

Abstract
Recent advances in sequencing technologies have made it possible to explore the influence of rare variants on complex diseases and traits. Meta-analysis is essential to this exploration because large sample sizes are required to detect rare variants. Several methods are available to conduct meta-analysis for rare variants under fixed-effects models, which assume that the genetic effects are the same across all studies. In practice, genetic associations are likely to be heterogeneous among studies because of differences in population composition, environmental factors, phenotype and genotype measurements, or analysis method. We propose random-effects models which allow the genetic effects to vary among studies and develop the corresponding meta-analysis methods for gene-level association tests. Our methods take score statistics, rather than individual participant data, as input and thus can accommodate any study designs and any phenotypes. We produce the random-effects versions of all commonly used gene-level association tests, including burden, variable threshold, and variance-component tests. We demonstrate through extensive simulation studies that our random-effects tests are substantially more powerful than the fixed-effects tests in the presence of moderate and high between-study heterogeneity and achieve similar power to the latter when the heterogeneity is low. The usefulness of the proposed methods is further illustrated with data from National Heart, Lung, and Blood Institute Exome Sequencing Project (NHLBI ESP). The relevant software is freely available.
Keywords: complex diseases, gene-level association tests, heterogeneity, next-generation sequencing, random-effects models, rare variants
Introduction
Recent technological advances have made it possible to conduct sequencing studies for complex diseases and traits. To enrich association signals with rare variants, it is customary to perform gene-level association tests by aggregating mutation information across variant sites within a gene. The simplest and most common approach is the burden test, which creates a burden score for each subject by collapsing the variants with minor allele frequencies (MAFs) below a certain threshold and then relates the burden score to the phenotype of interest (Lin and Tang, 2011; Madsen and Browning, 2009; Morgenthaler and Thilly, 2007; Morris and Zeggini, 2010; Price et al., 2010). As an extension, the Combined Multivariate and Collapsing (CMC) method divides the variants into subgroups according to their MAFs and collapses the variants within each subgroup (Li and Leal, 2008). A second approach is the variable threshold (VT) method, which performs a burden test at each MAF threshold and chooses the threshold that yields the largest test statistic (Lin and Tang, 2011; Price et al., 2010). A third approach is the variance-component (VC) test, such as sequence kernel association test (SKAT), for detecting variants with opposite effects within a gene (Neale et al., 2011; Tzeng and Zhang, 2007; Wu et al., 2011). A related test is SKAT-O, which is a weighted sum of the burden and SKAT statistics (Lee et al., 2012).
Gene-level association tests, although more powerful than single-variant tests, have limited power to detect rare variants because only a small percentage of study subjects carry any mutation within a gene and there is a high background rate of neutral variation even in a “causal” gene. Thus, identifying “causal” rare variants requires large-scale meta-analysis. Several research groups have recently developed meta-analysis methods for gene-level association tests under fixed-effects (FE) models, which assume that the genetic effects are the same in all participating studies (Hu et al., 2013; Lee et al., 2013; Liu et al., 2014; Tang and Lin, 2013).
If the populations or environmental factors differ among studies, then the effect sizes are likely unequal because the gene of interest may interact with other functional variants in the genome that may have different frequencies among populations and because environmental, dietary or lifestyle factors may modify the influence of the gene. This phenomenon is referred to as (between-study) heterogeneity, which may also be caused by different definitions or measurements of the phenotype and different collections or manipulations of genotype data (e.g., genotyping platforms, imputation accuracies, and genotyping errors) (Evangelou and Ioannidis, 2013; Han and Eskin, 2012; Ioannidis et al., 2007; McCarthy et al., 2008; Moonesinghe et al., 2008). Heterogeneity has been commonly observed in meta-analysis of genome-wide association studies (Heid et al., 2009, 2010; Ioannidis et al., 2007; Moonesinghe et al., 2008; Saxena et al., 2007; Scott et al., 2007; Waters et al., 2010; Zeggini et al., 2007), and is expected to be more severe in sequencing studies because of potential differences in sequencing targets, quality control criteria, gene annotation, selection of variants and calculation of MAFs. Indeed, rare variants tend to be population-specific (i.e., present in some populations but not in others) (Nelson et al., 2012; Tennessen et al., 2012), so different studies are likely to contribute different sets of rare variants to a given gene-level association test even if the same selection criteria are used. Consequently, the strengths of the gene-level association will likely be different among studies even when each variant has the same effect size. Although there are few publications on meta-analysis of sequencing studies, there is evidence of heterogeneity in reported results (Romeo et al., 2007; Slatter et al., 2008).
In this article, we propose simple meta-analysis methods for gene-level association tests under random-effects (RE) models, which allow the genetic effects to vary among studies. Our methods take score statistics, rather than individual participant data, as input and thus can accommodate any study designs (e.g., case-control, cross-sectional, cohort, and family studies) and any phenotypes (e.g., binary, quantitative, and censored). We produce the RE versions of all commonly used gene-level association tests, including burden, CMC, VT, VC, and SKAT-O. Each test statistic provides a joint test of the mean and the variation of the genetic effects among the studies and thus has high power when the average effect is large or the heterogeneity is strong or both. We demonstrate through extensive simulation studies that our RE methods are substantially more powerful than the FE methods in the presence of moderate and high heterogeneity and are nearly as powerful as the latter when the heterogeneity is low. We illustrate the usefulness of the proposed methods through an application to the NHLBI ESP.
Methods
Suppose that we are interested in the effects of d genetic variables on a particular phenotype. For the burden test, the genetic variable is the burden score. For the CMC test, the genetic variables consist of the burden scores for rare variants and the genotypes for common variants. For the VT test, the genetic variables are the burden scores at the observed MAF thresholds. For the VC test, the genetic variables are the genotypes of individual variants.
We wish to perform meta-analysis of K independent studies. For k = 1, …, K, let βk = (βk1, …, βkd)T denote the effects of the d genetic variables in the kth study. It is natural to postulate the following random-effects model:
| (1) |
where μ = (μ1, …, μd)T represents the average genetic effects among the studies, and ξk = (ξk1, …, ξkd)T is a set of random effects representing the deviations of the genetic effects of the kth study from the average effects. It is assumed that ξk follows a multivariate normal distribution with mean 0 and covariance matrix Σ.
We are interested in testing the null hypothesis that the d genetic variables are not associated with the phenotype in any of the K studies, i.e., β1 = β2 = ··· = βK = 0. This null hypothesis corresponds to H0 : μ = 0 and Σ = 0 under model (1). When the dimension d is large, the statistic for testing H0 with an arbitrary Σ will have many degrees of freedom and thus have limited power. To increase power, we impose some structure on Σ by writing Σ = σB, where σ is an unknown constant, and B is a pre-specified matrix. Because σ = 0 is equivalent to Σ = 0, the null hypothesis H0 can be written as H0 : μ = 0 and σ = 0.
In practice, the true structure of B is unknown. It is reasonable to assume compound symmetry such that
where (b1, ··· bd) controls the relative degrees of heterogeneity for the d genetic effects, and r specifies the correlation of heterogeneity. If we believe that heterogeneity is higher for rarer variants, then we let the bjs be inversely related to the MAFs. If the variations of the d effects are independent, then r = 0. In constructing the test statistics, we may set r to a certain value, say 0, or vary r from 0 to 1. It is important to point out that the choice of B affects the power but not the type I error because σ = 0 entails Σ = 0 regardless of the value of B. As will be seen later, B is involved only in the CMC and VC tests.
For the kth study, we obtain the d-dimensional score statistic Uk for testing the null hypothesis that βk = 0 and the corresponding information matrix Vk. We describe below how to use the Uks and Vks to construct the RE versions of the burden, VT, VC, and related tests. The derivations are given in the Appendix.
For the simple burden test, there is only one genetic variable, which is the burden score. The score statistic for testing the null hypothesis H0 : μ = 0 and σ = 0 is
| (2) |
The first term, denoted by FE-BS, pertains to the score statistic for testing μ = 0 under the fixed-effect model (σ = 0) and the second term to the score statistic for testing σ = 0 given μ = 0. The two statistics are combined through direct summation because they are uncorrelated. Because it is a joint test of the mean and heterogeneity of the effects, RE-BS will have high power when the mean effect size is large or/and when the between-study heterogeneity is strong.
For the CMC (Li and Leal, 2008) and other tests involving multiple burden scores, the test statistic takes a multivariate form
| (3) |
where , and tr stands for trace. If d = 1, then (3) reduces to (2). When d > 1, we set r = 0. Alternatively, we may choose the value of r that yields the smallest p -value for RE-CMC. The resulting test statistic is denoted by RE-CMC-O, where O means that the test statistic is “optimized” over r. The calculation of the p-value for RE-CMC- O needs to account for the fact that multiple values of r have been tried.
The asymptotic approximations to the distributions of RE-BS, RE-CMC, and RE-CMC-O require large K and may not be accurate for small K. Thus, we use Monte Carlo simulation to obtain the p-values for these tests and all subsequent ones. To be specific, we repeatedly generate Uk from the d-variate normal distribution with mean 0 and covariance matrix Vk for k = 1, …, K and recalculate the test statistic. The p-value is set to be the proportion of the simulated test statistics that are greater than the observed test statistic. To improve computational efficiency, we employ an adaptive procedure which uses a small number of simulations for a large p-value and a large number of simulations for an extreme p-value. Specifically, we use 1,000 simulations for p-values greater than 0.1, 100,000 simulations for p-values between 0.001 and 0.1, 1 million simulations for p-values less than 0.001. This adaptive strategy makes Monte Carlo simulation almost as fast as the asymptotic approximation because most genes have large p-values.
For the VT method, the genetic variables correspond to the burden scores at d MAF thresholds. We perform a burden test at each MAF threshold and choose the threshold that produces the largest test statistic. Thus, the VT test statistic is defined by
where uj and ukj are the j th components of Uμ and Uk, respectively, and vj and vkj are the j th diagonal elements of Vμ and Vk, respectively. The FE counterpart is .
For the VC test, the genetic variables consist of the individual genotypes of d variants. We assume that the set of average genetic effects μ is a d-variate normal random vector with mean 0 and covariance matrix τW, where τ is an unknown constant, and W is a prespecified matrix. We impose compound symmetry such that
where (w1, ··· wd) controls the relative magnitudes of the d average genetic effects, and ρ indicates the correlation of the d effects. Note that W measures the within-study random effects of individual variants whereas B measures the between-study heterogeneity.
Because τ = 0 is equivalent to μ = 0, the null hypothesis H0 becomes τ = σ = 0. The score statistic for testing H0 takes the form
where , Uμ, Vμ, and Uσ were defined below equation (3) but now pertain to individual variants instead of burden scores, and
which is the covariance matrix of (Uτ, Uσ). The FE version is . As in the case of RE-BS, RE-CMC, and RE-VT, both the mean and heterogeneity contributes to RE-VC; however, the two contributions are correlated and thus cannot be directly added.
In original VC tests, ρ is set to 0 to allow the multiple effects within a gene to vary independently. By default, we set ρ = 0 for FE-VC and ρ = r = 0 for RE-VC. If ρ = r = 1, then RE-VC would become RE-BS. We can choose the value of ρ that yields the smallest p -value for FE-VC and the combination of ρ and r that yields the smallest p -value for RE-VC. The resulting test statistics are denoted by FE-VC-O and RE-VC-O, respectively. FE-VC-O is a standardized version of SKAT-O, and RE-VC-O can be viewed as a RE version of SKAT-O.
RE-BS is optimal if the effects of individual variants are similar within each study. RE-VT allows the choice of the MAF threshold to be data-dependent. RE-VC is desirable if the effects of individual variants within a study are different. RE-VC-O allows the data to suggest how the effects of individual variants vary within and between studies.
Recently, Lee et al. (2013) proposed two test statistics to allow heterogeneous effects:
where Skj is the score statistic for testing the j th variant in the kth study, wkj is a weight for the j th variant, and ϱ is chosen to minimize the p-value. Note that Uk = (Sk1, …, Skd)T (when the genetic variables are the genotypes of individual variants). Het-SKAT is the Uσ part of our RE-VC with r = 0 (up to a scaling constant and a centering constant) and is a test of heterogeneity at the variant level. This test will not have good power if the average effect size is large or the heterogeneity exhibits at the burden score instead of the variant level. By heterogeneity at the burden score level, we mean that the between-study variations are the same for all the variants within a gene; by heterogeneity at the variant level, we mean that the effects of individual variants vary independently of one another across studies. Het-SKAT-O is a weighted sum of FE-BS (under the additive mode of inheritance) and Het-SKAT and thus is a joint test of the mean effect at the burden score level and the heterogeneity at the variant level. Het-SKAT-O will be less powerful than RE-BS if both the mean effects and heterogeneity exhibit at the burden score level and less powerful than RE-VC if both the mean effects and heterogeneity exhibit at the individual variant level. The p-values of Het-SKAT and Het-SKAT-O are based on asymptotic distributions. Consequently, the type I error may not be well-controlled, the burden scores can only be calculated under the additive mode of inheritance, and the same set of weights has to be used for the two components of Het-SKAT-O.
Results
Simulation Studies
We conducted extensive simulation studies to evaluate the performance of the proposed and existing methods. We considered meta- analysis of five studies with sample sizes of 800, 1,000, 1,200, 1,400, and 1,600. Following Liu et al. (2014), we generated 12,000 haplotypes of length 1,000 kb under a calibrated coalescent model (Hudson, 2002) mimicking a sample of three European populations (Kryukov et al., 2009). The model includes an ancient bottleneck, recent exponential growth, differentiation, and migration. For each simulated dataset, we randomly selected ten 300 base-pair regions to construct a 3 kb region, which is the average size of the coding region of a gene (Pruitt et al., 2012). The MAFs were < 1% for 97% of the polymorphic sites. We removed variants with MAFs>5%.
We considered both quantitative and binary traits. For the quantitative trait, we generated data from the linear regression model
where Gki consists of the genotypes of the variants in the gene for the ith subject of the kth study, Zki consists of 1 and a normal random variable with unit variance and with mean being the total minor allele count for the ith subject of the kth study, and εki is standard normal. The normal covariate represents a principal component for ancestry or a different genetically related variable. For the binary trait, we generated case-control data with an equal number of cases and controls from the logistic regression model
We set the intercepts in the linear and logistic regression models to 0 and −2, respectively, and set the regression coefficients for the normal covariate to 0.3. We compared ten meta-analysis methods: FE-BS, FE-VT, FE-VC, and FE-VC-O pertain to fixed-effect models; RE-BS, RE-VT, RE-VC, and RE-VC-O are our proposed methods under random-effects models; Het-SKAT and Het-SKAT-O are Lee et al. (2013)’s tests for heterogeneous effects. For the burden tests (FE-BS and RE-BS), the burden score was a weighted sum of the mutation counts with the j th variant receiving the weight Beta(MAFj ; 1, 25), where MAFj is the MAF of the j th variant estimated from all study subjects. (The beta function gives more weights to rarer variants.) We set the wjs and bjs involved in the VC tests (FE-VC, RE-VC, FE-VC-O, and RE-VC-O) according to Beta(MAFj ; 1, 25). For FE-VC-O, we did a grid search over ρ = (0, 0.5, 1). For RE-VC-O, we added a grid search over r = (0, 0.5, 1). We implemented Het-SKAT and Het-SKAT-Ovia the MetaSKAT software (Lee et al., 2013).
We used 1 million replicates to evaluate the type I error at the nominal significance level α = 10−2, 10−3, and 10−4 by setting β1 = β2 = …= β5 = 0. The results are shown in Table 1. All our tests have accurate control of the type I error, although the RE-VT test appears to be slightly conservative for the binary trait. Het-SKAT and Het-SKAT-O tend to be conservative for both the quantitative and binary traits.
Table 1.
Type I error divided by the nominal significance level α for various meta-analysis methods
| Phenotype | α | FE-BS | RE-BS | FE-VT | RE-VT | FE-VC | RE-VC | FE-VC-O | RE-VC-O | Het-SKAT | Het-SKAT-O |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Quantitative | 10−2 | 0.99 | 0.99 | 0.99 | 0.99 | 1.00 | 0.98 | 0.99 | 1.00 | 0.95 | 1.00 |
| 10−3 | 0.98 | 0.95 | 0.97 | 0.97 | 0.96 | 0.96 | 1.03 | 1.04 | 0.86 | 0.89 | |
| 10−4 | 0.96 | 1.02 | 1.05 | 0.94 | 0.97 | 0.95 | 0.92 | 1.04 | 0.82 | 0.78 | |
| Binary | 10−2 | 1.00 | 0.99 | 0.99 | 0.96 | 0.98 | 0.93 | 1.00 | 0.96 | 0.85 | 0.96 |
| 10−3 | 0.98 | 0.93 | 0.94 | 0.87 | 0.95 | 0.91 | 0.97 | 0.99 | 0.77 | 0.87 | |
| 10−4 | 0.93 | 0.92 | 0.93 | 0.81 | 1.00 | 0.95 | 0.96 | 0.90 | 0.69 | 0.61 |
We used 10,000 replicates to evaluate the power at α = 10−4. In each replicate, we randomly selected 80%, 50% or 20% of the variants to be potentially causal. Let m denote the total number of potentially causal variants. We determined the genetic effects βk = (βk1, …, βkm)T by specifying the average effects μ = (μ1, …, μm)T and the random effects ξk = (ξk1, …, ξkm)T in model (1). The genetic effects were allowed to exhibit at the burden score or individual variant level. Because rarer variants tend to have larger effects on complex diseases (Gorlov et al., 2008; Pritchard, 2001), we set the effect sizes of the m variants according to their MAFs through a beta function. Specifically, we generated three different structures of genetic effects: (a) set μj = aj θ and ξkj = aj δk (j = 1, …, m), where aj is given by the Beta(MAFj ; 1, 25) function, θ is a constant, and δk is a normal random variable with mean 0 and variance σ; (b) set μj and ξkj to be the same as under structure (a) if MAFj < 1% and set μj = ξkj = 0 otherwise; (c) set μj to be a normal random variable with mean 0 and variance , and set ξkj to be a normal random variable with mean 0 and variance (j = 1, …, m). Under structures (a) and (b), the genetic effects exhibit at the burden score level for variants with MAFs< 5% and < 1%, respectively, and the degree of (between-study) heterogeneity is measured by the coefficient of variation . Under structure (c), the genetic effects exhibit at the individual variant level, and the degree of heterogeneity is measured by the ratio of variances σ/τ. For each percentage of potential causal variants and each genetic structure, we varied the degree of heterogeneity (i.e., or σ/τ) from 0 to 2 with the increment of 0.5 and tuned the value of θ or τ such that the power is high enough to compare different methods.
Figures 1 and 2 display the power as a function of the degree of heterogeneity for the quantitative and binary traits, respectively. When the (between-study) heterogeneity is low, the FE tests (FE-BS, FE-VT, FE-VC, and FE-VC-O) are more powerful than their RE counterparts (RE-BS, RE-VT, RE-VC, and RE-VC-O), although the power loss of the latter is typically small. When the heterogeneity is high, the RE tests are much more powerful than the FE tests. Among the RE tests, RE-BS and RE-VT are the most powerful tests under structures (a) and (b), respectively, when the percentage of causal variants is high. Under structures (a) and (b) with low percentages of causal variants and under structure (c), RE-VC tends to be more powerful than RE-BS. The power of RE-VC-O is near the top in all scenarios. Under structures (a) and (b) with low percentages of causal variants and under structure (c), RE-VC and RE-VC-O are considerably more powerful than Het-SKAT and Het-SKAT-O when the heterogeneity is low or moderate and have similar power to the latter when the heterogeneity is high.
Figure 1.

Power as a function of the between-study heterogeneity for the quantitative trait. The left, middle, and right panels correspond to three different genetic structures: (a) genetic effects exhibit at the burden score level for variants with MAFs< 5%, (b) genetic effects exhibit at the burden score level for variants with MAFs< 1%, and (c) genetic effects exhibit at the individual variant level. For each structure, 80%, 50% or 20% of the variants in ten 300 base-pair regions were randomly selected to be potentially causal.
Figure 2.

Power as a function of the between-study heterogeneity for the binary trait. The left, middle, and right panels correspond to three different genetic structures: (a) genetic effects exhibit at the burden score level for variants with MAFs < 5%, (b) genetic effects exhibit at the burden score level for variants with MAFs < 1%, and (c) genetic effects exhibit at the individual variant level. For each structure, 80%, 50% or 20% of the variants in ten 300 base-pair regions were randomly selected to be potentially causal.
We conducted another set of simulation studies by allowing genetic effects to exist in only a subset of the five studies. In such scenarios, it is sensible to test the association for each study and adjust the smallest p-value by the Bonferroni correction. Thus, we included this method, to be referred to as minP, in the simulation studies. We varied the number of studies with genetic effects from 1 to 5 and set βk = μ for those studies, where μ was generated under structure (a), (b) or (c). Figure 3 displays the results for the continuous trait when 50% of the variants are potentially causal. When the number of studies with genetic effects is 4 or 5, the RE tests are slightly less powerful than their FE counterparts. When the number is 1, 2 or 3, the RE tests are more powerful than the FE tests. The minP tests are less powerful than the RE tests except when the association exists in only one study. We also considered the binary trait and different percentages of causal variants, and the conclusions remain unchanged (data not shown).
Figure 3.

Power as a function of the number of studies with genetic effects on the quantitative trait. The upper, middle, and lower panels correspond to three different genetic structures: (a) genetic effects exhibit at the burden score level for variants with MAFs < 5%, (b) genetic effects exhibit at the burden score level for variants with MAFs< 1%, and (c) genetic effects exhibit at the individual variant level. For each structure, 50% of the variants in ten 300 base-pair regions were randomly selected to be potentially causal.
NHLBI ESP
The goal of the NHLBI ESP is to identify genetic variants in all protein-coding regions of the human genome that are associated with heart, lung, and blood diseases. The project consists of seven phenotype groups: low-density lipoprotein (LDL), body mass index (BMI), blood pressure (BP), early-onset myocardial infarction (EOMI), stroke, asthma, and chronic obstructive pulmonary disease (COPD). In addition, there is a random sample of subjects who had measurements on a set of core variables (i.e., phenotypes, traits), which is referred to as deeply phenotyped reference (DPR). The DNA samples were sequenced on the Roche NimbleGen SeqCap EZ or Agilent SureSelect Human All Exon 50 MB at the University of Washington and the Broad Institute (Lang et al., 2014; Tennessen et al., 2012). The variants were called jointly at the University of Michigan. We set the individual genotype values to missing if the genotype depth was lower than 10. We restricted our attention to missense, nonsense, and splice-site variants with call rates > 90% and MAFs< 5%. We excluded any gene whose total minor allele count was less than 5 and ended up with a total of 14, 878 genes.
We considered LDL as the trait of interest and included several covariates in the linear regression: top two principal components for ancestry, age, age2, gender, cohorts, and sequencing targets. The principal components were calculated from the sequencing data. The adjustment for sequencing targets was intended to remove potential batch effects. LDL was measured in the LDL, BMI, BP, EOMI, stroke, and DPR groups, but not in the asthma and COPD groups. For each phenotype group, we treated the African American (AA) and European American (EA) samples separately. After excluding subjects with sex mismatch or relatedness, there were 296, 526, 214, 351, 75, and 240 AA subjects and 331, 0, 325, 484, 123, and 700 EA subjects in the LDL, BMI, BP, EOMI, stroke, and DPR groups, respectively. In the meta-analysis, the score statistics for the eleven studies (i.e., phenotype group × race combinations) were obtained from SCORE-SeqTDS (Lin et al., 2013) and then combined to produce gene-level association tests. For the burden tests, we used the MAF thresholds of 1% and 5%, the corresponding tests being T1 and T5. The matrices B and W involved in the VC tests were specified in the same manner as in the simulation studies. We used 100,000 million Monte Carlo simulations to estimate the extreme p-values.
The results for T1, VT, VC, and VC-O are displayed in Figure 4. (The results for T5 are similar to T1 and thus not shown. The burden scores for T1 and VT tests were unweighted; the weighted results are similar and not shown.) It is instructive to examine LDLR, which is the top gene in RE-T1. Several common variants in this gene were previously identified to be associated with lipid traits and coronary heart diseases, and heterogeneous associations among ethnic groups were reported (Zhang et al., 2013). In our data, there are 54 rare variants in LDLR, all with MAFs < 1%, so the T1 and T5 tests are the same. In the T1 and VC-O tests, the RE meta-analysis provides stronger evidence of association than the FE meta-analysis: the RE-T1 and RE-VC-O p-values are 5.4 × 10−5 and 8.0 × 10−5, respectively, whereas the FE-T1 and FE-VC-O p-values are 6.3 × 10−3 and 5.7 × 10−4, respectively. The trend is reversed for the VT tests: the FE-VT and RE-VT p- values are 4.6 × 10−8 and 1.0 × 10−5, respectively. For both FE-VT and RE-VT, the maxima of the test statistics occur at the MAF threshold of 0.02%. The forest plots shown in Figure 5 provide helpful insights. If we collapse variants with MAFs < 0.02%, the effects of the burden scores are largely similar among the 11 studies; if we collapse variants with MAFs < 1%, the effects of the burden scores are quite heterogeneous. As shown in Figure 6, for the variants with MAFs < 0.02%, the carriers of mutations tend to have higher LDL levels than the noncarriers in all studies; for the variants with MAFs > 0.02%, the distributions of the LDL values for the carriers are very different among studies. Figures 5 and 6 show that the heterogeneity is largely driven by the variability of genetic effects between AA and EA.
Figure 4.

Meta-analysis of the eleven studies in the NHLBI ESP: the left and middle panels are the quantile-quantile plots for the FE and RE tests, and the right panel compares the RE and FE results. The red dot indicates the gene LDLR.
Figure 5.

Forest plots for the burden tests with two MAF thresholds for the gene LDLR in the NHLBI ESP. For each study, the estimate of the genetic effect is shown by the square and the corresponding 95% confidence interval is shown by the line. The meta-estimate of the genetic effect and the corresponding 95% confidence interval are shown by the diamond.
Figure 6.

Standardized LDL values for the carriers of the LDLR mutations in the NHLBI ESP. Each point represents an individual who carries a mutation. There are 54 polymorphic sites with the chr:position IDs and MAFs labeled on the x-axis. The variants are ordered by the MAFs. The vertical line pertains to the MAF threshold at which the test statistics of FE-VT and RE-VT are maximized. The five phenotype groups are indicated by different colors. AA and EA subjects are shown in circles and triangles, respectively. The horizontal line pertains to the average LDL value among individuals who do not carry any mutation.
For LDLR, the RE-VC test (p -value = 6.3 × 10−4) is slightly less significant than the FE-VC test (p -value = 3.1 × 10−4). This is due to the fact that almost all the mutations of each variant are from one race group (see Fig. 6), so the heterogeneity between the two races can be fully captured by the FE-VC test and the RE-VC test does not gain further information. By contrast, the RE-T1 test is more powerful than the FE-T1 test because there is considerable heterogeneity at the burden score level. The Het-SKAT and Het-SKAT-O p-values are 2.9 × 10−3 and 1.2 × 10−3, which are much less significant than any of our RE tests. The minP p-values for T1, VT, VC, and VC-O are 2.7 × 10−4, 6.7 × 10−4, 7.1 × 10−2, and 4.0 × 10−4, respectively, which are less significant than their RE counterparts.
Discussion
In this article, we provide simple RE methods for all commonly used gene-level association tests, including the burden, VT, and VC tests. Each test statistic contains contributions from both the mean and heterogeneity of the same type of genetic effect (i.e., at the burden score level for the burden and VT tests and at the individual variant level for the VC test). This is important because different tests are optimal for different scenarios. The RE tests are generally preferable to their FE counterparts because they are more powerful than the latter in the presence of moderate and high heterogeneity and have similar power to the latter when the heterogeneity is low, as demonstrated in the simulated and empirical data. The proposed methods are numerically stable and computationally efficient. They have been incorporated into the software MASS. It takes only a few minutes to conduct meta-analysis of several sequencing studies with thousands of genes.
For ethical and logistical reasons, summary statistics are more readily available than individual participant data. The proposed methods are based on score statistics and are as efficient as joint analysis of individual participant data (Lin and Zeng, 2010). Because it inputs score statistics rather than individual participant data, our framework can accommodate any phenotype and any study design or studies with different designs. For sequencing studies, score statistics are preferable to Wald and likelihood ratio statistics (Lin and Tang, 2011). The latter would entail estimation of the between-study variance (in the univariate case) or covariance matrix (in the multivariate case), which is numerically unstable or infeasible for rare variants.
The conventional RE approach is focused on the mean effect size (DerSimonian and Laird, 1986). That approach is not suitable for association testing for several reasons. First, it tests the null hypothesis of no mean effect while allowing between-study heterogeneity. This is not the relevant hypothesis for association testing because the existence of heterogeneity implies association in at least some studies. For this reason, conventional RE tests are almost always less significant than FE tests and thus have rarely been used in genetic association studies (Han and Eskin, 2011). Second, the conventional RE approach is based on the asymptotic distribution, which requires a large number of studies, but the number of sequencing studies is usually small. Third, existing multivariate RE methods leave the between-study covariance matrix completely unstructured (Chen et al., 2012; Jackson et al., 2010) and thus may lose power due to the large number of degrees of freedom.
Our approach reflects the spirit of Han and Eskin (2011) in that it tests the joint null hypothesis that there is no mean effect and no between-study heterogeneity. Our framework differs from Han and Eskin’s in three major aspects. First, their method is restricted to single-variant analysis of common SNPs whereas our methods deal with gene-level tests of rare variants. Second, their test statistic is univariate whereas our framework accommodates both univariate and multivariate test statistics. Third, their method is based on the likelihood ratio statistic whereas our methods are based on the score statistic.
Our RE tests were derived under random-effects models and may appear to rely on the normality of random effects, which is an untestable assumption. However, the random-effects models were only used to motivate the forms of the test statistics. By using Monte Carlo simulation rather than asymptotic approximation to obtain the p-values, the proposed tests have correct type I error even if the underlying random-effects models are completely wrong.
Our framework can be readily extended to handle multiple levels of between-study heterogeneity. Suppose that there are several (ancestry) groups of studies such that the genetic effects are homogeneous within each group but heterogeneous across groups. In that case, we will sum the score statistics and information matrices over the studies within each group and then construct the RE test statistics to account for the between-group heterogeneity.
If the burden score is created under the additive mode of inheritance as the sum or a weighted sum of the mutation counts over the variant sites (Lin and Tang, 2011; Madsen and Browning, 2009; Morris and Zeggini, 2010; Price et al., 2010), then the score statistics and information matrices for the burden and VT tests can be generated from the score vector and information matrix for testing individual invariants used in the VC tests (Hu et al., 2013). Specifically, the score statistic for the burden test is the sum or a weighted sum of the score statistics for testing the effects of individual variants. Under the dominant mode of inheritance, the burden score indicates whether there is anymutation among the variant sites (Li and Leal, 2008; Morgenthaler and Thilly, 2007). Then the above conversion can no longer be used. Our framework allows any mode of inheritance because the creation of the burden scores is external to the construction of the test statistics.
In meta-analysis, it is wise to have consistency across studies in terms of quality control criteria, gene annotation, variant selection and MAF estimation. This requirement is less essential for the RE tests than for the FE tests because heterogeneity (of genetic effects) is allowed for the former but not for the latter. For studies that use different exome capturing kits or studies in which some use whole-exome sequencing while others use exome chips, the variants captured can be quite different among studies. In such situations, the RE tests should be used because the effects are expected to be heterogeneous.
Acknowledgments
This research was supported by the National Institutes of Health grants R01 CA082659 and P01 CA142538. We acknowledge the support of the NHLBI and the contributions of the research institutions, study investigators, field staff, and study participants in creating the ESP data for biomedical research. We thank Drs. D. J. Liu and M. Zawistowski for providing the demographic parameters used in the simulation studies.
Appendix
Derivations of Test Statistics
Let β̂k denote the maximum likelihood estimator (MLE) of βk. By the MLE theory, β̂k is approximately normal with mean βk and covariance matrix
, where
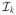 is the (profile) information matrix for βk. Under model (1) with fixed μ, β̂k is approximately normal with mean μ and covariance matrix
. For rare variants, the effect estimator β̂ks are unstable and may not be computable. Thus, we construct test statistics based on the score statistics rather than the Wald or likelihood ratio statistics. We use the scaled score statistic
as a surrogate for β̂k. Note that Vk is the same as
except that the former is the (profile) information matrix evaluated at βk = 0 and the latter at β̂k. For small βk, the statistic Xk is approximately normal with mean μ and covariance matrix
.
is the (profile) information matrix for βk. Under model (1) with fixed μ, β̂k is approximately normal with mean μ and covariance matrix
. For rare variants, the effect estimator β̂ks are unstable and may not be computable. Thus, we construct test statistics based on the score statistics rather than the Wald or likelihood ratio statistics. We use the scaled score statistic
as a surrogate for β̂k. Note that Vk is the same as
except that the former is the (profile) information matrix evaluated at βk = 0 and the latter at β̂k. For small βk, the statistic Xk is approximately normal with mean μ and covariance matrix
.
The log-likelihood function for μ and σ based on the statistics Xk (k = 1, …, K) can be written as
By tedious but straightforward matrix differentiation, we can show that the score function consists of
and the corresponding information matrix is
The score statistic for testing H0 : μ = 0 and σ = 0 is
where
, Vμσ =
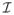 (0, 0),
, and
.
(0, 0),
, and
.
We now assume that μ is normal with mean 0 and covariance matrix τW. Write . The statistic X is approximately normal with mean 0 and covariance matrix , where JK is a K × K matrix composed of 1, IK is a K-dimensional identity matrix, and ⊗ is the Kronecker product. Then the log-likelihood function for τ and σ can be written as
The score function consists of
and the corresponding information matrix is
The score statistic for testing H0 : τ = σ = 0 is
where , and
References
- Chen H, Manning AK, Dupuis J. A method of moments estimator for random effect multivariate meta-analysis. Biometrics. 2012;68:1278–1284. doi: 10.1111/j.1541-0420.2012.01761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DerSimonian R, Laird N. Meta-analysis in clinical trials. Contr Clin Trials. 1986;7:177–188. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- Evangelou E, Ioannidis JP. Meta-analysis methods for genome-wide association studies and beyond. Nat Rev Genet. 2013;14:379–389. doi: 10.1038/nrg3472. [DOI] [PubMed] [Google Scholar]
- Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI. Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms. Am J Hum Genet. 2008;82:100–112. doi: 10.1016/j.ajhg.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Eskin E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 2012;8:e1002555. doi: 10.1371/journal.pgen.1002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid IM, Huth C, Loos RJF, Kronenberg F, Adamkova V, Anand SS, Ardlie K, Biebermann H, Bjerregaard P, Boeing H, et al. Meta-analysis of the INSIG2 association with obesity including 74,345 individuals: does heterogeneity of estimates relate to study design? PLoS Genet. 2009;5:e1000694. doi: 10.1371/journal.pgen.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, Thorleifsson G, Zillikens MC, Speliotes EK, Mägi R, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YJ, Berndt SI, Gustafsson S, Ganna A, Hirschhorn J, North KE, Ingelsson E, Lin DY. Meta-analysis of gene-level associations for rare variants based on single-variant statistics. Am J Hum Genet. 2013;93:236–248. doi: 10.1016/j.ajhg.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Patsopoulos NA, Evangelou E. Heterogeneity in meta-analyses of genome-wide association investigations. PLoS ONE. 2007;2:e841. doi: 10.1371/journal.pone.0000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson D, White IR, Thompson SG. Extending DerSimonian and Laird’s methodology to perform multivariate random effects meta-analyses. Stat Med. 2010;29:1282–1297. doi: 10.1002/sim.3602. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Shpunt A, Stamatoyannopoulos JA, Sunyaev SR. Power of deep, all-exon resequencing for discovery of human trait genes. Proc Natl Acad Sci USA. 2009;106:3871–3876. doi: 10.1073/pnas.0812824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange LA, Hu Y, Zhang H, Xue C, Schmidt EM, Tang ZZ, Bizon C, Lange EM, Smith JD, Turner EH, et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genet. 2014;94:233–245. doi: 10.1016/j.ajhg.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, Christiani DC, Wurfel MM, Lin X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Teslovich T, Boehnke M, Lin X. General framework for meta-analysis of rare variants in sequencing association studies. Am J Hum Genet. 2013;93:42–53. doi: 10.1016/j.ajhg.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet. 2011;89:354–367. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Zeng D. On the relative efficiency of using summary statistics versus individual-level data in meta-analysis. Biometrika. 2010;97:321–332. doi: 10.1093/biomet/asq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Zeng D, Tang ZZ. Quantitative trait analysis in sequencing studies under trait-dependent sampling. Proc Natl Acad Sci USA. 2013;110:12247–12252. doi: 10.1073/pnas.1221713110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DJ, Peloso GM, Zhan X, Holmen OL, Zawistowski M, Feng S, Nikpay M, Auer PL, Goel A, Zhang H, et al. Meta-analysis of gene-level tests for rare variant association. Nat Genet. 2014;46:200–204. doi: 10.1038/ng.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Moonesinghe R, Khoury MJ, Liu T, Ioannidis JP. Required sample size and nonreplicability thresholds for heterogeneous genetic associations. Proc Natl Acad Sci USA. 2008;105:617–622. doi: 10.1073/pnas.0705554105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) Mutat Res. 2007;615:28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Rivas MA, Voight BF, Altshuler D, Devlin B, Orho-Melander M, Kathiresan S, Purcell SM, Roeder K, Daly MJ. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Kryukov GV, de Bakker PIW, Purcell SM, Staples J, Wei LJ, Sunyaev SR. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:130–135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S, Pennacchio LA, Fu Y, Boerwinkle E, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PIW, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter TL, Jones GT, Williams MJA, Van Rij AM, McCormick SPA. Novel rare mutations and promoter haplotypes in ABCA1 contribute to low-HDL-C levels. Clin Genet. 2008;73:179–184. doi: 10.1111/j.1399-0004.2007.00940.x. [DOI] [PubMed] [Google Scholar]
- Tang ZZ, Lin DY. MASS: meta-analysis of score statistics for sequencing studies. Bioinformatics. 2013;29:1803–1805. doi: 10.1093/bioinformatics/btt280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng JY, Zhang D. Haplotype-based association analysis via variance-components score test. Am J Hum Genet. 2007;81:927–938. doi: 10.1086/521558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters KM, Stram DO, Hassanein MT, Le Marchand L, Wilkens LR, Maskarinec G, Monroe KR, Kolonel LN, Altshuler D, Henderson BE, et al. Consistent association of type 2 diabetes risk variants found in Europeans in diverse racial and ethnic groups. PLoS Genet. 2010;6:e1001078. doi: 10.1371/journal.pgen.1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JRB, Rayner NW, Freathy RM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yuan F, Liu P, Fei L, Huang Y, Xu L, Hao L, Qiu X, Le Y, Yang X, et al. Association between PCSK9 and LDLR gene polymorphisms with coronary heart disease: case-control study and meta-analysis. Clin Biochem. 2013;46:727–732. doi: 10.1016/j.clinbiochem.2013.01.013. [DOI] [PubMed] [Google Scholar]