

Abstract
The TRIpartite Motif (TRIM) family of RING-domain-containing proteins participate in a variety of cellular functions. The β-transducin repeat-containing protein (β-TrCP), a component of the Skp-Cullin-F-box-containing (SCF) E3 ubiquitin ligase complex, recognizes the NF-κB inhibitor IκBα and precursor p100 for proteasomal degradation and processing respectively. β-TrCP thus plays a critical role in both canonical and non-canonical NF-κB activation. Here, we report TRIM9 is a negative regulator of NF-κBactivation. Interaction between the phosphorylated degron motif of TRIM9 and the WD40 repeat region of β-TrCP prevented β-TrCP from binding its substrates, stabilizing IκBα and p100 and thereby blocking NF-κB activation. Consequently, expression or depletion of the TRIM9 gene significantly affected NF-κB-induced inflammatory cytokine production. This study not only elucidates a mechanism for TRIM9-mediated regulation of the β-TrCP SCF complex activity, but also identifies TRIM9 as a brain-specific negative regulator of the NF-κB pro-inflammatory signaling pathway.
Introduction
The nuclear factor-κB (NF-κB) transcription factor is a critical regulator of immediate responses to pathogens and also plays an important role in regulating cell proliferation and survival1–3. Since unchecked regulation of NF-κBis linked to inflammation, cancer, autoimmune diseases, and viral infection, both positive and negative regulation of the NF-κB pathway have been the subjects of intense study4–6. NF-κB signaling is divided into the canonical and non-canonical pathways7, 8. Most NF-κBactivating stimuli that engage distinct cell-surface receptors (e.g. Toll like receptors, Interleukin-1 receptor and tumor necrosis factor receptor) or cytoplasmic sensors (RIG-I-like receptors and nucleotide-binding oligomerization domain receptors) induce the canonical response, which is dependent on the IκB kinase (IKK) complex. This IKK complex contains the catalytic subunits IKKα and IKKβ, and the scaffold protein NF-κB essential modulator (NEMO; also called IKK-γ)9. In non-stimulated cells, NF-κB is held in the cytoplasm in its latent form by a family of inhibitory factors, IκB. Upon stimulation, the IKK complex is activated and phosphorylates the two N-terminal serine residues (S32 and S36) of the inhibitory IκBα protein, triggering recognition and ubiquitination by a complex composed of SKP1–CUL1–F-box protein (SCF) and β-transducin repeat-containing protein (β-TrCP). UbiquitinatedIκBα is consequently degraded through the 26S-proteasome pathway10, 11. Degradation of IκB releases the NF-κB p50/p65 complex from its latent form, allowing nuclear translocation to ultimately result in NF-κB-mediated gene expression. The non-canonical NF-κB pathway involves different signaling molecules that depend on NF-κB2 (p100) processing12. Genetic studies show that NF-κB inducing kinase (NIK) and IKKα 13 are critical kinases for phosphorylation of the C-terminal S866 and S870 residues of p100 to generate the binding motif for β-TrCP14. Subsequently, p100 undergoes ubiquitination at its C-terminus and degradation, which not only generates the p52 subunit, but also leads to the nuclear translocation of NF-κB to turn on a large number of target genes. Therefore, the SCF-β-TrCP complex plays a central role in both the canonical and non-canonical NF-κB activation pathways.
β-TrCP belongs to the F-box protein family. It contains seven C-terminal WD40 repeats that recognize substrates, and an N-terminal F-box that recruits SKP1 to form the so-called SCF E3 complex. β-TrCP is a critical regulator of many cellular processes such as cell cycle, proliferation and development11. Mammals have two β-TrCPparalogues with indistinguishable biochemical properties: β-TrCP1 (also known as FBXW1, FBW1A and FWD1) and β-TrCP2 (also known as FBXW11, FBW11, FBXW1B, FBX1B and HOS) and herein β-TrCP is used to represent both of them. β-TrCP is known to recognize the consensus degron (DSGX(2+n)S) motif and its variants in which the serine residues are phosphorylated by specific kinases. A number of growth controlling factors have been identified as β-TrCP substrates15, including IκBα NF-κB inhibitor, p100 NF-κB precursor, FOXO3 tumor suppressor, Cdc25A cell cycle regulator, β-catenin, and Mdm2 oncogenes11. Additionally, a number of viral proteins are reported to directly or indirectly target β-TrCP. These include the rotavirus protein NSP116, human papilloma virus E717, JC virus large T antigen18 and human immunodeficiency virus-1 (HIV-1) Vpu19. Specifically, HIV-1 Vpu accessory protein downregulates the cell surface expression of host proteins CD4 and BST-2/tetherin, and subsequently induces their proteolysis via a mechanism involving a β-TrCP-SCF E3 ubiquitin ligase complex. While the host regulates β-TrCP activity by modulating its expression, localization or substrate abundance20, the direct regulatory mechanisms of β-TrCP activity have not been well elucidated.
TRIpartite motif proteins (TRIMs) are an expanding family containing more than 70 members in humans and mice that are characterized by the presence of a conserved RBCC region: a really interesting new gene (RING) domain, one or two B-Box domains and a predicted Coiled-Coil (CC) domain. They have variable domains at their C-termini. TRIM family proteins have been shown to be involved in multiple biological processes and are related to some disorders and diseases21, 22. The N-terminal RING domain is thought to be critical for their ubiquitin E3 ligase activity, and the central B-Box is structurally similar to RING, suggesting that it is also involved in ubiquitination, while the CC domains contribute to homo-oligomeric and hetero-oligomeric interactions. Recent studies have shown that several TRIM proteins play important roles in innate and adaptive immunity, ultimately contributing to intracellular anti-viral restriction and regulation of type I IFN and NF-κB signaling23–25. Among the most interesting and well-characterized TRIMs, TRIM5 is the most well-characterized restriction factor of HIV in old world monkeys26; TRIM25 is essential for retinoic acid-inducible gene 1 (RIG-I) signal transduction27; TRIM56 positively regulates STING-mediated, double-strand DNA-induced IFN production28; TRIM30 negatively regulates TLR-mediated NF-κB activity29 and NLRP3 inflammasome30; and TRIM27 negatively regulates IKK-mediated NF-κB activation31. Thus, TRIM family proteins play either positive or negative roles in infection-induced host immunity.
In order to explore the roles of TRIM family proteins in host innate immune regulation, we screened 62 different TRIM proteins for their activity in the NF-κB signaling pathway and identified TRIM9 as a potent negative regulator of NF-κB activation. TRIM9 is predominantly expressed in the proximal dendrites of neurons in the cerebral cortex and hippocampus, and the Purkinje cells of the cerebellum. Its expression is severely reduced in the affected brain areas in Parkinson's disease and dementia32, 33. Consistent with its potential roles in axonal development, TRIM9 is also highly expressed in the mantle layer of the spinal cord33, and its homologs in Caenorhabditis elegans and Drosophila. melanogaster have been shown to contribute to neuron development34, 35. In this report, we demonstrate that TRIM9 interacts with the WD repeat region of β-TrCP through its N-terminal degron motif depending on the phosphorylation status. This ultimately stabilizes β-TrCP substrates (IκBα and p100), thereby blocking both canonical and non-canonical NF-κB activation. Our study identifies a novel brain-specific TRIM9-mediated regulatory mechanism of β-TrCP activity that turns off the NF-κB signaling pathway to potentially contribute to brain immune surveillance.
Results
Efficient inhibition of NF-κB activation by TRIM9
We initially investigated the roles of TRIM family proteins in nucleotide-binding oligomerization domain-containing protein 2 (NOD2)-mediated NF-κB activation. To do this, we co-transfected expression vectors for individual TRIM proteins with a NF-κB-luciferase reporter into HEK293-NOD2 cells, followed by stimulation with the NOD2 ligand (L18-MDP) for 24hrs. While none of the TRIM proteins robustly stimulated NOD2-mediated NF-κB activation, several of them showed apparent inhibition of NF-κB activation to various degrees (Fig. 1a). Specifically, TRIM9 expression showed nearly 10-20-fold reduction of NOD2-mediated NF-κB activation (Fig. 1a). Further analysis showed that TRIM9 also blocked NF-κB activation induced by various stimulations such as PMA/Ionomycin, IL-1β and TNF-α in a dosage dependent manner (Fig. 1b), as well as upon expression of the constitutively active form of intracellular viral RNA receptor RIG-I (RIG-I 2CARD) (Fig. 1c). In contrast, it showed little or no effect on PMA/Ionomycin-induced AP-1 or NF-AT activation, nor on RIG-I-mediated ISRE activation (Supplementary Fig. 1). These results indicate that TRIM9 specifically blocks NF-κB activation from various stimuli.
Figure 1. TRIM9 expression inhibits NF-κB activation.
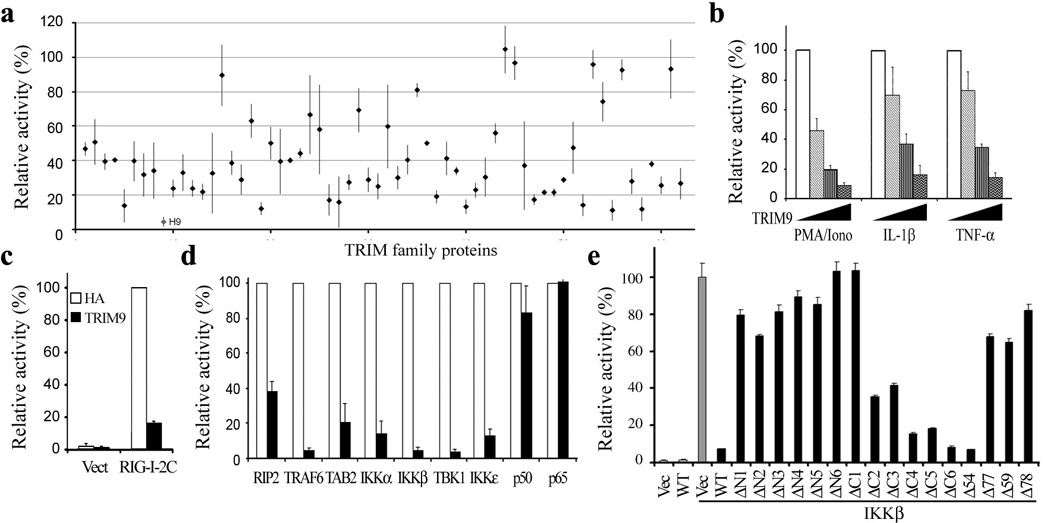
(a) NOD2-HEK293 cells were transfected with NF-κB and TK-Renilla reporter plasmids together with individual TRIM expression vector for 24hrs, stimulated with 10 ng/ml L18-MDP for 16 hrs, and subjected to dual-luciferase assay. H9: Human TRIM9. (b) At 24h post transfection with TK-Renilla luciferase transfection control plasmid, NF-κB reporter plasmid and increasing amounts of TRIM9 plasmid (0, 10, 50, 200ng), HEK293T cells were stimulated with 50ng/ml PMA plus 1µg/ml Ionomycin (PMA/Iono), 100pg/ml IL-1 β or 1ng/ml TNF-α overnight and then used for dual-luciferase assay. (c and d)) At 24h post transfection with TK-Renilla luciferase transfection control plasmid, NF-κB reporter plasmid and TRIM9 or vector control, plus plasmids expressing RIG-I 2 CARD (c), or RIP2, TRAF6, TAB2, IKKα, IKK β, TBK1, IKKε, p50, p65 (d). HEK293T cells were used for dual-luciferase assay. White bar: Vector control; Black bar: TRIM9. (e and f) At 24h post transfection with TK-Renilla luciferase transfection control plasmid, NF-κB reporter plasmid and TRIM9 WT or mutants together with IKK β, HEK293T cells were used for dual-luciferase assay. Results are presented as a percent relative to the activity of cells transfected with vector only. Data (mean and s.e.m.) are representative of at least three independent experiments.
Canonical NF-κB signaling converges on the NEMO/IKKα /IKKβ complex that phosphorylates IκBα 4, so over-expression of some critical signaling molecules in this pathway strongly activates NF-κB activity. To determine at which step of the NF-κB activation pathway TRIM9 blocks signaling, we co-expressed TRIM9 with several known mediators of canonical NF-κB signaling, including RIP2, TRAF6, TAB2, TBK1, IKKα, IKKβ, IKKε, p50 and p65, and then measured the effect of TRIM9 expression on NF-κB activation. This showed that TRIM9 expression effectively blocked NF-κB activation induced by RIP2, TRAF6, TAB2, TBK1, IKKα, IKKβ or IKKε, but it showed nearly no effect on p50- or p65-mediated NF-κB activation (Fig. 1d), suggesting that TRIM9 may function upstream of the p65/p50 NF-κB factor, but downstream of the NEMO/IKK complex.
TRIM9 N-terminal required for inhibition of NF-κB activation
TRIM9 contains a RING domain at its N-terminus, two B-box domains (B-box1 and B-box2) and a CC region in its central region, and a C-terminal subgroup one signature(COS) box, a Fibronectin III (FN3) domain, and a PRY-SPRY domain (also known as B30.2) at its C-terminus (Supplementary Fig. 2a)32, 33. To map which domain(s) of TRIM9 are required for inhibition of NF-κB activation, various TRIM9 mutants were generated and tested for their ability to block IKKβ-mediated NF-κB activation (Fig.1e and Supplementary Fig 2a). The N-terminal deletion mutants (ΔN1-ΔN6) of TRIM9 lost their ability to block NF-κB activation, whereas most of the C-terminal deletion mutants (ΔC2-ΔC6) of TRIM9, with the exception of the ΔC1 mutant, were able to block NF-κB activation as strongly as TRIM9 wild type (WT) (Fig. 1e). Furthermore, the Δ54, ΔB1, and ΔB2 mutants showed similar levels of NF-κB inhibitory activity to WT (Fig. 1e). By contrast, the Δ77 mutant (deletion of 1-77aa), Δ59 mutant (deletion of 59-77aa), and Δ78 mutant (deletion of 78-136aa) lost their ability to block NF-κB activation (Fig. 1e). Similar levels of NF-κB inhibition were observed upon stimulation with PMA/Ionomycin, IL-1β or TNF-α, or co-expression with TRAF6, IKKε or TBK1 for TRIM9 WT and Δ54 mutant, but not for Δ77, Δ59 and Δ78 mutants (Supplementary Fig. 2b, c). Surprisingly, the E3 ligase-dead CA mutant carrying the C30A/C33A mutations at the N-terminal RING domain still significantly inhibited NF-κB activation, suggesting that the TRIM9 E3 ligase activity is not required for the inhibition of NF-κB activity (Supplementary Fig. 2d). These indicate that the N-terminal 54-136aa region of TRIM9 is necessary for its inhibition of NF-κB activation.
TRIM9 interacts with β-TrCP
Mass spectrometry analysis of the V5-TRIM9 complex identified β-TrCP1 (also called FBW1A) and β-TrCP2 (also called FBW1B, FBW11) as binding partners (Fig. 2a). Co-immunoprecipitation with anti-β-TrCP antibody that reacts with both β-TrCP1 and β-TrCP2 showed the specific interaction between TRIM9 and β-TrCP (Fig. 2b). Furthermore, endogenous TRIM9 and β-TrCP interaction was readily detected in neuroblastoma SK-N-AS cells whereas their interactions were reduced upon depletion of TRIM9 expression (Fig. 2c, d). Precise inspection revealed that the N-terminal 54-136aa region of TRIM9 contains the 75DSGYGS80 sequence that matches very well with the degron motif (DSGXXS) recognized by β-TrCP where the two serines are phosphorylated (Fig. 2e). Specifically, the C-terminal region of β-TrCP containing seven WD40 repeats recognizes the phosphorylated degron motif of IκBα, and results in its degradation, thereby inducing NF-κB activation11. Indeed, TRIM9 WT and Δ54 mutant, which efficiently inhibited NF-κB activity, apparently bound β-TrCP, whereas the TRIM9 mutants (Δ59, Δ78, and ΔN1) that were not able to inhibit NF-κB activity, did not bind β-TrCP (Supplementary Fig. 3a). Furthermore, the TRIM9 SA mutant, in which the serine residues of the putative degron motif were replaced with alanines (S76→A and S80→A), neither bound to β-TrCP nor inhibited NF-κB activation induced by PMA/Ionomycin, IL-1β or TNF-α (Fig. 2e-g). As also seen with p100, the non-canonical NF-κB path way substrate of β-TrCP whose S866D/S870Dmutant loses β-TrCP binding activity36, the S76D/S80D mutant (SD) of TRIM9 showed the lack of β-TrCP binding activity, thereby lack of inhibition of NF-κB activation (Supplementary Fig. 4). The C-terminal region of β-TrCP containing seven WD40 repeats bound to TRIM9 WT but not the SA mutant, while the truncated WD40 mutants failed to interact with TRIM9 (Fig. 2h and Supplementary Fig. 3b, c). Immunofluorescence microscopy showed that as previously reported37, both the TRIM9 WT and the SA mutant appeared to be associated with intracellular cytoskeletons through its COS box region, while β-TrCP was diffuse in the cytoplasm (Fig. 2i). Upon co-expression, however, TRIM9 WT and β-TrCP were redistributed into punctate compartments, which were not observed upon with SA mutant expression (Fig. 2i). These results demonstrate that TRIM9 and β-TrCP interact through the N-terminal degron motif of TRIM9 and the C-terminal WD40 repeats region of β-TrCP.
Figure 2. TRIM9 interaction with β-TrCP.
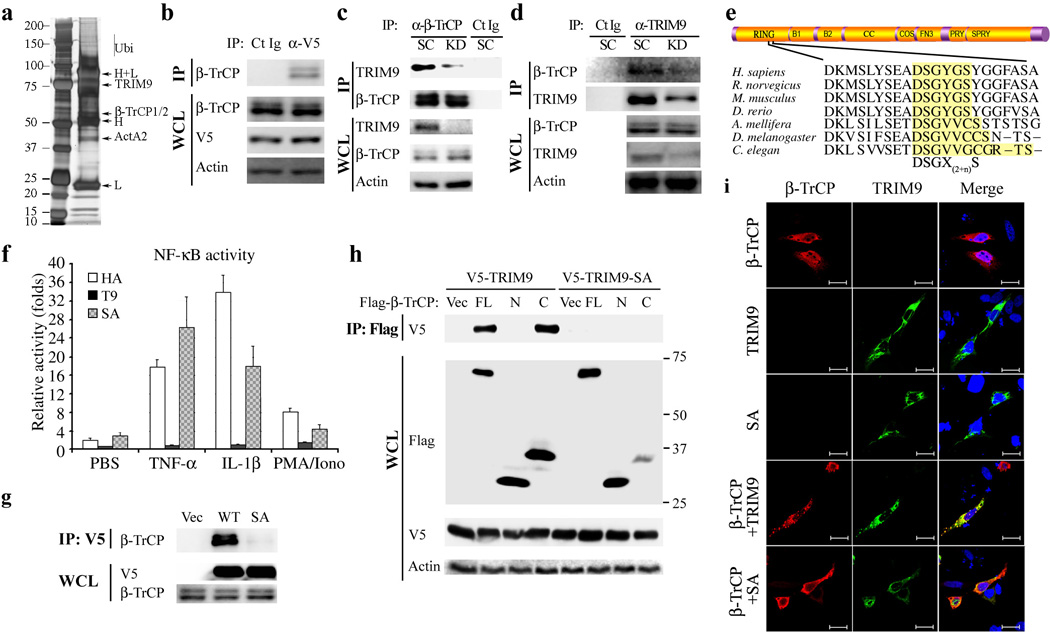
(a) Anti-V5 immunoprecipitates from 293T-V5-TRIM9 cells were separated by SDS-PAGE and visualized by silver staining. Specific bands were cut and analyzed by mass spectrometry. H, IgG heavy chain; L, IgG light chain; ActA2, actin A2. (b)Immunoblot analysis of V5-TRIM9 complexes with anti-β -TRCP. (c and d) SK-N-AS neuroblastoma cells were depleted for TRIM9 expression using scramble shRNA or TRIM9-specific shRNA lentivirus and then used for immunoprecipitation (IP) and immunoblotting (IB) with the indicated antibodies. Ct Ig: isotype control immunoglobulin. (e) Diagram of the conserved domains of TRIM9 (upper) and the degron DSGX(2+n)S motif of TRIM9 orthologs (lower). B1, B-Box1; B2, B-Box2; CC, coiled coil; COS, C-terminal subgroup one signature; FN3, fibronectin type 3; and PRY-SPRY, also known as B30.2. (f). At 24h post transfection with TK-Renilla luciferase transfection control plasmid, NF-κB reporter plasmid and TRIM9 WT (T9) or SA mutant, HEK293T cells were stimulated with PBS, PMA/Ionomycin, IL-1 β or TNF-α overnight and cell lysates were then used for dual-luciferase assay. (g) At 48h post transfection with Flag-β-TrCP and HA-TRIM9 WT or SA mutant, HEK293T cells were used for immunoprecipitation (IP) and immunoblot (IB) with the indicated antibodies. (h) At 48h post transfection with V5-TRIM9 WT or SA mutant together with Flag-β -TrCP full-length (FL), N-terminal region (N) or C-terminal region (C), HEK293T cells were used for IP and IB with the indicated antibodies. (i) Confocal immunofluorescence microscopy of Hela cells transfected with HA-β -TrCP (Green) together with HA-TRIM9 (Red) WT or SA mutant. DAPI (Blue) was used for nuclear staining. Scale bar=10µm. The data are representative of three independent experiments.
Degron motif phosphorylation is critical for TRIM9 function
To test whether serine phosphorylation of the TRIM9 degron motif is required for its interaction with β-TrCP, immunopurified HA-TRIM9 complexes were treated with mock or λ-phosphatase, incubated with lysates containing Flag-β-TrCP, and then subjected to immunoblotting with anti-Flag antibody. This showed that treatment of immunopurified TRIM9 with λ-phosphatase drastically abrogated its β-TrCP-binding ability (Fig. 3a). Phosphoserine (Sep), the most abundant phosphoamino acid in the eukaryotic phosphoproteome, is not encoded in the genetic code, but is synthesized post translationally. Since our attempts to generate the pS76pS80 phospho-specific TRIM9 antibodies failed twice, we utilized the specific cotranslational Sep incorporation system38, 39to study the Ser76 residue of TRIM9. An Escherichia coli strain was genetically engineered to harbor a Sep-accepting transfer RNA (tRNASep), its cognate Sep–tRNAsynthetase (SepRS), and an engineered EF-Tu (EF-Sep) of Methanocaldococcus jannaschii, and then transformed with bacterial expression vector containing the N-terminal HA-tagged RING domain (1-160aa) of TRIM9 WT, the SA mutant, and the non-natural Sep-incorporated pS76(directed by UAG) mutant (Fig. 3b). These HA-TRIM9 proteins were purified from genetically engineered E. coli and used for in vitro pulldown (PD) assay with Flag-β-TrCP. This showed that the pS76 TRIM9 protein specifically and efficiently bound β-TrCP in vitro, whereas the WT and SA TRIM9 proteins did not bind (Fig. 3c, d), suggesting that the serine phosphorylation of the TRIM9 degron motif is necessary for binding the WD40 domain of β-TrCP.
Figure 3. TRIM9 interacts with β-TrCP in a phosphorylation dependent manner.

(a) Immunopurified HA-TRIM9 from HEK293T cells was treated with or without λ-phosphatase (500U) for 30 min at 37C. 10% of untreated or λ-phosphatase-treated HA-TRIM9 protein was used for IB with anti-HA or anti-phospho-serine-specific antibody (p-Ser). The remaining untreated or λ-phosphatase-treated HA-TRIM9 protein was mixed with HEK293T cell lysates containing Flag-β -TrCP for 2h at 4°C, washed several times with 1% Tween-PBS washing buffer and subjected to IB with anti-Flag. Data are representative of at least three independent experiments. (b) E. coli BL21ΔSerB strain was engineered with tRNASep (derived from M. jannaschii tRNACys), SerRS (Sep-tRNA synthetase derived from M. maripaludis SepRS) and EF-Sep (Engineered phosphoserine specific EF-Tu). Italicized codon of the TRIM9 mRNA indicates the mutation site (ACG→UAG). Sep: Phosphoserine. (c and d) In vitro binding assay of bacterially purified HA-TRIM9 WT, the Ser76 phosphorylated (pS76) form or the Ser76-to-alanine (SA) mutated form with mammalian Flag-β-TrCP purified from 293T cells. IP with anti-HA and IB with anti-Flag (c) and vise versa (d).(e and f) 293T-TRIM9-V5 cells were stimulated with TNF-α for the indicated times (e) or pretreated with the indicated inhibitors (f), followed by TNF-α stimulation. Cell lysates were used for IP and IB with the indicated antibodies. DM (DMSO control), Fos (Fostriecin, PP2A, 4, 5 inhibitor, 100µM for 6hrs), FK (FK-506. PP2B inhibitor, 100µM for 6hrs), San (Sanguinarine, PP2C inhibitor, 10mM for 6hrs), Cal (Calyculin A, PP1, 2A, 4, 5 inhibitor, 10nM for 1hr). Ct Ig: isotype control immunoglobulin. (g) At 48 hr post-infection with lentivirus containing scrambled shRNA (SC) or TRIM9-specific shRNA (KD), 293T cells expressing V5-TRIM9 were used for IP with anti-V5, followed by IB with the indicated antibodies. The data are representative of three independent experiments.
Upon TNFα stimulation, the phosphorylated IκBα protein is recognized and ubiquitinated by the SCF-β-TrCP complex and consequently degraded through a proteasome pathway11. To investigate the kinetics of TRIM9 and β-TrCP interaction, HEK293T cells stably expressing V5-TRIM9 were stimulated with TNFα, followed by immunoprecipitation for TRIM9 and β-TrCP interaction. While the TRIM9 and β-TrCP interaction was readily detected under no stimulation, it was immediately diminished at 5-15 min following TNFα stimulation and then recovered at 30-60 min following TNFα stimulation (Fig. 3e). Conversely, the IκBα phosphorylation was initially undetected without stimulation, but robustly increased at 15-30 min after TNFα stimulation (Fig. 3e). Ultimately, IκBα degradation was observed at 30-60 min following TNFα stimulation (Fig. 3e), Finally, treatment with Calyculin A, an inhibitor for PPase1, 2A, and 4/5, considerably increased the TRIM9 and β-TrCP interaction, while other PPase inhibitors, such as FK-506 (PP2B inhibitor), Sanguinarine (PP2C inhibitor) or Fostriecin (PP2A, 4/5 inhibitor) did not do so (Fig. 3f). Furthermore, even a marginal depletion of PP1 expression led to an apparent increase of the TRIM9 and β-TrCP interaction (Fig 3g), suggesting that PPase 1 is a primary candidate phosphatase to remove the phosphorylation from the TRIM9 degron motif upon TNF-α stimulation. These data further support that TRIM9 interacts with β-TrCP in a phosphorylation-dependent manner.
Effects of TRIM9 on IκBα binding and degradation of β-TrCP
To address whether TRIM9 competes with IκBα for β-TrCP binding, TRIM9, β-TrCP, and IκBα were expressed in HEK293T cells, followed by an examination of the interaction of β-TrCP with IκBα and TRIM9. This showed that TRIM9 WT, but not the TRIM9 SA mutant, impaired β-TrCP interaction with IκBα in a dosage-dependent manner (Fig. 4a). On the contrary, increased expression of IκBα did not significantly block the β-TrCP and TRIM9 interaction (Supplementary Fig 5). Similarly, when endogenous β-TrCP was immunopurified, its interaction with endogenous phosphorylated IκBα was also diminished in the presence of the TRIM9 WT, but not the SA mutant (Fig. 4b). Moreover, this reduction was more pronounced upon TNF-α stimulation. Indeed, expression of the TRIM9 WT suppressed the TNF-α -induced ubiquitination of IκBα, whereas expression of the SA mutant did not do so (Fig. 4c).
Figure 4. TRIM9 competes with IκBα for β-TrCP binding and thereby blocks IκBα degradation.

(a) At 48h post transfection with Flag-β-TrCP and increasing amounts of HA-TRIM9 (µg) or HA-TRIM9 SA mutant, HEK293T cell lysates were used for IP with anti-Flag, followed by IB with anti-IκBα, anti-phospho-specific IκBα, or anti-HA antibody. HA-TRIM9 SA mutant was included as a control. (b) HEK293T-HA-TRIM9 or HA-SA mutant cells were treated with TNF-α and 25 µM MG132 for 1h and cell lysates were used for IP and IB with indicated antibodies. (c) HEK293T cells expressing Vector, HA-TRIM9 WT or the SA mutant were used for IP with anti-IκBα, followed by IB with anti-ubiquitin (Ubi) or anti-β-TrCP antibody. (d and e) SK-N-AS cells expressing HA-TRIM9 WT or the SA mutant (d) or carrying lentivirus containing scrambled shRNA (SC) or TRIM9-specific shRNA (KD) (e) were stimulated with IL-1 β for the indicated times, followed by IB with the indicated antibodies. The IκBα and Actin bands were quantified and the relative IκBα/Actin ratios were used to demonstrate the degradation rate of IκBα (bottom panel).The data are representative of three independent experiments.
To further delineate the mechanism of TRIM9-mediated inhibition of β-TrCP function, we examined IκBα degradation and phosphorylation upon expression of the TRIM9 WT, the SA mutant or genetic depletion of the TRIM9 expression. The IL-1 β-induced degradation of IκBα in SK-N-AS neuroblastoma cells was readily suppressed by expression of the TRIM9 WT, but not the SA mutant (Fig. 4d). It should be noted that the IL-1β stimulation-induced phosphorylation of IκBα was not significantly changed by expression of either the TRIM9 WT or the SA mutant, suggesting that the IKK activity is not affected by the TRIM9 expression. Conversely, the IL-1β-induced degradation of IκBα was facilitated by the specific shRNA-mediated depletion of the TRIM9 expression in human SK-N-AS cells (Fig. 4e). The S536 phosphorylation levels of p65, a NF-κB active subunit, were suppressed by TRIM9 expression but enhanced by TRIM9 depletion (Fig. 4d, e). Similar results were obtained with human HEK293T cells upon expression of TRIM9 WT or SA mutant (Supplementary Fig. 6a) and with human A549 lung epithelial cells upon depletion of the TRIM9 expression (Supplementary Fig. 6b, c). These data show that TRIM9 inhibits NF-κB activity by suppressing IκBα degradation.
TRIM9 inhibits non-canonical pathways of NF-κB activation
Because β-TrCP also plays a critical function in non-canonical NF-κB activation by converting the NF-κB precursor p100 to p5214, we tested the effect of TRIM9 expression on the master NF-κB inducing kinase (NIK)-induced activation of non-canonical NF-κB signaling. In correlation with β-TrCP interaction, TRIM9 WT and the ΔN54 mutant effectively blocked NIK-induced NF-κB activation, but the SA and ΔN1 mutants failed to do so (Fig. 5a). As previously reported12, NIK expression induced β-TrCP-mediated processing of p100 to p52 in HEK293T cells, but TRIM9 expression considerably blocked this process in a β-TrCP-binding-dependent manner (Fig. 5b, c). Finally, β-TrCP also bound to p100 in the presence of NIK expression, but this was abrogated by the expression of TRIM9 WT and enhanced by expression of the SA mutant (Fig. 5c). Furthermore, upon stimulation of Raji cells with α -CD40 antibody (Fig. 5d) or stimulation of A549 cells with LT β (Fig. 5e), overexpression of WT TRIM9, but not the SA mutant, decreased IL-6 production while depletion of TRIM9 expression increased IL-6 production. These results collectively demonstrate that TRIM9 effectively blocks β -TrCP-mediated canonical and non-canonical NF-κB activation.
Figure 5. TRIM9 inhibits NIK-mediated non-canonical NF-κB processing.
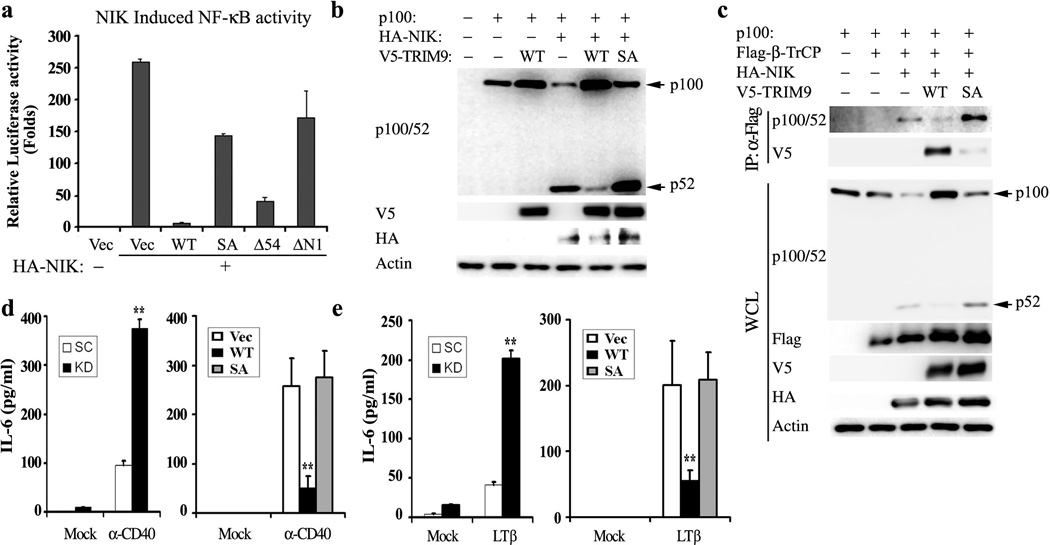
(a) At 24h post transfection with NF-κB reporter plasmid, NIK, and HA-TRIM9 WT or its mutants, HEK293T cells were used for dual-luciferase assay. (b and c) At 48h post transfection with p100, HA-NIK and TRIM9 WT or SA mutant, HEK293T cell lysates were used for IB with the indicated antibodies. (c) At 48h post transfection with p100, Flag-β-TrCP, HA-NIK, TRIM9 WT or SA mutant, HEK293T cell lysates were used for IP with anti-Flag and IB with anti-p100/52 or anti-TRIM9 antibody. WCL were used for IB with the indicated antibodies. Arrows indicate the precursor p100 protein and the processed p52 protein. (d and e) Upon depletion of TRIM9 expression (left panel) or expression of TRIM9 WT or SA mutant (right panel), human Raji B cells were treated with α -CD40 (d) and A549 epithelial cells were treated with LT β (e) for 24h and their supernatants were subjected to IL-6 ELISA. The mean ± s.d. is shown:**p < 0.01.The data are representative of three independent experiments.
TRIM9 blocks NF-κB-induced inflammatory cytokine production
We then tested whether TRIM9 expression affected NF-κB-mediated production of inflammatory cytokine IL-6. HEK293T cells were transfected with TRIM9 or the SA mutant, stimulated with TNF-α, IL-1β or PMA/Ionomycin for 24hrs, followed by ELISA to detect IL-6 production. This showed that TRIM9 WT but not the SA mutant inhibited stimulation-induced IL-6 production (Fig. 6a). Conversely, shRNA-mediated depletion of TRIM9 expression in A549 cells increased IL-6 production upon IL-1β, TNF-α, or PMA/Ionomycin stimulation (Fig. 6b). Similar results were obtained with SK-N-AS cells upon TRIM9 expression or depletion (Supplementary Fig. 7).
Figure 6. TRIM9 inhibits IL-6 inflammatory cytokine production.
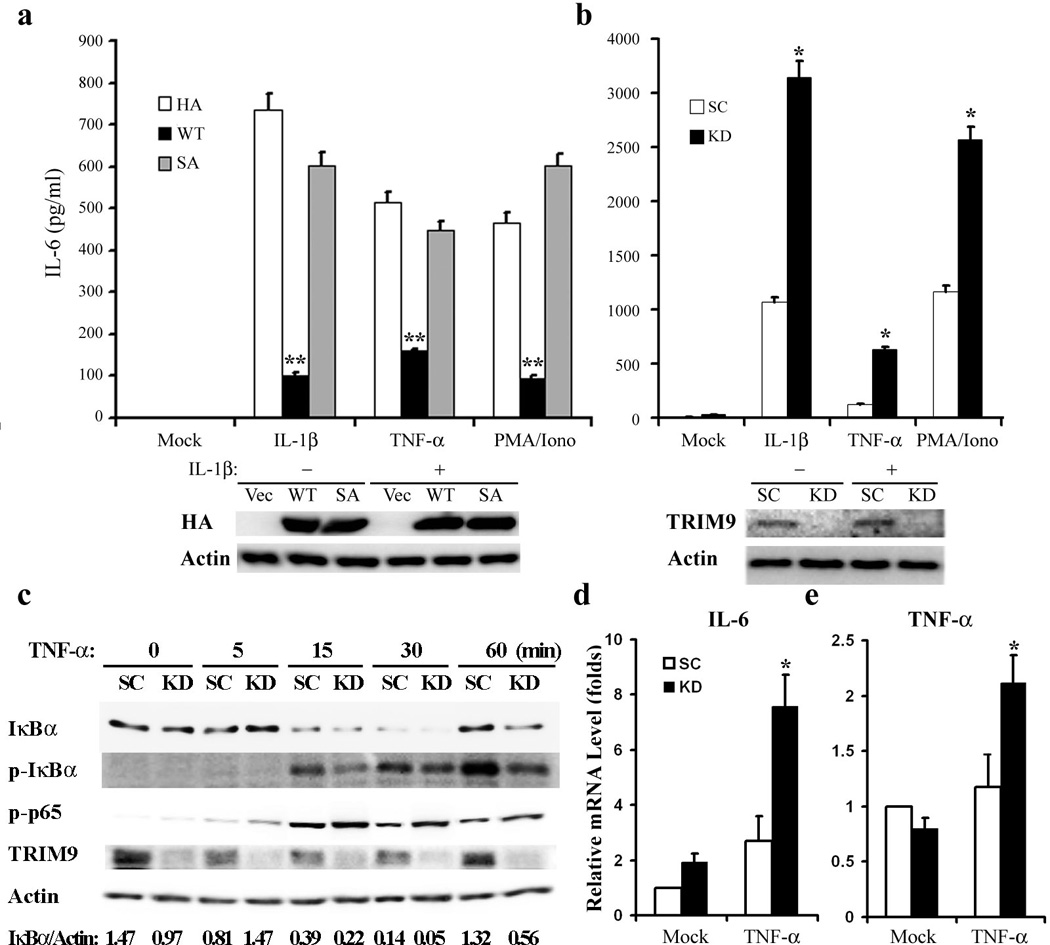
(a) At 48h post transfection with vector, TRIM9 WT or SA mutant, HEK293T cells were stimulated with IL-1 β, TNF-α or PMA/Iono overnight. The supernatants were then subjected to ELISA to measure IL-6 production. (b) A549 cells carrying lentivirus containing scrambled shRNA (SC) or TRIM9-specific shRNA (KD) were stimulated with IL-1 β, TNF-α or PMA/Iono overnight. The supernatants were subjected to ELISA to measure IL-6 production. WCL were used for IB with the indicated antibodies. (c) Primary rat neurons were harvested and infected with scrambled shRNA (SC) or TRIM9-specific shRNA lentivirus (KD), followed by TNF-α stimulation for 2hrs. Primary neuron cells were used for IB with the indicated antibody (c) or for real-time RT-PCR to measure IL-6 mRNAs (d) and TNF-α mRNAs (e). The IκBα and Actin bands were quantified and the relative IκBα/Actin ratios are presented at the bottom of (c). The mean ± s.d. is shown: *p < 0.05, **p < 0.01. The data are representative of three independent experiments.
TRIM9 is predominantly expressed in neurons in the cerebral cortex and hippocampus33, and its homologs in C. elegans and D. melanogaster have been shown to contribute to neuron development34, 35. To test whether TRIM9 expression also blocks NF-κB activation in neurons, primary neurons from rat brains were isolated and infected with lentivirus containing scrambled shRNA or TRIM9-specific shRNA (Fig. 6c). The shRNA-mediated depletion of TRIM9 expression in primary rat neural cells led to the reduced accumulation of p-IκBα, the enhanced degradation of IκBα, the augmented S536 phosphorylation of p65, and the increased expression of IL-6, TNF-α, and neuronal nitric oxide synthase 1 (NOS1) mRNAs upon TNF-α stimulation (Fig. 6c-e and Supplementary Fig. 8). These results indicate that TRIM9 expression effectively suppresses the stimulation-induced production of inflammatory cytokines in neurons, which may contribute to brain immune surveillance.
Discussion
Due to its central role in NF-κB signaling, activity of the β-TrCP SCF complex has to be tightly regulated. So far, expression, localization and substrate abundance have been identified to be the primary means of regulating β-TrCP SCF complex activity. Over 40 proteins have been identified as substrates recognized by the β-TrCP SCF complex15; however, it remains to be determined whether these substrates compete with each other for β-TrCP binding and whether these substrates or other molecules regulate β-TrCP SCF complex E3 enzymatic activity. Here, we identify TRIM9 as a negative regulator of β-TrCP SCF complex activity, comprehensively blocking canonical and non-canonical NF-κB activation. Thus, similar to other TRIM family members, TRIM9 contributes to constrains on host immunity; otherwise, continuous NF-κB-mediated signaling could lead to deleterious effects on the host such as inflammation, cancer, autoimmune diseases, and viral infection.
TRIM9 is highly expressed in embryonic and adult stages of the brain32 and its expression is not significantly affected by TNF-α, IL-1β LPS, influenza virus or type I IFN challenge40 (Fig 4e and Supplementary Fig 6b, c). Although TRIM9 is recognized as a substrate by β-TrCP, rather than functioning as a substrate, it competes with other substrates for β-TrCP binding, thereby deregulating β-TrCP SCF complex activity. This feature of TRIM9 is similar to that of hnRNP-U41 and HIV-1 Vpu accessory protein19, which are also recognized by β-TrCP through their degron motifs. In fact, Vpu has also been shown to inhibit IκBα degradation in a manner similar to TRIM942. A common feature of TRIM9 and β-TrCP is that their interaction did not lead to a reduction of their protein levels but rather an alteration in the intracellular localization of TRIM9 and β-TrCP: specifically, upon binding, TRIM9 and β-TrCP were redistributed into punctate cytoplasmic compartments. TRIM9 has been shown to be a neuron-specific component of a SNARE (soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptor) complex associated with synaptic vesicle release control43 as well as with the cytoskeleton through its COS box region37. These associations may ultimately contribute to their intracellular relocalizations in a manner reminiscent of RIG-I, which is also associated with the actin cytoskeleton. It is tempting to speculate that TRIM9-mediated recruitment of the β-TrCP SCF complex to cytoplasmic bodies plays two complementary roles. On one hand, it physically separates the β-TrCP SCF complex from its known substrates such as IκBα and p100 NF-κB inhibitors. On the other hand, movement enables β-TrCP SCF complex to encounter a different set of substrates recruited by TRIM9 for ubiquitination, as HIV-1 Vpu accessory protein does19. Further study is necessary to prove or disprove this hypothesis.
Phosphorylation of the serine residues of the TRIM9 degron is required for β-TrCP interaction since the dephosphorylation (SA) mutation of TRIM9 impaired its ability to bind β-TrCP. In vitro treatment of phosphatase completely eliminated the interaction between TRIM9 and β-TrCP. These suggest that the phosphorylation and dephosphorylation of the degron motif may be potential regulatory mechanisms for TRIM9 function. Indeed, while the interaction between TRIM9 and β-TrCP was detected under normal conditions, it was rapidly diminished and recovered upon stimulation. It is intriguing that the dissociation of TRIM9 and β-TrCP complex occurred prior to the IκBα phosphorylation and degradation, suggesting that the dephosphorylation of TRIM9 appears to play an important role in its function as a NF-κB regulator. In fact, Calyculin A (inhibitor of PP1, PP2A, PP4 and PP5) or PPase 1 KD dramatically enhanced the interaction of TRIM9 and β-TrCP. This is correlated with the previous findings44, 45, 46 that PP1 activity is increased after TNFα stimulation, and that depleted expression of PP1 α decreases NF-κB activity while increased expression of PP1 α augments NF-κB activity upon T cell receptor engagement. Thus, the phosphorylated TRIM9 may be a substrate for PP1 α, which ultimately contributes to the regulation of NF-κB activation.
Interestingly, TRIM67, which is also selectively expressed in the cerebellum, shows significant structural and sequence homology to TRIM947: both TRIMs have the same structural organization and exhibit 62% identity and 73% homology. More interestingly, it also carries a degron motif at its N-terminal RING domain and efficiently blocks NF-κB activation upon stimulation with PMA/Ionomycin, IL-1 β or TNF-α as we showed with TRIM9 (unpublished results). This suggests that targeting and blocking β-TrCP function in the brain may be a common feature of TRIM9 and TRIM67 as checkpoints for NF-κB activation.
TRIM9 expression is decreased in the damaged brains of patients with Parkinson's disease and Lewy bodies dementia32, 33. Consistently, NF-κB plays important roles in disorders such as epilepsy, stroke, Parkinson's disease and Alzheimer's disease48 and in inflammation49. Indeed, NF-κB activity has been reported to be dramatically increased in Parkinson's disease patients and Alzheimer disease patients compared to that of age-matched healthy controls50, 51. Furthermore, NF-κB signaling has been implicated in the regulation of neuronal axon initiation, elongation, guidance and branching, and dendrite arbor size and complexity52. In fact, the C. elegans TRIM9 homolog (MADD-2) was reported to be involved in axon attraction and branching34. Thus, the brain-specific expression of TRIM9, and likely TRIM67, may target the β-TrCP SCF complex to tightly regulate NF-κB activity, contributing to immune balance and neuronal axon growth in the brain. Future functional analysis of TRIM9 may provide potential therapeutic benefits for neurodegenerative diseases.
Methods
Plasmids
TRIM1-TRIM32 and TRIM37 were obtained from Dr. Andrea Ballabio(Texaschildren’s hospital)53; TRIM35, 39, 41, 43, 44, 46, 47, 52, 58, and 62 were from Dr. Walther Mothes (Yale University)54; and TRIM36, 48, 49, 56 59, 60, 61, 63, and 72 were from Open Biosystems and Invitrogen. Expression plasmid for RIG-I-2CARD and TRIM25 was described previously27 and TRIM34 was obtained from Dr. Paul D. Bieniasz(Howard Hughes Medical Institute)55., HA-NIK (Addgene plasmid 27554) was from Dr. Shao-Cong Sun(MD Anderson Cancer Center) 12. β-TrCP and NF-κB2 (p100) were received from Dr. Michele Pagano (New York University)56. TRIM9 and β -TrCP mutants were generated using a PCR site-directed mutagenesis kit (Stratagene) and subcloned into pCDH vector (System Biosciences) or pEBG vector. Lentivirus-shRNAs against TRIM9 were purchased from OpenBiosystem. The plasmid pRL-TK constitutively expressing Renilla luciferase was purchased from Promega. IKKα, IKK β, p65, p50, IκBα were gifted from Dr. EbrahimZandi (University of Southern California)57. Firefly luciferase reporter plasmids under the ISRE (ISG54), NF-AT, AP-1 or NF-κB promoters were purchased from Stratagene. TRAF6, TAB2, IKKε, TBK1, and ubiquitin-expressing plasmids were described previously29.
Cells and reagents
HEK293T, A549, SK-N-AS and HeLa cells were purchased from ATCC. HEK293-NOD2 stable cells were purchased from Invivogen. HEK293T, A549, and HeLa cells were maintained in Dulbecco'smodified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin-streptomycin. HEK293-NOD2 cells were maintained with 1µg/ml Blasticidin (Invivogen. Inc). SK-N-AS cells were maintained in Eagle’s Minimum Essential Medium (EMEM; ATCC). Polyclonal TRIM9 antibody (Dilution: 1/20000) was generated by immunization of rabbits with the TRIM9 N-terminal 1-261aa purified from Escherichia coli. Antibody sources are: TRIM9 mouse monoclonal antibody (6300-1F12; ABNOVA; Dilution: 1/2000), β-TrCP rabbit monoclonal antibody (D13F10; Cell Signaling; Dilution: 1/2000), IκBα mouse monoclonal antibody (L35A5; Cell Signaling; Dilution: 1/2000), phosphorylated IκBα (5A5; Cell Signaling, Dilution: 1/2000), NF-κB p65 (93H1; Cell Signaling; Dilution: 1/2000), IκBα rabbit polyclonal antibody (CalBiochem, Dilution: 1/2000), p100/p52 mouse monoclonal antibody (9D2; Abcam; Dilution: 1/1000), HA mouse monoclonal (16B12, Dilution: 1/2000) and rabbit polyclonal antibodies (Covance, Dilution: 1/2000), Flag mouse monoclonal (M2; Dilution: 1/5000) and rabbit polyclonal antibodies (Sigma; Dilution: 1/2000), V5 mouse antibody (Life Technologies; 2F11F7; Dilution: 1/5000)and rabbit polyclonal antibodies (Bethyl Laboratories; Dilution: 1/2000), GST polyclonal antibody (Sigma; Dilution: 1/2000), and Ubiquitin monoclonal antibody (P4D1; Santa Cruz; Dilution: 1/1000). IL-1 β and TNF-α were purchased from R&D; PMA and Ionomycin were purchased from Sigma; siRNA against PPase 1 was purchased from Santa Cruz.
Construction of TRIM9-expressing and knockdown cells
A549 and SK-N-AS cells were seeded into 6-well clusters overnight and culture medium was replaced with complete growth medium containing 8µg/mL polybrene (Sigma) and 5x105 infectious units (IU) of lentiviruses containing scrambled shRNA, TRIM9-specific shRNAs (target sequence for human: CGATGCCCTCAACAGAAGAAA; for mouse and rat: AATGTCTTTCTGTTTAAGTCG) or pCDH lentivirus expressing HA-TRIM9 WT or the SA mutant. At 48h post infection, cells were trypsinized and seeded into 60 mm dishes with complete growth medium containing 2µg/mL puromycin (Invivogen Inc). Puromycin-resistant cells were maintained in growth medium containing puromycin for a maximum of 15 passages.
Reporter Assays
Cells were transfected with the indicated reporters together with pRL-TK transfection control reporter. At 24h post transfection, cells were treated as indicated in the figure legends, followed by luciferase assay. For TRIM protein screening, HEK-293 NOD2 stable cells were seeded in 48-well plates at 2.5 × 104 cells/well. At 16 h post seeding, cells from each well were transfected with a total of 250 ng DNA containing 48 ng firefly luciferase reporter plasmid, 2 ng renilla luciferase reporter plasmid (pRL-TK), and individual TRIM plasmids using Lipofectamine 2000 (Invitrogen). All samples were transfected in triplicate. At 24h post transfection, cells were stimulated with 100pg/ml L18-MDP (Invivogen) for 16hrs and then lysed in passive lysis buffer (Promega), and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega)using a Beckman Coulter DTX880 plate reader. Firefly luciferase values were normalized to renilla luciferase values. Fold increase of the firefly reporter was calculated relative to the vector control in each experiment. In addition, HEK293T cells were transfected with the indicated plasmids combined with NF-κB-Luc, AP-1-Luc, NF-AT-Luc or pISRE-Luc reporter (Stratagene), and pRL-TK (Clontech). At 24h post transfection, cells were lysed or treated with IL-1 β, TNF-α and PMA/Ionomycin for 16hrs and then reporter luciferase activity was analyzed with the Dual-Luciferase Reporter Assay System (Promega).
Immunoprecipitation, GST pulldown and Immunoblot Analysis
For co-IPs, cells were lysed with RIPA minimum lysis buffer (Millipore). After clarification and preclearing, protein amounts were determined using BCA assay. Cell extracts were incubated for 12–16 h with the indicated antibodies, followed by additional incubation with protein A/G beads for 2–4 hr. Immune complexes were washed with lysis buffer with 400mMNaCl and subjected to immunoblot analysis. For endogenous co-IP, antibodies were conjugated to resin using Co-IP kit (Pierce). For ubiquitination assay, cells were initially lysed with RIPA buffer containing 1% SDS, and cell extracts were diluted with RIPA buffer to 0.1% SDS concentration and then subjected to IP and IB. Full scan image of the Western blots are provided in Supplementary figure 9.
Confocal Microscopy
At 24h post transfection, HeLa cells were fixed with 2% paraformaldehyde solution at room temperature for 20 mins, permeabilized, and stained with antibodies for confocal microscopy (Nikon Eclipse Ti). Anti-mouse HA (TRIM9), anti-rabbit Flag (β –TrCP), anti-mouse and/or anti-rabbit Alexa Fluor-488, and Alexa Fluor-568 antibodies (Invitrogen) were used. Hoechst dye (Invitrogen) was used to stain nuclei.
ELISA
For overexpression, HEK293T cells were transfected for 24 h with TRIM9 WT, TRIM9 SA or vector. Cells were then stimulated for 24hrs with IL-1 β, TNF-α or PMA/Ionomycin. SK-N-AS cells stably transfected with TRIM9 WT, SA mutant or vector were stimulated with IL-1 β for 24hrs. For knockdown assay, SK-N-AS and A549 stable knockdown TRIM9 or control cells were stimulated with IL-1 β, TNF-α or PMA/Ionomycin respectively. Supernatants were collected and their concentration of IL-6 or TNF-α was determined with a human-specific ELISA kit (BD Biosciences), followed by analysis with a Beckman Coulter DTX880 plate reader.
Q-PCR
Total RNAs were extracted from cultured cells with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Oligo(dT) priming and Superscript III reverse transcriptase (Invitrogen) were used for reverse transcription of purified RNA. All gene transcripts were quantified by quantitative PCR with iQ SYBR Green supermix and CFX96 real-time PCR system (BioRad). Primers for NOS1: Forward, GGCGTCCGTGACTACTGT, Reverse, ATGAAGGACTCCGTGGC; IL-6: Forward, TCCTACCCCAACTTCCAATGCTC, Reverse, TTGGATGGTCTTGGTCCTTAGCC ; TNF Forward, AAATGGGCTCCCTCTCATCAGTTC, Reverse, TCTGCTTGGTGGTTTGCTACGAC; HPRT Forward, CTCATGGACTGATTATGGACAGGAC, Reverse, GCAGGTCAGCAAAGAACTTATAGCC.
Protein purification and mass spectrometry
HEK293T cells stable expressing V5-TRIM9 were collected and lysed with NP40 buffer (50 mM HEPES, pH 7.4, 150mM NaCl, 1mM EDTA, 1% (v/v) NP40) supplemented with a complete protease inhibitor cocktail (Roche) and phosphatase inhibitors cocktail 3 (Sigma). Post-centrifuged supernatants were pre-cleared with protein A/G beads at 4°C for 2 hours and mixed with mouse anti-V5 and protein A/G beads for 4 hours at 4°C. Precipitates were washed extensively with lysis buffer and separated by SDS-PAGE. After silver staining, specific protein bands were excised and analyzed by ion-trap mass spectrometry at the Harvard Taplin Biological Mass Spectrometry facility, and amino acid sequences were determined by tandem mass spectrometry and database searches.
Unnatural amino acid protein expression
The production of HA-tagged TRIM9 N-terminal (1-160aa)-His6 WT, the Ser76 phosphorylated form or the SA mutant was followed as described previously in detail39. Briefly, the Ser76 (TCG) codon was replaced by the pSer76 (UAG) amber codon or the SA (CGG) mutation codon by site-directed mutategenesis (Stratagene). The expression clones were transformed into E. coli BL21ΔSerB with pKD-SepRS-EFTu. E. coli were grown at 25°C supplemented with 100 µg/ml of Amp, 50 µg/ml Kan, 12 µg/ml Tet, 2mM Sep, 5052 solution, and phosphate buffer for autoinduction. After induction, E. coli were harvested, lysed, subjected to Ni2+-NTA agarose column purification, and anion exchange column purification. Finally, purified proteins were separated through SDS-PAGE and stained with Coomassie brilliant blue.
Rat neural cell isolation, culture and transfection
Sprague-Dawley rats were raised, bred, and maintained in accordance with protocols approved by the USC Institutional Animal Care and Use Committee until euthanasia for neuron isolation. Rat neural precursor cells were obtained from the brain cortex as described elsewhere58. Briefly, telencephalons of E14 rat fetuses were dissected and the tissues were incubated with trypsin-EDTA solution (Gibco) for 10 minutes at 37°C and then dissociated mechanically. Neural precursor cells were cultured in passaging medium: DMEM/F12 (Gibco), 0.6% glucose, 10µg Insulin-Transferrin-Sodium Selenite Supplement (Roche), 1% penicillin/streptomycin, 20 ng/mL EGF (Sigma); 5 ng/mL FGF-2 (Sigma), 5 µg/mL heparin (Sigma) and 2% B27 supplement (Gibco) and grown as neurospheres. Then, neurospheres were digested with Accutase (Life Technologies) and plated onto coverslips coated with the poly-L-lysine (Sigma) and laminin (Sigma) in differentiation medium (Neurobasal, 2% B27 supplement from Gibco), allowing them to migrate and differentiate for 7 days. Cells were then maintained in an incubator at 37°C with a 5% CO2 atmosphere and 95% humidity. For TRIM9 knockdown, neurospheres were digested with Accutase and cultured in passaging medium containing 8µg/mL polybrene (Sigma) and 5x105 infectious units (IU) of lentiviruses containing scrambled shRNA or rat TRIM9-specific shRNA. After 48 hrs, differentiation medium was added to allow cells to differentiate for 7 days.
Statistics
One-way analysis of variance was used for multiple-group comparisons, followed by Bonferroni procedure for comparison of means. Student’s t-test was used for the comparison of two independent groups. For all tests, a p-value of less than 0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
This work was partly supported by grants CA082057, CA31363, CA115284, CA180779, DE023926, AI073099, AI083025, HL110609, GRL Program (K20815000001) from National Research Foundation of Korea, Hastings Foundation, and Fletcher Jones Foundation to JUJ, by grants 2012R1A2A2A02047051 and 2011-0020322 to H-SP, and by grants AI083025 and AI090935 to AG-S. We especially thank Drs. Michele Pagano, Paul D. Bieniasz, EbrahimZandi, Shao-Cong Sun and Walter Mothes for providing reagents and Ms. Rina Amatya for manuscript preparation.
Footnotes
Author contributions
M.S., J.J., B.Z., H.P. and A.G. designed the research; M.S, H.C., K.I. A.Y.Z.Z. Q.L., G.V., S.A. and L.W. performed the experiments and analyzed the data; M.S. and J.J. wrote the manuscript; all authors discussed the results and commented on the manuscript.
Conflict of interest statement
The authors declare no competing financial interests.
References
- 1.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol. 2011;12:709–714. doi: 10.1038/ni.2055. [DOI] [PubMed] [Google Scholar]
- 3.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 5.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–796. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 6.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 7.Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Razani B, Reichardt AD, Cheng G. Non-canonical NF-kappaB signaling activation and regulation: principles and perspectives. Immunol Rev. 2011;244:44–54. doi: 10.1111/j.1600-065X.2011.01059.x. [DOI] [PubMed] [Google Scholar]
- 9.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 10.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 13.Senftleben U, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 14.Liang C, Zhang M, Sun SC. beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal. 2006;18:1309–1317. doi: 10.1016/j.cellsig.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Skaar JR, D'Angiolella V, Pagan JK, Pagano M. SnapShot: F Box Proteins II. Cell. 2009;137:1358. doi: 10.1016/j.cell.2009.05.040. 1358 e1. [DOI] [PubMed] [Google Scholar]
- 16.Graff JW, Ettayebi K, Hardy ME. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 2009;5:e1000280. doi: 10.1371/journal.ppat.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spardy N, et al. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009;69:7022–7029. doi: 10.1158/0008-5472.CAN-09-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reviriego-Mendoza MM, Frisque RJ. Interaction and co-localization of JC virus large T antigen and the F-box protein beta-transducin-repeat containing protein. Virology. 2011;410:119–128. doi: 10.1016/j.virol.2010.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blanchet FP, Mitchell JP, Piguet V. beta-TrCP Dependency of HIV-1 Vpu-Induced Downregulation of CD4 and BST-2/Tetherin. Curr HIV Res. 2012;10:307–314. doi: 10.2174/157016212800792441. [DOI] [PubMed] [Google Scholar]
- 20.Kanarek N, Ben-Neriah Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol Rev. 2012;246:77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- 21.Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 22.Jefferies C, Wynne C, Higgs R. Antiviral TRIMs: friend or foe in autoimmune and autoinflammatory disease? Nat Rev Immunol. 2011;11:617–625. doi: 10.1038/nri3043. [DOI] [PubMed] [Google Scholar]
- 23.Kawai T, Akira S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med. 2011;3:513–527. doi: 10.1002/emmm.201100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNab FW, Rajsbaum R, Stoye JP, O'Garra A. Tripartite-motif proteins and innate immune regulation. Curr Opin Immunol. 2011;23:46–56. doi: 10.1016/j.coi.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 25.Ozato K, Shin DM, Chang TH, Morse HC., 3rd TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman RM, Johnson WE. A brief history of TRIM5alpha. AIDS Rev. 2007;9:114–125. [PubMed] [Google Scholar]
- 27.Gack MU, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 28.Tsuchida T, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. 2010;33:765–776. doi: 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Shi M, et al. TRIM30 alpha negatively regulates TLR-mediated NF-kappa B activation by targeting TAB2 and TAB3 for degradation. Nat Immunol. 2008;9:369–377. doi: 10.1038/ni1577. [DOI] [PubMed] [Google Scholar]
- 30.Hu Y, et al. Tripartite-motif protein 30 negatively regulates NLRP3 inflammasome activation by modulating reactive oxygen species production. J Immunol. 2010;185:7699–7705. doi: 10.4049/jimmunol.1001099. [DOI] [PubMed] [Google Scholar]
- 31.Zha J, et al. The Ret finger protein inhibits signaling mediated by the noncanonical and canonical IkappaB kinase family members. J Immunol. 2006;176:1072–1080. doi: 10.4049/jimmunol.176.2.1072. [DOI] [PubMed] [Google Scholar]
- 32.Berti C, Messali S, Ballabio A, Reymond A, Meroni G. TRIM9 is specifically expressed in the embryonic and adult nervous system. Mech Dev. 2002;113:159–162. doi: 10.1016/s0925-4773(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 33.Tanji K, et al. TRIM9, a novel brain-specific E3 ubiquitin ligase, is repressed in the brain of Parkinson's disease and dementia with Lewy bodies. Neurobiol Dis. 2010;38:210–218. doi: 10.1016/j.nbd.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hao JC, et al. The tripartite motif protein MADD-2 functions with the receptor UNC-40 (DCC) in Netrin-mediated axon attraction and branching. Dev Cell. 2010;18:950–960. doi: 10.1016/j.devcel.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song S, et al. TRIM-9 functions in the UNC-6/UNC-40 pathway to regulate ventral guidance. J Genet Genomics. 2011;38:1–11. doi: 10.1016/j.jcg.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J BiolChem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 37.Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J BiolChem. 2006;281:8970–8980. doi: 10.1074/jbc.M512755200. [DOI] [PubMed] [Google Scholar]
- 38.Lee S, et al. A facile strategy for selective incorporation of phosphoserine into histones. AngewChemInt Ed Engl. 2013;52:5771–5775. doi: 10.1002/anie.201300531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park HS, et al. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajsbaum R, Stoye JP, O'Garra A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur J Immunol. 2008;38:619–630. doi: 10.1002/eji.200737916. [DOI] [PubMed] [Google Scholar]
- 41.Davis M, et al. Pseudosubstrate regulation of the SCF(beta-TrCP) ubiquitin ligase by hnRNP-U. Genes Dev. 2002;16:439–451. doi: 10.1101/gad.218702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bour S, Perrin C, Akari H, Strebel K. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem. 2001;276:15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Chin LS, Weigel C, Li L. Spring, a novel RING finger protein that regulates synaptic vesicle exocytosis. J Biol Chem. 2001;276:40824–40833. doi: 10.1074/jbc.M106141200. [DOI] [PubMed] [Google Scholar]
- 44.Pribiag H, Stellwagen D. TNF-alpha downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J Neurosci. 2013;33:15879–15893. doi: 10.1523/JNEUROSCI.0530-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nie H, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med. 2013;19:322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- 46.Mock T. Identification and Characterisation of Protein Phosphatase 1, Catalytic Subunit Alpha (PP1alpha) as a Regulator of NF-kappaB in T Lymphocytes. Dissertation. 2012 http://www.ub.uni-heidelberg.de/archiv/13079.
- 47.Yaguchi H, et al. TRIM67 protein negatively regulates Ras activity through degradation of 80K-H and induces neuritogenesis. J Biol Chem. 2012;287:12050–12059. doi: 10.1074/jbc.M111.307678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 49.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunot S, et al. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci U S A. 1997;94:7531–7536. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gutierrez H, Davies AM. Regulation of neural process growth, elaboration and structural plasticity by NF-kappaB. Trends Neurosci. 2011;34:316–325. doi: 10.1016/j.tins.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reymond A, et al. The tripartite motif family identifies cell compartments. Embo J. 2001;20:2140–2151. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008;4:e16. doi: 10.1371/journal.ppat.0040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang F, Hatziioannou T, Perez-Caballero D, Derse D, Bieniasz PD. Antiretroviral potential of human tripartite motif-5 and related proteins. Virology. 2006;353:396–409. doi: 10.1016/j.virol.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 56.Busino L, et al. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012;14:375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zandi E, Chen Y, Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 58.Matsumura N, Yoshida N, Ohta A, Miyamoto Y, Hisatsune T. Neural precursor cells from adult mouse cerebral cortex differentiate into both neurons and oligodendrocytes. Cytotechnology. 2003;43:19–25. doi: 10.1023/B:CYTO.0000039909.28068.1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.