Lenz syndrome. Am. J. Med. Genet. A. 2014 doi: 10.1002/ajmg.a.36647. Published online June 12, 2014. [DOI] [PubMed] [Google Scholar]
- 15.Robertson S.P., Twigg S.R.F., Sutherland-Smith A.J., Biancalana V., Gorlin R.J., Horn D., Kenwrick S.J., Kim C.A., Morava E., Newbury-Ecob R., OPD-spectrum Disorders Clinical Collaborative Group Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat. Genet. 2003;33:487–491. doi: 10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida K., Furuya S., Osuka S., Mitoma J., Shinoda Y., Watanabe M., Azuma N., Tanaka H., Hashikawa T., Itohara S., Hirabayashi Y. Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J. Biol. Chem. 2004;279:3573–3577. doi: 10.1074/jbc.C300507200. [DOI] [PubMed] [Google Scholar]
- 17.de Koning T.J., Klomp L.W.J., van Oppen A.C., Beemer F.A., Dorland L., van den Berg I., Berger R. Prenatal and early postnatal treatment in 3-phosphoglycerate-dehydrogenase deficiency. Lancet. 2004;364:2221–2222. doi: 10.1016/S0140-6736(04)17596-X. [DOI] [PubMed] [Google Scholar]
- 18.de Koning T.J. Treatment with amino acids in serine deficiency disorders. J. Inherit. Metab. Dis. 2006;29:347–351. doi: 10.1007/s10545-006-0269-0. [DOI] [PubMed] [Google Scholar]
- 19.Vié N., Copois V., Bascoul-Mollevi C., Denis V., Bec N., Robert B., Fraslon C., Conseiller E., Molina F., Larroque C. Overexpression of phosphoserine aminotransferase PSAT1 stimulates cell growth and increases chemoresistance of colon cancer cells. Mol. Cancer. 2008;7:14. doi: 10.1186/1476-4598-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Possemato R., Marks K.M., Shaul Y.D., Pacold M.E., Kim D., Birsoy K., Sethumadhavan S., Woo H.-K., Jang H.G., Jha A.K. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amelio I., Markert E.K., Rufini A., Antonov A.V., Sayan B.S., Tucci P., Agostini M., Mineo T.C., Levine A.J., Melino G. p73 regulates serine biosynthesis in cancer. Oncogene. 2013 doi: 10.1038/onc.2013.456. Published online November 4, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Labuschagne C.F., van den Broek N.J.F., Mackay G.M., Vousden K.H., Maddocks O.D.K. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–1258. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 23.Tedeschi P.M., Markert E.K., Gounder M., Lin H., Dvorzhinski D., Dolfi S.C., Chan L.L.-Y., Qiu J., DiPaola R.S., Hirshfield K.M. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013;4:e877. doi: 10.1038/cddis.2013.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carss K.J., Hillman S.C., Parthiban V., McMullan D.J., Maher E.R., Kilby M.D., Hurles M.E. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum. Mol. Genet. 2014;23:3269–3277. doi: 10.1093/hmg/ddu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serra-Juhé C., Rodríguez-Santiago B., Cuscó I., Vendrell T., Camats N., Torán N., Pérez-Jurado L.A. Contribution of rare copy number variants to isolated human malformations. PLoS ONE. 2012;7:e45530. doi: 10.1371/journal.pone.0045530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.