

Abstract
Background
TAR DNA-binding protein 43 (TDP-43) has been identified as a major disease protein in frontotemporal lobar degeneration. More recently, TDP-43 proteinopathy has also been observed in Alzheimer's disease (AD) with a characteristic distribution of TDP-43 predominantly in the mesial temporal lobe, and to a lesser degree in the neocortical areas. AD subjects with psychotic symptoms (AD+P) represent a subgroup characterized by greater impairment of frontal cortex-dependent cognitive functions and more severe frontal cortical neuropathology. The aim of this study is to determine whether there is an association between TDP-43 pathology and AD+P. We hypothesized that TDP-43 pathology would be more frequent in AD+P than in AD without psychosis.
Methods
We studied the presence and distribution of TDP-43 pathology by immunohistochemistry in the dentate gyrus (DG) and prefrontal cortex (FC) of postmortem brain specimens from 68 subjects with a primary neuropathologic diagnosis of AD as determined by the Neuropathology Core of the University of Pittsburgh Alzheimer's Disease Research Center.
Results
Forty-five (66%) subjects were classified as AD+P. Fourteen (20.6%) subjects had TDP-43 pathology in DG, eight (11.8%) had TDP-43 pathology in FC, and six (8.8%) had TDP-43 pathology in both regions. TDP-43 in DG was not significantly associated with AD+P. However, TDP-43 in FC demonstrated a trend toward reduced likelihood of psychosis (p = 0.068). TDP-43 pathology in DG, but not FC, was significantly associated with greater age at death and longer duration of illness.
Conclusions
Our findings indicate that there was no association between concomitant TDP-43 pathology in DG or FC and AD+P.
Keywords: TDP-43, Alzheimer's disease, psychosis
Introduction
Alzheimer's disease (AD) is a progressive clinicopathological condition characterized by a dementia syndrome with specific neurodegenerative changes in the cerebral cortex. Psychotic symptoms, delusions, and hallucinations are common in AD, with a prevalence exceeding 40% (Ropacki and Jeste, 2005). AD with psychotic symptoms (AD+P) has been identified as a subtype of AD distinguishable from AD without psychosis (AD−P) (Sweet et al., 2003; Murray et al., 2013). The most consistent correlate of AD+P has been greater global cognitive impairment and more rapid cognitive decline (Ropacki and Jeste, 2005; Emanuel et al., 2011). AD+P is also associated with greater impairment of executive function (Jeste et al., 1992), greater functional impairment (Scarmeas et al., 2005), significant patient and caregiver distress (Kaufer et al., 1998), higher institutionalization rates (Lopez et al., 1999), and personality and behavioral changes (Deutsch et al., 1991; Flynn et al., 1991). Congruent with the clinical profile of AD+P, in vivo functional imaging studies have found that AD+P subjects demonstrated excessive impairment of neocortical blood flow and glucose metabolism in comparison with AD−P subjects, with the dorsolateral prefrontal cortex (DLPFC) particularly affected (Starkstein et al., 1994; Kotrla et al., 1995; Sultzer et al., 1995; Staff et al., 1999; Mega et al., 2000).
The clinical correlates of AD+P summarized above led researchers to hypothesize that AD+P would be associated with elevated frontal cortical neuropathology, particularly involving the DLPFC. Farber et al. (2000) found that AD+P subjects had a 2.3-fold greater density of neocortical neurofibrillary tangles than AD−P subjects, involving the middle frontal cortex, superior temporal cortex, and inferior parietal lobule (Farber et al., 2000). Sweet et al. (2002) examined neuronal markers of synaptic integrity and found reduced N-acetyl-L-aspartate concentrations and increased glycerophosphoethanolamine concentrations in multiple neocortical brain regions of AD+P subjects, including the DLPFC (Sweet et al., 2002). A recent study by Murray et al. (2012) showed reduced concentrations of soluble Aβ 1–40, and increased ratio of soluble Aβ 1–42/Aβ 1–40 (Murray et al., 2012). That study also examined DLPFC levels of kalirin (Murray et al., 2012), a neuronal guanine nucleotide exchange factor thought to function as a primary controller of neuronal plasticity at the level of dendritic spines (Penzes and Remmers, 2012). Multiple kalirin isoforms, kalirin-7, kalirin-9, and kalirin-12 had reduced expression in the DLPFC of AD+P subjects (Murray et al., 2012). These reports are consistent with the hypothesis that AD+P is associated with more severe frontal cortical neuropathology, although findings of no increased pathology have been reported (Sweet et al., 2000).
TAR DNA-binding protein 43 (TDP-43) has been identified as a major disease protein in the pathologic lesions in frontotemporal lobar degeneration (FTLD) with ubiquitin-positive inclusions, now renamed as FTLD-TDP (Mackenzie et al., 2009) and amyotrophic lateral sclerosis (Neumann et al., 2006). TDP-43 proteinopathy has also been observed in other neurodegenerative disorders, including AD (Uryu et al., 2008). TDP-43 is a highly conserved 414 amino acid nuclear protein with a molecular weight of approximately 43 kDa (Chen-Plotkin et al., 2010) and is predominantly nuclear under physiological conditions (Wang et al., 2002). TDP-43 is synthesized in the cytoplasm and shuttles between the nuclear and cytoplasmic compartments (Ayala et al., 2008; Winton et al., 2008). In neurodegenerative diseases, TDP-43 is poorly soluble, hyperphosphorylated, ubiquitinated, cleaved into small fragments, and localized in the cytoplasm or nucleus as inclusion bodies (Chen-Plotkin et al., 2010). The physiological function of TDP-43 is not completely understood; however, current evidence suggests that it has several roles in the regulation of gene expression, including nuclear transcription, and splicing and stability of RNA transcripts (Buratti and Baralle, 2008).
Many previous studies have identified AD subjects with TDP-43 pathology in the mesial temporal lobe, e.g. dentate gyrus (DG), entorhinal cortex, and amygdala, and in temporal and frontal neocortex (Amador-Ortiz et al., 2007; Uryu et al., 2008; Bigio et al., 2010). Several studies have examined the clinical relevance of concomitant TDP-43 pathology in AD; however, the results were inconsistent (Josephs et al., 2008; Uryu et al., 2008; Bigio et al., 2010; Davidson et al., 2011). One study reported that TDP-43 pathology in AD was associated with significantly longer disease duration, but not with the clinical presentation (Uryu et al., 2008). However, this study was limited as it did not examine TDP-43 pathology in frontal and limbic cortices separately. To our knowledge, there have been no previous studies which have specifically investigated the relationship between TDP-43 and AD+P. Based on the current data indicating an association between AD+P and frontal dysfunction, we hypothesized that concomitant TDP-43 pathology in AD, especially in the prefrontal cortex (FC), might be associated with AD+P. We therefore undertook to evaluate TDP-43 pathology in prefrontal cortex (TDP-43-FC) and DG (TDP-43-DG) of AD+P subjects in comparison with AD−P subjects.
Methods
Subjects
Subjects were identified through the brain bank of the Alzheimer's Disease Research Center (ADRC) at the University of Pittsburgh. Sixty-eight subjects underwent neurologic, neuropsychologic, and psychiatric diagnostic evaluations at successive time points as part of their participation in the Clinical Core of the ADRC, with methods described previously (Sweet et al., 2000; 2001). All the subjects received a primary neuropathologic diagnosis of AD. Dates of death for the 68 subjects ranged from January 1997 to February 2011.
Characterization of psychosis
The presence or absence of delusions and hallucinations were indicated as part of semi-structured examinations conducted by research psychiatrists and rated on the Consortium to Establish a Registry for Alzheimer's disease (CERAD) Behavioral Rating Scale (Tariot et al., 1995). Delusions were defined as fixed, false beliefs based on incorrect inference about external reality, not attributable to membership in a social or cultural group. Hallucinations were defined as sensory perceptions for which there was no basis in reality. Psychosis was defined as the presence of any hallucination or delusion. No subject had a history of schizophrenia, schizoaffective disorder, or other idiopathic psychosis.
Tissue processing
All samples were obtained through the brain tissue bank of the ADRC at the University of Pittsburgh. At the time of brain removal, postmortem interval (PMI) was recorded and the brain was divided in the midsagittal plane. The left hemisphere was immersion-fixed in 10% buffered formalin for at least one week, sectioned into 1.0–2.0-cm coronal slabs, and sampled according to the CERAD protocol (Mirra et al., 1991). The diagnosis of AD was made using the recommendations of the National Institute on Aging and the Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease (1997). All but one subject (intermediate probability) had a high probability that the dementia was due to AD using these criteria.
Immunohistochemical staining
Immunohistochemical staining for TDP-43 was performed on 6 μm-thick sections of paraffin-embedded samples of the middle frontal gyrus (FC) and the mesial temporal lobe at the level of the lateral geniculate body. Following heat antigen retrieval in pH 6.0 citrate buffer and blocking for 10 minutes (Power Block, BioGenex, Fremont, CA), slides were incubated with primary antibody against TDP-43 (TARDBP #10782–2-AP, 1:200; Proteintech Group, Chicago, IL) (Montine et al., 2012) for one hour at room temperature followed by incubation with biotinylated rabbit IgG secondary antibody (1:200; Vector Laboratories, Burlingame, CA). Slides were developed with ABC Elite and Nova Red kits (Vector Laboratories) following the manufacturer's directions and counterstained with hematoxylin. The DG and FC were evaluated for the absence or presence of TDP-43 positive neuronal cytoplasmic inclusions, neuronal intranuclear inclusions, and dystrophic neuritis (Figures 1 and 2 respectively). Data were extracted from the database of the ADRC neuropathology core.
Figure 1.
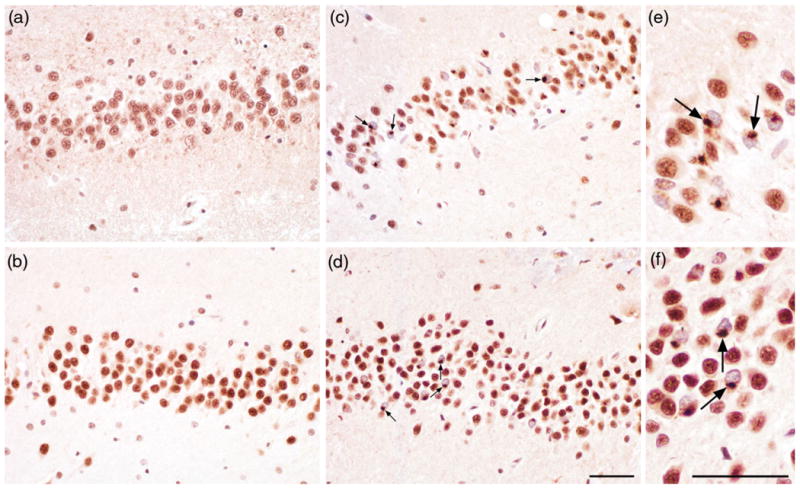
TAR DNA-binding protein 43 (TDP-43) pathology in dentate gyrus (DG) in AD. Subjects without TDP-43 inclusions in DG ((a) and (b)). Immunohistochemistry with anti-TDP-43 showing neuronal cytoplasmic inclusions in DG ((c) and (d), arrows). Inclusions are shown at higher magnification ((e) and (f)). The scale bar represents 50 μm.
Figure 2.
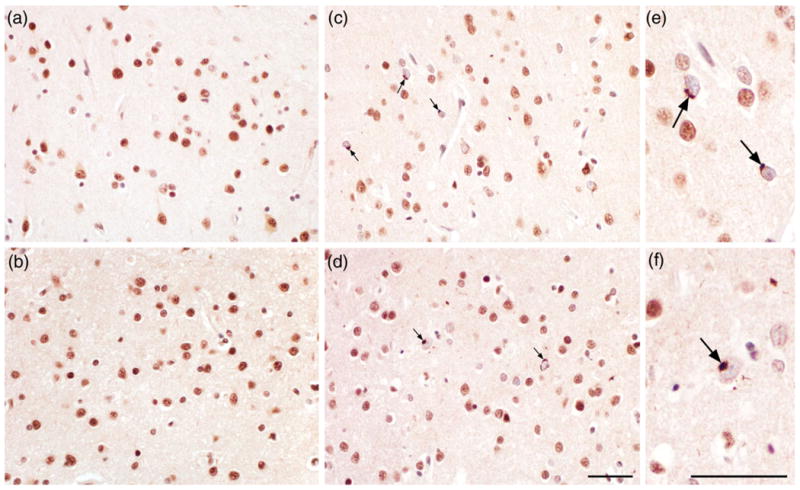
TAR DNA-binding protein 43 (TDP-43) pathology in frontal cortex (FC) in AD. Subjects without TDP-43 inclusions in FC ((a) and (b)). Immunohistochemistry with anti-TDP-43 showing neuronal cytoplasmic inclusions in FC ((c) and (d), arrows). Inclusions are shown at higher magnification ((e) and (f)). The scale bar represents 50 μm.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics 20. Demographic, clinical, and pathological variables were compared between groups using Pearson's χ2, Fisher's exact test, and analysis of variance where appropriate. The following groups were described: AD+P, AD−P; TDP-43-DG, and TDP-43-FC status (positive and negative).
Results
Demographic and clinical characteristics of the 68 subjects with primary diagnosis of AD are summarized in Table 1. Forty-five (66.2%) subjects were classified as AD+P. Women comprised 52.9% of all the subjects, and there was no association of gender with psychosis. All the subjects were Caucasian. The mean (SD) age at onset of the group is 70.1 (11.4) years, which did not differ significantly between AD+P and AD−P subjects. AD+P and AD−P subjects did not differ in age at death, duration of AD, PMI, and presence of comorbid Lewy body pathology. The mean (SD) Mini-Mental State Examination (MMSE) score in AD+P and AD−P subjects was 10.13 (6.7) and 14.04 (8.3) respectively, which significantly differed between the groups (p = 0.040). Although AD+P and AD−P subjects differed in the interval between last clinic visit and death, accounting for this covariate only modestly affected the association of AD+P with lower MMSE score (AD+P mean (standard error (SE)) 10.09 (1.1); AD−P 13.40 (1.6); F(1,63) = 2.77, p = 0.1).
Table 1. Demographic and clinical characteristics and their association with AD+P.
| variable | psychosis status | total n (%) or mean (sd) | χ2*or F† | df | p-value | |
|---|---|---|---|---|---|---|
|
| ||||||
| negative n (%) or mean (sd) | positive n (%) or mean (sd) | |||||
| Sex | 2.661* | 1 | 0.103 | |||
| Male | 14 (60.9) | 18 (40.0) | 32 (47.1) | |||
| Female | 9 (39.1) | 27 (60.0) | 36 (52.9) | |||
| Age at onset | 70.7 (11.6) | 69.8 (11.5) | 70.1 (11.4) | 0.092† | 1 | 0.763 |
| Age at death | 80.22 (10.8) | 80.33 (10.9) | 80.29 (10.8) | 0.002† | 1 | 0.967 |
| Duration of illness (years) | 9.52 (4.4) | 10.53 (4.7) | 10.19 (4.6) | 0.740† | 1 | 0.393 |
| Interval between last clinic visit and death (years)‡ | 4.39 (4) | 2.67 (1.9) | 3.3 (2.8) | 5.086† | 1 | 0.028 |
| PMI (hr) | 8.19 (5.6) | 6.90 (5.7) | 7.33 (5.6) | 0.723† | 1 | 0.399 |
| Lewy body neuropathology | 0.322* | 2 | 0.851 | |||
| None | 10 (43.5) | 20 (44.4) | 30 (44.1) | |||
| Limbic | 8 (34.8) | 13 (28.9) | 21 (30.9) | |||
| Neocortical | 5 (21.7) | 12 (26.7) | 17 (25) | |||
| MMSE | 14.04 (8.3) | 10.13 (6.7) | 11.46 (7.5) | 4.381† | 1 | 0.040 |
Notes: AD+P: Alzheimer's disease with psychosis; PMI: postmortem interval; MMSE: Mini-Mental State Examination.
Pearson's χ2 test: χ2 values are presented.
One-Way Analysis of Variance: F-values are presented.
Data unavailable for two subjects.
The associations of TDP-43-DG and TDP-43-FC with psychosis, demographic factors, and clinical characteristics of the 68 AD subjects are presented in Tables 2 and 3 respectively. Fourteen subjects (20.6%) demonstrated pathologic TDP-43 deposition in DG, eight (11.8%) subjects had pathologic TDP-43 deposition in FC. Six (8.8%) subjects were positive in both regions, while 52 (76.5%) subjects were negative for TDP-43 in both DG and FC. There was no significant association between TDP-43-DG and psychosis. However, TDP-43 in FC demonstrated a trend toward reduced likelihood of psychosis (p = 0.068). Entering the interval between last clinic visit and death in a logistic regression model only modestly affected the association of TDP-43 in FC with psychosis (B (SE) = −1.54 (0.9); Wald χ2 = 2.70, p = 0.1). When subjects positive for both TDP-43-DG and TDP-43-FC were contrasted with subjects without TDP-43 pathology in either region, no association was observed with psychosis (Exact p = 0.657, data not shown). Similarly, when subjects with TDP-43 pathology in either region were contrasted with subjects without TDP-43 pathology in either region, no association was observed with psychosis (Exact p = 0.478, data not shown).
Table 2. Demographic and clinical characteristics and their association with TDP-43-DG.
| variable | tdp-43-dg status | total n (%) or mean (sd) | χ2* or F† | df | p-value | |
|---|---|---|---|---|---|---|
|
| ||||||
| negative n (%) or mean (sd) | positive n (%) or mean (sd) | |||||
| Sex | 0.911* | 1 | 0.340 | |||
| Male | 27 (50) | 5 (35.7) | 32 (47.1) | |||
| Female | 27 (50) | 9 (64.3) | 36 (52.9) | |||
| Age at onset | 69.09 (12) | 74 (8.2) | 70.10 (11.4) | 2.077† | 1 | 0.154 |
| Age at death | 78.69 (10.7) | 86.50 (9.1) | 80.29 (10.8) | 6.312† | 1 | 0.014 |
| Duration of illness (years) | 9.59 (4.2) | 12.50 (5.3) | 10.19 (4.6) | 4.733† | 1 | 0.033 |
| Interval between last clinic visit and death (years) ‡ | 3.48 (3.1) | 2.58 (1.4) | 3.3 (2.8) | 1.058† | 1 | 0.307 |
| PMI (hr) | 7.25 (5.5) | 7.67 (6.6) | 7.33 (5.6) | 0.051† | 1 | 0.822 |
| Lewy body neuropathology | 0.738* | 2 | 0.691 | |||
| None | 23 (42.6) | 7 (50.0) | 30 (44.1) | |||
| Limbic | 18 (33.3) | 3 (21.4) | 21 (30.9) | |||
| Neocortical | 13 (24.1) | 4 (28.6) | 17 (25.0) | |||
| MMSE | 11.85 (7.9) | 9.93 (5.5) | 11.46 (7.5) | 0.734† | 1 | 0.395 |
| Psychosis status | 1.210* | 1 | 0.271 | |||
| Negative | 20 (37) | 3 (21.4) | 23 (33.8) | |||
| Positive | 34 (63) | 11 (78.6) | 45 (66.2) | |||
Notes: TDP 43-DG: TAR-DNA binding protein 43 in dentate gyrus; PMI: postmortem interval; MMSE: Mini-Mental State Examination.
Pearson's χ2 test: χ2 values are presented.
One-Way Analysis of Variance: F-values are presented.
Data unavailable for two subjects.
Table 3. Demographic and clinical characteristics and their association with TDP-43-FC.
| variable | tdp-43-fc status | total n (%) or mean (sd) | χ2* or F† | df | p-value | |
|---|---|---|---|---|---|---|
|
| ||||||
| negative n (%) or mean (sd) | positive n (%) or mean (sd) | |||||
| Sex | 1.771* | 1 | 0.183 | |||
| Male | 30 (50.0) | 2 (25) | 32 (47.1) | |||
| Female | 30 (50.0) | 6 (75) | 36 (52.9) | |||
| Age at onset | 69.47 (11.9) | 74.87 (5.6) | 70.10 (11.4) | 1.590 | 1 | 0.212 |
| Age at death | 79.38 (10.8) | 87.13 (8.1) | 80.29 (10.8) | 3.796† | 1 | 0.056 |
| Duration of illness (years) | 9.92 (4.4) | 12.25 (5.5) | 10.19 (4.6) | 1.857† | 1 | 0.178 |
| Interval between last clinic visit and death (years) ‡ | 2.99 (2.2) | 5.88 (5.8) | 3.3 (2.8) | 7.011 t | 1 | 0.01 |
| PMI (hr) | 7.50 (5.9) | 6.00 (2.9) | 7.33 (5.6) | 0.435† | 1 | 0.512 |
| Lewy body neuropathology | 1.630* | 2 | 0.443 | |||
| None | 26 (43.3) | 4 (50.0) | 30 (44.1) | |||
| Limbic | 20 (33.3) | 1 (12.5) | 21 (30.9) | |||
| Neocortical | 14 (23.3) | 3 (37.5) | 17 (25.0) | |||
| MMSE | 11.48 (7.6) | 11.25 (7.2) | 11.46 (7.5) | 0.007† | 1 | 0.935 |
| Psychosis status | 3.331* | 1 | 0.068 | |||
| Negative | 18 (30) | 5 (62.5) | 23 (33.8) | |||
| Positive | 42 (70) | 3 (37.5) | 45 (66.2) | |||
Notes: TDP 43-FC: TAR-DNA binding protein 43 in frontal cortex; PMI: postmortem interval; MMSE: Mini-Mental State Examination.
Pearson's χ2 test: χ2 values are presented.
One-Way Analysis of Variance: F-values are presented.
Data unavailable for two subjects.
TDP-43-DG was significantly associated with greater age at death and longer duration of illness (p = 0.014 and 0.033 respectively), but no such associations were observed with TDP-43-FC. No significant associations of gender, age at onset, PMI, Lewy body pathology, and MMSE scores with TDP-43-DG and TDP-43-FC were identified.
Discussion
In this study we found that there was no association between concomitant TDP-43 pathology in either DG or FC and AD+P. AD subjects with concomitant TDP-43-DG pathology demonstrated significantly higher age at death and longer duration of illness.
Some prior studies have examined the clinical relevance of concomitant TDP-43 pathology in AD. (Geser et al. 2010), although not examining subjects with AD, similarly found no association of TDP-43 burden with psychotic disorders (predominantly schizophrenia) in older adults (Geser et al., 2010). In regard to age at death and duration of illness, our findings concurred with some prior reports. (Josephs et al. 2008) found that AD subjects with abnormal TDP-43 immunoreactivity were older at disease onset and death (Josephs et al., 2008). (Uryu and colleagues 2008) found that TDP-43 pathology in AD was associated with significantly longer disease duration (Uryu et al., 2008). Significant increase in age at death and longer duration of illness in AD subjects with concomitant TDP-43-DG might suggest that TDP-43 pathology in DG, by an unknown mechanism, protects the individual from the neurotoxic effects of amyloid and tau pathology. Alternatively, TDP-43 pathology in DG might be an epiphenomenon in AD subjects with a relatively milder course. Unlike the current study where we did not show an association of TDP-43 pathology with greater global cognitive impairment, (Josephs et al. 2008) reported that AD subjects with TDP-43 pathology performed worse on the Clinical Dementia Rating scale and MMSE (Josephs et al., 2008).
In this study, AD+P subjects demonstrated significantly lower mean MMSE score in comparison with AD−P subjects. Although this difference in MMSE was reduced by controlling for differences between the groups in the interval between last clinic visit and death, substantial in vivo data indicate that AD+P is associated with greater cognitive impairment than AD−P (Ropacki and Jeste, 2005). A cross-sectional autopsy study cannot identify whether the lower MMSE in AD+P is a general consequence of globally greater neuropathology (as opposed to a psychosis-specific effect). However, data indicating that AD+P subjects have a more rapid rate of cognitive decline than AD−P subjects, even in the earliest stages of disease (Paulsen et al., 2000; Ropacki and Jeste, 2005; Wilkosz et al., 2010; Emanuel et al., 2011), suggest that any greater pathologic burden in AD+P would result from an association of AD+P with factors that accelerate AD progression.
A significant strength of this study was our cohort of subjects. The individuals were followed and evaluated during the progression of their disease, allowing for an extensive characterization of late-life behavior and cognition with corresponding postmortem analysis. Subjects were matched on a range of variables, including age, gender, illness duration, PMI, and Lewy body pathology. This stringency ensured that the identified postmortem correlates of psychosis were not due to having sampled AD+P subjects with more advanced pathologies than AD−P subjects, but rather were due to a discrete pathogenic process. One important limitation of our study is low number of TDP-43 positive subjects with psychosis. However, the failure to detect the hypothesized association of TDP-43 pathology with an increased risk of psychosis cannot be attributed to limited power due to small sample size, as the observed trend was in the opposite direction. It remains possible, however, that with a larger cohort of subjects, a significantly decreased risk of psychosis in subjects positive for TDP-43 in FC would have been detected.
In conclusion, our findings do not support the hypothesis that TDP-43 pathology is more frequent in AD+P than in AD−P. Thus, TDP-43 pathology does not appear to mediate the phenotypic expression of AD+P. Moreover, TDP-43-FC may have a protective effect regarding the risk of psychosis, since the majority of TDP-43-FC positive subjects were without psychosis. Alternatively, reduced TDP-43 pathology might be an epiphenomenon in AD+P. Additional studies exploring possible mechanisms whereby concomitant TDP-43 pathology in AD may be protective and lead to longer duration of illness and increase in age at death are recommended.
Acknowledgments
This work was supported in part by the National Institute of Aging grants AG05133 (OLL) and AG027224 (RAS), and the VA Pittsburgh Healthcare System (VAPHS) grant BX000452 (RAS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health, the National Institutes of Health, the Department of Veterans Affairs, or the United States Government.
Footnotes
Conflicts of interest: All authors have no conflict of interest to report.
Description of authors' roles: Dr. Vatsavayi formulated the research question, designed the study, and wrote the manuscript. Dr. Kofler processed the brain tissue, prepared the tissue for immunohistochemistry, collected the data, and assisted in writing the manuscript. Dr. DeMichele-Sweet did the statistical analysis and assisted in writing the manuscript. Drs. Murray, Lopez, and Sweet contributed to the conception of the project, critically reviewed drafts of the manuscript, and approved the final version of the paper. Dr. Robert Sweet was responsible for overseeing the entire project, including formulating the research question, directing the conduct of the analysis, and assisting in writing the manuscript.
References
- The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiology of Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- Amador-Ortiz C, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Annals of Neurology. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, et al. Structural determinants of the cellular localization and shuttling of TDP-43. Journal of Cell Science. 2008;121:3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- Bigio EH, et al. TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer's disease. Acta Neuropathologica. 2010;120:43–54. doi: 10.1007/s00401-010-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Frontiers in Bioscience. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nature Reviews Neurology. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson YS, et al. TDP-43 pathological changes in early onset familial and sporadic Alzheimer's disease, late onset Alzheimer's disease and Down's syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathologica. 2011;122:703–713. doi: 10.1007/s00401-011-0879-y. [DOI] [PubMed] [Google Scholar]
- Deutsch LH, Bylsma FW, Rovner BW, Steele C, Folstein MF. Psychosis and physical aggression in probable Alzheimer's disease. American Journal of Psychiatry. 1991;148:1159–1163. doi: 10.1176/ajp.148.9.1159. [DOI] [PubMed] [Google Scholar]
- Emanuel JE, et al. Trajectory of cognitive decline as a predictor of psychosis in early Alzheimer's disease in the cardiovascular health study. American Journal of Geriatric Psychiatry. 2011;19:160–168. doi: 10.1097/JGP.0b013e3181e446c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber NB, et al. Increased neocortical neurofibrillary tangle density in subjects with Alzheimer's disease. Archives of General Psychiatry. 2000;57:1165–1173. doi: 10.1001/archpsyc.57.12.1165. [DOI] [PubMed] [Google Scholar]
- Flynn FG, Cummings JL, Gornbein J. Delusions in dementia syndromes: investigation of behavioral and neuropsychological correlates. Journal of Neuropsychiatry & Clinical Neurosciences. 1991;3:364–370. doi: 10.1176/jnp.3.4.364. [DOI] [PubMed] [Google Scholar]
- Geser F, et al. Pathological 43-kDa transactivation response DNA-binding protein in older adults with and without severe mental illness. Archives of Neurology. 2010;67:1238–1250. doi: 10.1001/archneurol.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeste DV, Wragg RE, Salmon DP, Harris MJ, Thal LJ. Cognitive deficits of patients with Alzheimer's disease with and without delusions. American Journal of Psychiatry. 1992;149:184–189. doi: 10.1176/ajp.149.2.184. [DOI] [PubMed] [Google Scholar]
- Josephs KA, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70:1850–1857. doi: 10.1212/01.wnl.0000304041.09418.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufer DI, et al. Assessing the impact of neuropsychiatric symptoms in Alzheimer's disease: the Neuropsychiatric Inventory Caregiver Distress Scale. Journal of the American Geriatrics Society. 1998;46:210–215. doi: 10.1111/j.1532-5415.1998.tb02542.x. [DOI] [PubMed] [Google Scholar]
- Kotrla KJ, Chacko RC, Harper RG, Jhingran S, Doody R. SPECT findings on psychosis in Alzheimer's disease. American Journal of Psychiatry. 1995;152:1470–1475. doi: 10.1176/ajp.152.10.1470. [DOI] [PubMed] [Google Scholar]
- Lopez OL, Wisniewski SR, Becker JT, Boller F, DeKosky ST. Psychiatric medication and abnormal behavior as predictors of progression in probable Alzheimer's disease. Archives of Neurology. 1999;56:1266–1272. doi: 10.1001/archneur.56.10.1266. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathologica. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mega MS, Lee L, Dinov ID, Mishkin F, Toga AW, Cummings JL. Cerebral correlates of psychotic symptoms in Alzheimer's disease. Journal of Neurology, Neurosurgery and Psychiatry. 2000;69:167–171. doi: 10.1136/jnnp.69.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Montine TJ, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathologica. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PS, et al. Beta-amyloid 42/40 ratio and kalirin expression in Alzheimer's disease with psychosis. Neurobiology of Aging. 2012;33:2807–2816. doi: 10.1016/j.neurobiolaging.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PS, Kumar S, DeMichele-Sweet MA, Sweet RA. Psychosis in Alzheimer's disease. Biological Psychiatry. doi: 10.1016/j.biopsych.2013.08.020. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, et al. Incidence of and risk factors for hallucinations and delusions in patients with probable Alzheimer's disease. Neurology. 2000;54:1965–1971. doi: 10.1212/wnl.54.10.1965. [DOI] [PubMed] [Google Scholar]
- Penzes P, Remmers C. Kalirin signaling: implications for synaptic pathology. Molecular Neurobiology. 2012;45:109–118. doi: 10.1007/s12035-011-8223-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropacki SA, Jeste DV. Epidemiology of and risk factors for psychosis of Alzheimer's disease: a review of 55 studies published from 1990 to 2003. American Journal of Psychiatry. 2005;162:2022–2030. doi: 10.1176/appi.ajp.162.11.2022. [DOI] [PubMed] [Google Scholar]
- Scarmeas N, et al. Delusions and hallucinations are associated with worse outcome in Alzheimer's disease. Archives of Neurology. 2005;62:1601–1608. doi: 10.1001/archneur.62.10.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff RT, Shanks MF, Macintosh L, Pestell SJ, Gemmell HG, Venneri A. Delusions in Alzheimer's disease: spet evidence of right hemispheric dysfunction. Cortex. 1999;35:549–560. doi: 10.1016/s0010-9452(08)70818-9. [DOI] [PubMed] [Google Scholar]
- Starkstein SE, et al. A SPECT study of delusions in Alzheimer's disease. Neurology. 1994;44:2055–2059. doi: 10.1212/wnl.44.11.2055. [DOI] [PubMed] [Google Scholar]
- Sultzer DL, et al. The relationship between psychiatric symptoms and regional cortical metabolism in Alzheimer's disease. Journal of Neuropsychiatry and Clinical Neurosciences. 1995;7:476–484. doi: 10.1176/jnp.7.4.476. [DOI] [PubMed] [Google Scholar]
- Sweet RA, et al. Psychotic symptoms in Alzheimer's disease are not associated with more severe neuropathologic features. International Psychogeriatrics. 2000;12:547–558. doi: 10.1017/s1041610200006657. [DOI] [PubMed] [Google Scholar]
- Sweet RA, Nimgaonkar VL, Devlin B, Jeste DV. Psychotic symptoms in Alzheimer's disease: evidence for a distinct phenotype. Molecular Psychiatry. 2003;8:383–392. doi: 10.1038/sj.mp.4001262. [DOI] [PubMed] [Google Scholar]
- Sweet RA, et al. Psychosis in Alzheimer's disease:postmortem magnetic resonance spectroscopy evidence of excess neuronal and membrane phospholipid pathology. Neurobiology of Aging. 2002;23:547–553. doi: 10.1016/s0197-4580(02)00009-x. [DOI] [PubMed] [Google Scholar]
- Sweet RA, et al. The 5-HTTPR polymorphism confers liability to a combined phenotype of psychotic and aggressive behavior in Alzheimer's disease. International Psychogeriatrics. 2001;13:401–409. doi: 10.1017/s1041610201007827. [DOI] [PubMed] [Google Scholar]
- Tariot PN, et al. The behavior rating scale for dementia of the consortium to establish a registry for Alzheimer's disease. American Journal of Psychiatry. 1995;152:1349–1357. doi: 10.1176/ajp.152.9.1349. [DOI] [PubMed] [Google Scholar]
- Uryu K, et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer's disease and corticobasal degeneration but not in other tauopathies. Journal of Neuropathology & Experimental Neurology. 2008;67:555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proceedings of the National Academy of Sciences USA. 2002;99:13583–13588. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkosz PA, et al. Trajectories of cognitive decline in Alzheimer's disease. International Psychogeriatrics. 2010;22:281–290. doi: 10.1017/S1041610209991001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. Journal of Biological Chemistry. 2008;283:13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]