

Microseed matrix screening is increasingly successful in the optimization of protein crystallization leads.
Keywords: microseed matrix screening, optimization, crystallization
Abstract
Protein crystals obtained in initial screens typically require optimization before they are of X-ray diffraction quality. Seeding is one such optimization method. In classical seeding experiments, the seed crystals are put into new, albeit similar, conditions. The past decade has seen the emergence of an alternative seeding strategy: microseed matrix screening (MMS). In this strategy, the seed crystals are transferred into conditions unrelated to the seed source. Examples of MMS applications from in-house projects and the literature include the generation of multiple crystal forms and different space groups, better diffracting crystals and crystallization of previously uncrystallizable targets. MMS can be implemented robotically, making it a viable option for drug-discovery programs. In conclusion, MMS is a simple, time- and cost-efficient optimization method that is applicable to many recalcitrant crystallization problems.
1. Introduction
Seeding is a crystallization method in which crystals or crystalline material generated in one experiment is transferred to a new experiment. The crystalline material acts as a nucleation site for crystal growth in the new experiment, removing the need for de novo nucleation. This approach is rationalized by the hypothesis that the optimal conditions for crystal nucleation and for crystal growth can be quite different. Typically, the new experiment uses the same conditions that produced the initial crystal seeds, but at lower levels of supersaturation (for a review, see Bergfors, 2003 ▶).
In a variation of seeding termed microseed matrix screening (MMS), seeds are transferred from an initial crystallization experiment into a variety of new and often unrelated crystallization conditions. This method was introduced by Ireton & Stoddard (2004 ▶) for yeast cytosine deaminase, resulting in a dramatic improvement in crystal quality. Microseeds made from initial crystals were introduced into a new condition distinguished from the original by the addition of calcium acetate. Attempts to produce crystals in the new conditions without microseeds were unsuccessful, confirming that MMS was essential for crystallization.
In 2007, D’Arcy and coworkers implemented a modified version of the method, using a robotic liquid-handling device to seed into sparse-matrix screens (D’Arcy et al., 2007 ▶). This transformed the method into a generally applicable extended screening method requiring minimal manual intervention. The use of MMS led to a dramatic increase in the number of crystallization conditions that produced crystals. There were also many examples of improvements in crystal quality, in particular the reduction or elimination of twinning and an improvement in diffraction properties.
Crystals obtained in initial screens are usually not of sufficient quality for structure determination by X-ray diffraction and the conditions must be optimized in subsequent rounds of experiments to produce the desired quality crystals. The classical approach to optimization would be to use hit conditions identified in the initial coarse screening phase as a guide for fine screening. In this approach, crystal nucleation and crystal growth are refined in the same conditions. MMS represents a paradigm shift in optimization. MMS can be viewed as the second stage of a two-step process that sequentially screens a wide range of conditions first for crystal nucleation and then for crystal growth.
Since the introduction of MMS in 2004, a number of reports (Obmolova et al., 2010 ▶; Persson et al., 2010 ▶; Newman et al., 2011 ▶) have confirmed that it is a viable optimization alternative. It can produce high-quality crystals from poor starting points that are not amenable to the classical optimization strategies. Here, we review some recent developments in our own work and that of others with MMS. On the basis of the successes from MMS, we offer recommendations for wider applications of MMS to crystallization problems.
2. Methods
2.1. Producing seed stocks and dilutions
Our method for the preparation of seeds for MMS is based on the ‘seed bead’ method of Luft & DeTitta (1999 ▶). It is particularly important that a critical mass of suitable seeds is introduced in the initial optimization. Once the optimized conditions have been identified, further optimization of crystal number, size and quality is possible simply by adjusting the concentration of the seed stock. The detailed procedure is described below.
(i) Select the best quality crystals you can afford to use. If no crystals are available, use any crystalline material. Take as many crystals as possible to generate the seed stock, as a higher initial concentration of seeds allows more flexibility in optimization.
(ii) For vapour-diffusion setups, add 10 µl of the reservoir solution to the drop containing the crystals. In our experience, it is not necessary to add protein to the solution to stabilize the seeds as recommended by Luft & DeTitta (1999 ▶).
(iii) Crystals should be crushed using a spade-like tool (Hampton Research, USA) or a rounded glass probe made from a glass pipette or capillary stretched during heating. (This is very easy to do yourself, but probes prepared in this way are also available on request from Douglas Instruments, UK or Hampton Research, USA.)
(iv) Pipette the 10 µl of crushed seeds into an Eppendorf tube containing a seed bead (Hampton Research, USA)
(v) Add another 10 µl of reservoir solution to the drop and mix thoroughly to recover more seeds, then add this amount into the seed bead tube.
(vi) Continue in this way until there is a total of 50 µl in the seed bead tube. This ensures a high recovery of crushed seeds.
(vii) Vortex the seed bead for 2–3 min to further crush the seeds. We do not recommend sonication (as used by Luft & DeTitta, 1999 ▶) as it increases the risk of overheating the seed stock.
(viii) Make 1:10 serial dilutions in the reservoir solution with the concentrated seed stock. Store concentrated and diluted seed stocks at −80°C.
Seeds prepared in this way are usually stable in the original reservoir solution. We have found that frozen seed stocks (both concentrated and diluted) can undergo multiple cycles of freezing and thawing without any adverse effect on their ability to nucleate crystallization. An in-depth study of the stability of seed crystals has been made by Shaw Stewart et al. (2011 ▶).
2.2. Performing MMS using a robot
MMS experiments are set up in essentially the same way as standard vapour-diffusion experiments. The difference is that a seed stock is added to each drop, in addition to protein and reservoir solution. We offer the following guidelines for setting up MMS with a liquid-handling robot.
(i) Use any crystallization screen. A good starting point might be to use the screen that contained the conditions in which the seed crystals grew, or any general-purpose sparse-matrix screen.
(ii) Use any crystallization robot that employs contact dispensing and has fluidics with a sufficiently wide bore to accommodate seed stocks without clogging. We have successfully used both the Douglas Instrument Oryx and the TTP Labtech Mosquito for MMS.
(iii) Resuspend the seed stock immediately before setup to ensure a homogeneous suspension (vortex or repeatedly aspirate with a pipette).
(iv) For most purposes a crystallization drop volume ratio of 3 parts protein:2 parts reservoir solution:1 part seed stock is suitable, but this can be varied. We have routinely used a total drop volume of 600 nl consisting of 300 nl protein, 200 nl reservoir and 100 nl seed stock.
(v) Mixing of the crystallization drops after adding the seed stock is not recommended.
2.3. Effect of seed stock dilution
We suggest that a new and untested seed stock first be used without dilution. In later experiments, the number and size of crystals can be controlled by diluting the seed stock with reservoir solution. Lowering the concentration of the seed stock lowers the level of nucleation in the drop, giving fewer but larger crystals. Fig. 1 ▶ shows an example of how different concentrations of seed stocks affected crystal growth for a tyrosine kinase. In this case the seed stock was made and MMS carried out into The PEGs Suite (Qiagen) screen using the methods described in §§2.1 and 2.2. The seed stock was then diluted 1:100 and 1:1000 with additional reservoir solution and MMS was repeated.
Figure 1.
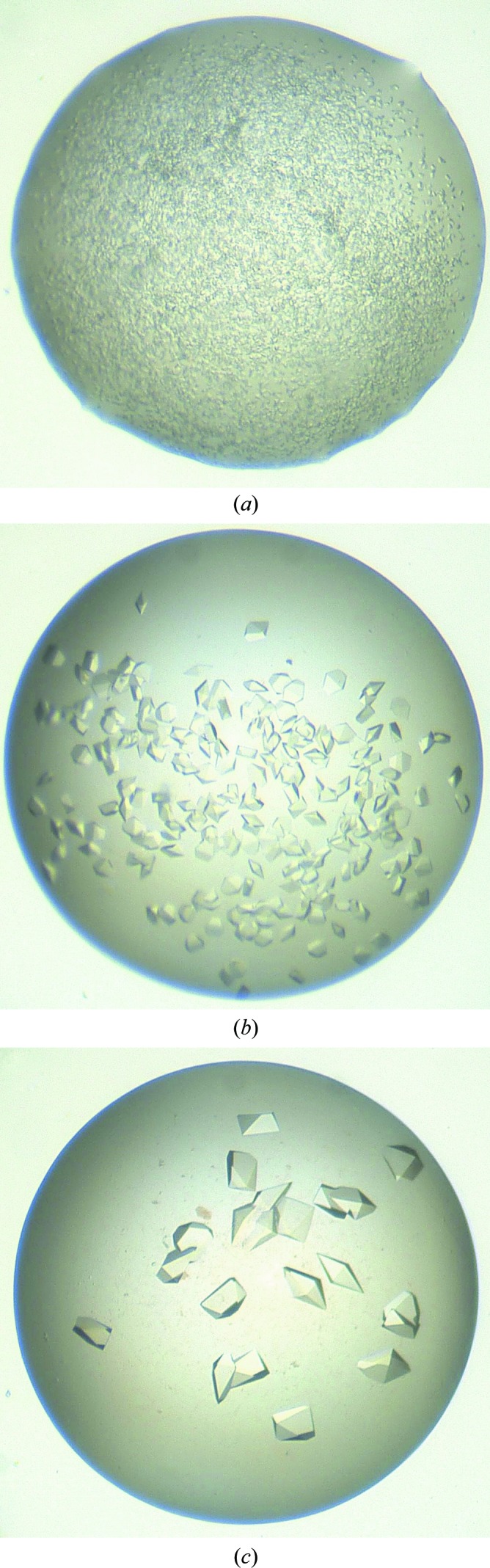
The effect of adjusting seed stock concentration. An initial seed stock was made from crystals of a tyrosine kinase grown in 100 mM HEPES pH 7.0, 8%(w/v) PEG 8000, 7%(v/v) ethylene glycol. MMS into The PEGs Suite screen (Qiagen) was performed with an Oyrx8 robot (Douglas Instruments, UK). Sitting drops were set up in Innovaplate SD-2 crystallization plates, with droplets consisting of 300 nl protein solution, 200 nl reservoir solution and 100 nl seed stock. The figures show the effect of seed concentration on the crystallization in a single condition [0.1 M MES pH 6.5, 25%(w/v) PEG 550 monomethyl ether (MME) from The PEGs Suite]. Drop (a) was seeded with undiluted seed stock, (b) with a 1:100 dilution and (c) with a 1:1000 dilution (Marsh, 2008 ▶; Cowan-Jacob, unpublished results).
2.4. Iterative rounds of seeding
Crystal quality can often be improved by successive rounds of seeding (referred to as ‘iterative seeding’). For this reason, we recommend seeding from the best quality crystals available, in the confidence that even better crystals are likely to be obtained after seeding. An example of improvement in crystal quality for a helicase protein achieved by iterative seeding is illustrated in Fig. 2 ▶. Crystals were grown in the The PEGs Suite screen (Fig. 2 ▶ a), a seed stock was made from these crystals and MMS was carried out. Fig. 2 ▶(b) shows the improved morphology of the crystals produced by MMS. These crystals were made into a second seed stock, which was used to perform a second round of MMS. This resulted in further improvement in crystal morphology (Fig. 2 ▶ c).
Figure 2.
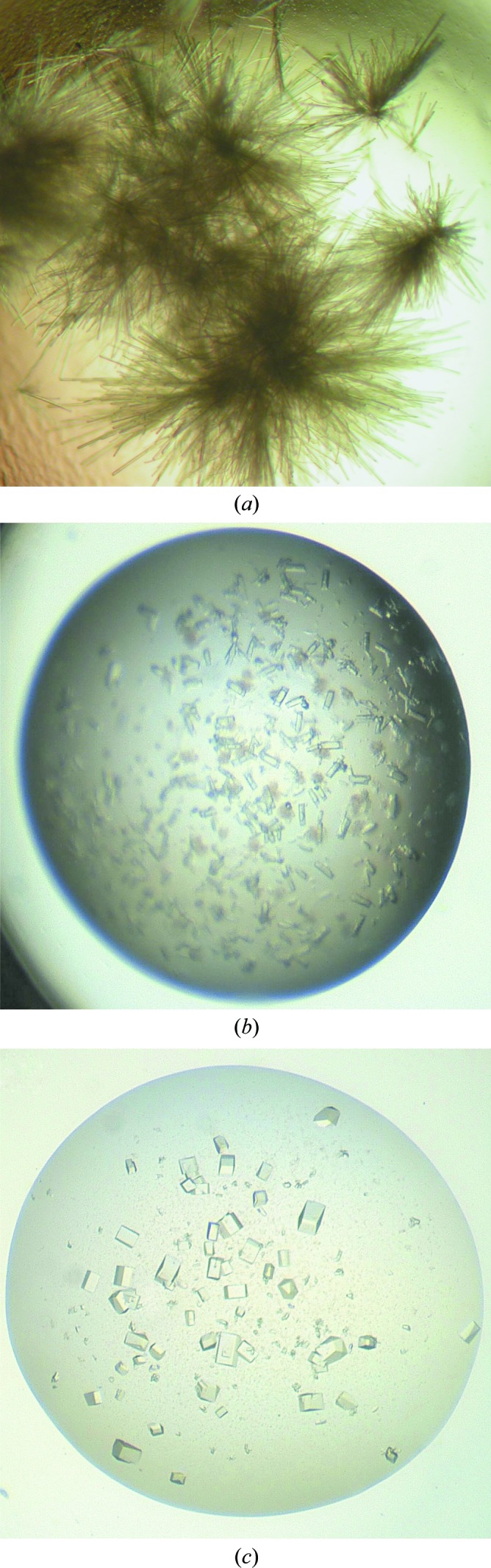
Improving crystal quality by iterative MMS. Initial crystals of a helicase were grown in the The PEGs Suite screen (Qiagen). A seed stock was prepared and MMS into The PEGs Suite screen was performed using the same procedure as in Fig. 1 ▶. Additional rounds of MMS into The PEGs Suite screen were then performed to optimize the crystals. All figures are taken from Marsh (2008 ▶) and Cowan-Jacob (unpublished results). (a) Crystals used to make an initial seed stock for MMS; these grew in 14%(w/v) PEG 8000, 0.1 M Tris–HCl pH 7.5. (b) Crystals obtained from the first round of MMS. These grew in 20%(w/v) PEG 3350, 0.2 M magnesium sulfate and were used to make the second seed stock. (c) Crystals obtained from the second round of MMS. These grew in 40%(v/v) PEG 200, 0.1 M Tris–HCl pH 8.5.
2.5. Cross-linking of seeds
During our initial studies, methods to optimize seed stability by cross-linking seeds with glutaraldehyde were also investigated, following a procedure described by Lusty (1999 ▶). Our experiments demonstrated that the cross-linked seeds retained their capacity to induce crystal formation. This has also been confirmed by Newman et al. (2011 ▶) in work on human arginase.
2.6. Effects of the reservoir solution containing the seeds
Introduction of the seeds into the new drops necessarily includes the reservoir solution in which the seeds are contained. It has been suggested by St John et al. (2008 ▶) that the additional crystallization hits generated by MMS arise primarily (but not exclusively) from changes in the drop composition, rather than the seeds.
However, in our own experiments on more than 15 different proteins (D’Arcy, Cowan-Jacob, Marsh & Villard, unpublished results; Marsh, 2008 ▶), MMS crystallization experiments using either the reservoir solution from an initial hit condition alone or the reservoir solution containing a seed stock were set up in parallel. In every case, the seed stock was necessary to induce crystallization. Therefore, although addition of the reservoir solution alone may be sufficient to induce crystallization in some cases, we have experienced much greater success for minimal extra effort by using a seed stock.
3. Applications of MMS in drug-discovery platforms
3.1. Early results with MMS
MMS studies performed at Novartis between 2006 and 2007 showed that the technique had a positive outcome for the crystallization of 21 of the 26 proteins tested (D’Arcy et al., 2007 ▶; Marsh, 2008 ▶; D’Arcy, Cowan-Jacob, Marsh & Villard, unpublished results). These effects included an increase in the number of crystal hits, the generation of new crystal forms, crystals with improved diffraction quality and structures for previously uncrystallizable complexes. Examples of the improvement in crystal quality are shown in Fig. 3 ▶. Some of these studies used MMS in combination with cross-seeding between complexes of a protein with different inhibitors or between mutants of a protein to increase the probability of crystallization for difficult complexes or mutants. These results inspired further studies that are discussed in more detail in the following sections.
Figure 3.

Improving crystal morphology by MMS. (a) Crystals of a cysteine protease used to make seed stock; these grew in 30%(w/v) PEG 3350, 0.1 M HEPES pH 7.0 (D’Arcy et al., 2007 ▶). (b) Optimized crystals of the cysteine protease grown in 20%(w/v) PEG 3350, 0.2 M magnesium hexahydrate through the use of MMS with the seed stock from the crystals in (a) (D’Arcy et al., 2007 ▶). (c) Crystals of a serine protease used to make seed stock; the conditions were 30%(w/v) PEG 3350, 0.1 M Tris–HCl pH 8.5. (d) Optimized crystals of the serine protease grown in 30%(w/v) PEG 3350, 0.2 M magnesium chloride, 0.1 M Tris–HCl pH 8.5 after MMS with the crystals from (c) (D’Arcy et al., 2007 ▶, unpublished results). (e) These small, unpromising crystalline aggregates of a serine protease complexed with a natural product inhibitor were grown in 20%(w/v) PEG 10 000, 0.1 M Tris–HCl pH 8.5 and used to make a seed stock. (f) Optimized crystals of the same complex after MMS with the seed stock from the original conditions. The final conditions were 25%(w/v) PEG 3350, 0.2 M lithium sulfate, 0.1 M HEPES pH 7.5. (g) A multiple-layer crystal of a serine/threonine kinase obtained through MMS in 2.2 M ammonium sulfate, 0.2 M cadmium sulfate using a seed stock of crystals from a different serine/threonine construct. This crystal was then made into a new seed stock (Marsh, 2008 ▶; Cowan-Jacob, unpublished results). (h) Crystals obtained through MMS using this new seed stock. These crystals grew in 2 M ammonium sulfate, 0.1 M MES pH 6.5, 4.4%(v/v) PEG 400 (Marsh, 2008 ▶; Cowan-Jacob, unpublished results).
3.2. MMS is efficient and cost-effective
In the pharmaceutical industry efficiency and cost-effectiveness are of prime importance, and the most expensive part of crystallization/optimization for medically relevant human proteins is the protein itself. One of the advantages of MMS optimization is that as little as 15–30 µl of protein and between 5 and 10 µl of seed stock is sufficient to set up 96 crystallization conditions. Another advantage is that seeding can reduce the waiting time for crystals to appear.
An example showing the effectiveness of the MMS approach for the crystallization of a medically relevant protein is described by Rak (2013a ▶,b ▶). A classical, manual optimization was used to optimize an initial hit. The optimization process required 4.5 mg protein solution, approximately 9 d of effort and six weeks to grow the crystals, which diffracted to only 3.2 Å resolution. Subsequently, an MMS optimization was performed in which with the crystals from the initial screen were introduced as seeds into four unrelated screens: two sparse-matrix and two grid screens. Crystals grew within 1–2 d. The best crystals from the MMS optimization diffracted to between 2.2 and 1.7 Å resolution (Fig. 4 ▶).
Figure 4.

A comparison of classical and MMS optimizations. (a) Initial crystals requiring optimization. (b) Crystals diffracting to 2.7 Å resolution obtained using classical optimization techniques without seeding. (c) Crystals diffracting to 1.7 Å resolution obtained by MMS into unrelated conditions. The MMS was performed using a seed stock from the crystals in (a). Figures provided by Rak (2013b ▶).
3.3. MMS for crystallization with compounds and co-solvents
Another major challenge for structural biology in the context of drug discovery is that robust crystallization systems must be established that allow efficient soaking or co-crystallization with small-molecule inhibitors. A frequent stumbling block in co-crystallization is the presence of ligand molecules or co-solvents at high concentrations; these may or may not inhibit crystallization in otherwise productive conditions. It is our experience that MMS can improve the chances of overcoming this hurdle. MMS has frequently produced crystals of apo or inhibitor-bound states of proteins that were otherwise recalcitrant to crystallization despite extensive screening. The increased probability of crystal formation with MMS can apparently compensate to some degree the inhibitory effects caused by the ligand compounds and their co-solvents. Fig. 3 ▶(e) shows an example of a serine protease drug target (complexed with a natural product inhibitor) that only produced poor-quality crystalline aggregates after initial screening. These spherulites could be reproduced and were used as a seed stock for MMS; the crystals that were subsequently obtained (Fig. 3 ▶ f) were of high quality and led to the structure determination of this important complex.
3.4. MMS for crystal engineering
MMS proved to be particularly useful in a fragment-based drug-discovery effort screening for inhibitors of the protein–protein interaction between the human recombinase RAD51 and hub protein BRCA2 (Valkov et al., 2012 ▶). Human RAD51 cannot be expressed in a stable, monomeric, unliganded form, so RadA, the archaeal homologue of RAD51, was used as a surrogate protein. More than 20 multiple-point mutants of RadA were engineered that were unable to self-associate, were free of protein or peptide ligands, and had humanized interaction surfaces around the binding site. These mutants crystallized in a number of different crystal forms and space groups, reflecting the differences in structure. Unfortunately, the most humanized mutants tended to crystallize with obstructed binding sites. The use of MMS, combined with cross-seeding between different mutants, made it possible to rationally engineer the crystal forms of the most humanized mutants so that they had unobstructed binding sites optimal for soaking (Fig. 5 ▶). This was achieved by seeding the crystallization drops of the most humanized mutants with seed stocks from crystals of other mutants that had the desired crystal form. Furthermore, this approach allowed the crystallization of various mutants that had previously been uncrystallizable (M. Marsh & M. Hyvonen, manuscript in preparation).
Figure 5.

Crystal engineering of RadA by MMS. Crystal structures of mutants of RadA showing variation in crystal contacts and space groups. These different structures were achieved through the use of MMS and cross-seeding. The structures are arranged (top to bottom) from the most-open to the least-open binding sites (Valkov et al., 2012 ▶). Atoms involved in crystal contacts are coloured red and those comprising the ligand-binding site are shown in blue. The crystal symmetry mates closest to the binding sites are shown as cartoons. Space groups are indicated. The wild-type RadA (WT) structure is shown in its native crystal form; RadA mutant 1 is shown with the crystal form obtained through seeding with mutant 2; RadA mutants 2 and 3 are shown in their native crystal forms obtained without the use of seeding. Using MMS and cross-seeding, it was possible to engineer the crystal forms of the different mutants. For example, it was not possible to crystallize mutant 1 without the seeds from mutant 2. Seeding mutant 2 with the wild-type crystals produced the wild-type crystal form. Seeding mutant 3 with crystals of mutant 2 produced the mutant 1 crystal form.
4. Expanded applications of MMS: recent developments and wider applications
4.1. MMS for many protein classes
The original experiments using automated MMS were performed exclusively with proteases (D’Arcy et al., 2007 ▶). In subsequent studies performed at Novartis, MMS was successfully extended to include kinases and helicases (Cowan-Jacob, 2007 ▶, 2014 ▶; Marsh, 2008 ▶). Obmolova and coworkers have performed some of the most convincing and elegant examples of MMS to obtain and optimize crystals of Fabs and Fab–antigen complexes (Obmolova et al., 2010 ▶, 2014 ▶; Obmolova, 2011 ▶, 2013 ▶).
However, to our knowledge, seeding and therefore MMS have not yet been used to a great extent with membrane proteins. Although the nucleation event is equally important with this class of proteins, the complexity of the crystallization systems and the lack of large quantities of stable protein have hindered the systematic implementation of MMS. Another problem is the difficulty of introducing seeds into the lipid cubic phase that is often used to crystallize membrane proteins.
We are aware of only one published report, from the Maier group (Biocenter, Basel, Switzerland), describing the successful use of seeding with a membrane protein. For TamA, a protein involved in autotransporter biogenesis, a seed stock was made from plate-like crystals and added in a 1:9 ratio to the TamA solution in bicelles before the crystallization setup. This led to an improvement in crystal quality and a 2.25 Å resolution data set was obtained (Gruss et al., 2013 ▶).
This work should encourage other attempts to use MMS with membrane proteins. Indeed, Professor O. Einsle (Freiburg University, Germany) has observed an increased number of hits from MMS for the membrane proteins FocA and NirC (Gerhardt, 2014 ▶, unpublished results), although there was no improvement in the diffraction quality of the crystals.
4.2. MMS to promote crystallization in different space groups
Multiple crystal forms may be necessary for improving diffraction quality, facilitating data collection or obtaining a suitable crystal packing for inhibitor-soaking studies. A common question is whether MMS can generate crystals in a space group different from that of the original seeds. One recent example, arylamine N-acetyltransferase (NAT; Table 1 ▶), shows that MMS can indeed lead to a space-group change (Abuhammad et al., 2013 ▶). In general, though, it is difficult to provide a universal answer to this question, as in many cases the crystals used for the seed source are of such poor quality that no reliable information concerning space group and unit-cell parameters is available. At the Recent Advances in Macromolecular Crystallization 2013 meeting, a request for examples of changes in space group or unit-cell parameters in crystals obtained by MMS was presented to the community. The authors are grateful to the researchers who have kindly submitted their examples, which are summarized here in Table 1 ▶ (Newman, 2013 ▶; Oganesyan, 2013 ▶; Rak, 2013a ▶).
Table 1. Examples of changes in space group or unit-cell parameters in crystals obtained by MMS.
Unit-cell lengths are all given in Å and angles are given in degrees.
| Space group and unit-cell parameters | |||
|---|---|---|---|
| Investigator | Protein code | Crystals used for seeding (MMS) | Crystals resulting from the MMS experiment |
| A. Abuhammad | NAT | P41212; a = 51.94, b = 51.9, c = 176.6, α = β = γ = 90 | P21; a = 96.5, b = 139.2, c = 96.5, α = 90, β = 91.2, γ = 90 |
| V. Oganesyan | V12 | P6122; a = b = 108.1, c = 140.3 | C2; a = 126.2, b = 64.0, c = 82.4, β = 129.1 |
| J. Newman & T. Peat | Arginase (round 1) | P3; a = 90.4, b = 90.4, c = 69.5, α = 90, β = 90, γ = 120 | P21; a = 53.3, b = 281.9, c = 67.0, α = 90, β = 89.6, γ = 90 |
| J. Newman & T. Peat | Arginase (round 2) | P21; a = 53.3, b = 281.9, c = 67.0, α = 90, β = 89.6, γ = 90 | P212121; a = 52.5–53.1, b = 67.3, c = 245.2–261.2 |
| J. Newman & T. Peat | CA, mutant 1 | Mutant 1 crystals | P21; a = 45.3, b = 141.1, c = 77.2, α = 90, β = 90.1, γ = 90 |
| J. Newman & T. Peat | CA, mutant 2 | Mutant 2 crystals | P21; a = 41.8, b = 69.3, c = 44.6, α = 90, β = 107.8, γ = 90 |
| J. Newman & T. Peat | CA, mutant 3 | Mutant 3 crystals | P21; a = 42.2, b = 134.3, c = 46.8, α = 90, β = 104, γ = 90 |
| J. Newman & T. Peat | CA, mutant 4 | Mutant 2 crystals | P21; a = 41.6, b = 67.9, c = 43.1, α = 90, β = 103.1, γ = 90 |
| J. Newman & T. Peat | Truncated AtzF | P222; a = 113, b = 158, c = 184, α = 90, β = 90, γ = 90 | P21; a = 82.4, b = 179.2, c = 112.6, α = 90, β = 106.6, γ = 90 |
| J. Newman & T. Peat | AtzD | P2; a = 85, b = 117.1, c = 159.6, α = 90, β = 88.8, γ = 90 | H3; a = 131, b = 131, c = 233, α = 90, β = 90, γ = 120 |
| J. Newman & T. Peat | Protein V | P312; a = 102, b = 102, c = 133.5, α = 90, β = 90, γ = 120 | P43212; a = 119.4, b = 119.4, c = 220.9, α = 90, β = 90, γ = 90 |
| A. Rak | F1 | P61; a = b = 87.4, c = 174.0 | P65; a = b = 145.7, c = 163.6 |
| P622; a = b = 131, c = 328 | |||
4.3. MMS in combination with cross-seeding
In general, MMS experiments use seeds of the same protein to generate more crystal hits or crystals of better quality (self-seeding). Cross-seeding and MMS can be used in the crystallization of proteins that differ slightly in their physicochemical properties or for related proteins with sequence differences (but see also §4.4). This is illustrated by the following examples.
(i) Walter et al. (2008 ▶) describe the use of cross-seeding to obtain crystals of selenomethionine-labelled Vaccinia virus CrmE. The selenomethionine-derivatized protein could only be crystallized by seeding with the crystals of the native protein (i.e. unlabelled with selenomethionine). Another example is that of native and selenomethionine-labelled SHARPIN (Stieglitz et al., 2012 ▶).
(ii) As detailed in §3.4, Valkov et al. (2012 ▶) obtained crystals of different mutants of a protein by cross-seeding and MMS.
(iii) In experiments performed at Novartis, seeds from apo-form crystals were used in MMS to facilitate crystallization of inhibitor complexes and vice versa (D’Arcy, Cowan-Jacob, Marsh & Villard, unpublished results).
(iv) Cross-seeding has been used to crystallize homologous proteins. Crystals of Mycobacterium marinum arylamine N-acetyltransferase (NAT; Table 1 ▶; Abuhammad et al., 2013 ▶) were used to cross-seed the M. tuberculosis homologue. The two proteins share 74% sequence homology. Whereas the M. marinum protein crystallized easily, thousands of crystallization trials were unsuccessful for the M. tuberculosis homologue until cross-seeding was used. We also routinely cross-seed with M. smegmatis IspD crystals to provide a steady and reliable source of M. tuberculosis IspD crystals (Björkelid et al., 2011 ▶). Without cross-seeding, the M. tuberculosis IspD crystallizes in a single condition in the MORPHEUS screen (Gorrec, 2009 ▶), whereas cross-seeding increases the number of hit conditions tenfold.
(v) MMS has been successfully applied to the crystallization of antibody–antigen complexes. Obmolova et al. (2010 ▶) performed MMS using seeds derived from crystals of IL13 in complex with a humanized antibody H2L6 to obtain diffraction-quality crystals of IL13 in complex with either an affinity-matured human variant (M1295/IL13) or a mouse antibody (C836/IL13). Coarse screens without MMS were originally set up for all three complexes (192 conditions each), but only H2L6/IL13 produced needle-like microcrystals (Fig. 6 ▶ a). These crystals were optimized and used to cross-seed the crystallization of M1295/IL13 and C836/IL13 (Figs. 6 ▶ b–6 ▶ d).The Fab antibodies H2L6 and C836 have identical constant domains but different variable domains. The authors observed an intermolecular crystal contact common to both crystal forms: a β-bridge between the light and heavy chains of contacting Fab constant domains. In contrast, the crystal contacts involving either the Fab variable domains or IL-13 are quite different in the two crystal forms. It is therefore possible that the interactions between the constant domains formed the basis of the crystal lattice to nucleate cross-seeded crystal growth.
Figure 6.

MMS and cross-seeding between FAB complex H2L6/IL13 and FAB complexes M1295/IL13 and C836/IL13. (a) Microcrystals of H2L6/IL13 [grown in 28%(w/v) PEG 8000, 0.1 M MES pH 6.5] used as seed stock for the MMS crystallization of H2L6/IL13. (b) Optimized crystals of H2L6/IL13 [grown in 20%(w/v) PEG 3350, 0.2 M ammonium citrate pH 5.1] used to generate a seed stock for MMS crystallization of M1295/IL13 and C836/IL13. (c) Crystals of M1295/IL13 obtained after MMS in 20%(w/v) PEG 3350, 0.2 M sodium citrate pH 8.3. (d) Crystals of C836/IL13 obtained by MMS in 20%(w/v) PEG 3350, 0.2 M ammonium citrate. All figures taken from Obmolova et al. (2010 ▶).
4.4. Cross-seeding between unrelated proteins
The group at Novartis (D’Arcy, Cowan-Jacob, Marsh & Villard, unpublished results) tested the feasibility of using crystals of different proteins for cross-seeding. Porcine pancreatic elastase and bovine trypsin were selected as they share the same fold, have a sequence identity of 36% and both crystallize in space group P212121 with similar unit-cell parameters: a = 51, b = 58, c = 76 Å for porcine pancreatic elastase and a = 54, b = 58, c = 67 Å for bovine trypsin. Highly concentrated seed stocks were used for both proteins in an attempt to induce cross-nucleation between them. Although the seed stocks could nucleate the crystallization of their parent protein, the cross-seeding was unsuccessful. Analysis of the crystal packing of the two proteins shows that there is no apparent commonality of crystal contacts between the two lattices that might support epitaxial nucleation (Schiering, 2014 ▶). Further experiments would be necessary to determine what degree of sequence or structural similarity that two proteins must share before cross-seeding between them can be successful.
5. Future perspectives
As the number of MMS users increases, there is mounting evidence that cross-seeding is a viable strategy. It can be used to improve existing crystals, to optimize crystalline leads and to screen for polymorphs. Another potential application may be for generating homogeneous crystals for free-electron laser (FEL) measurements. To better implement MMS and extend its areas of application, an understanding of how it works would be beneficial.
5.1. Identifying nonspecific nucleants for MMS
A careful and systematic examination of crystal contacts in structures from crystals produced by cross-seeding may elucidate the mechanisms and structural determinants of epitaxial nucleation and the relationship between the source and the final crystals. This is a challenge for structural bioinformaticians and may provide important insights into the crystallization process.
However, it may be that in many cases the nucleation is heterogeneous rather than epitaxial, i.e. the seeds (or a contaminant) work by introducing a generic nucleation surface rather than specific lattice interactions. If heterogeneous nucleation is the mechanism, this opens the exciting possibility of performing MMS with a mixture of nucleation agents when seeds from the target protein or closely related protein constructs are not available. There is mounting evidence that a combination of agents can be used to influence nucleation through both specific and nonspecific interactions during initial screening. Thakur et al. (2008 ▶) have reported that a combination of heterogeneous nucleation agents had a positive effect on the number of hits obtained in screening. It might be possible to construct a generic ‘magic pot’ of various protein crystals combined with a selection of heterogeneous nucleants described in the literature (McPherson & Shlichta, 1987 ▶; Punzi et al., 1991 ▶; Chayen et al., 2001 ▶; Fermani et al., 2001 ▶; Pechkova & Nicolini, 2001 ▶, 2002 ▶; D’Arcy et al., 2003 ▶; Rong et al., 2004 ▶; Saridakis et al., 2011 ▶). This mixture might then be used in a final attempt to induce nucleation when normal screening has failed to produce any material suitable as seeds.
5.2. Improving identification of specific nucleants for MMS
We and many other authors have shown that even unpromising small crystalline aggregates can be suitable starting points for MMS. In some cases, the keys to unlock a crystallization deadlock may be present in apparently ‘failed’ screens, where they remain undetected or unrecognized. Successful application of MMS will therefore benefit greatly from improvements in methods for detecting crystalline material that is not immediately apparent with standard white-light microscopy. Promising detection methods include the following.
(i) Ultraviolet fluorescence microscopy relies on the fluorescing absorbance of tryptophans (and to a lesser degree tyrosines) to identify protein crystals. Among other things, it can identify microcrystals obscured by heavy precipitates and differentiate salt from protein crystals. However, its usefulness is restricted to proteins that contain fluorescent amino acids.
(ii) The method developed by Pusey (2011 ▶, 2013 ▶) circumvents the requirement for the protein to contain tryptophans. His method covalently binds a trace fluorescent label to the protein before crystallization trials; the fluorescent signal originates from the bound label and not the amino acids. It allows the detection of small regions of unusually concentrated protein in drops where there are no visible crystals (Pusey, 2011 ▶, 2013 ▶). The material in these conditions may represent a favourably high concentration of protein (including gel formation) that is often a necessary pre-condition for crystal optimization. An example is shown in Fig. 7 ▶.
(iii) Second-harmonic generation (SHG) is a phenomenon of nonlinear optics where excitation of a crystal by two low-frequency (low-energy) photons at very high intensity gives rise to emission of a photon at the second harmonic of the excitation frequency (twice the frequency, twice the energy).This phenomenon has been exploited in second-order nonlinear optical imaging of chiral crystals (SONICC; Wampler et al., 2008 ▶). An instrument is commercially available as the ‘SONICC’ imager from Formulatrix. This instrument offers another alternative for early detection of microcrystalline material and can differentiate between salt and protein crystals. Imaging the resulting fluorescence emission in the far UV provides additional information for distinguishing between chiral salt crystals and protein crystals, both of which can give rise to signals in SONICC imaging.
Figure 7.

Advances in the detection of crystalline material. (a) White-light image of canavalin [screening condition 30%(w/v) PEG monomethyl ether (MME) 2000, 0.2 M ammonium sulfate, 0.1 M sodium acetate pH 4.6]. (b) Fluorescence image of canavalin trace-labelled with carboxyrhodamine in the same condition. Despite the unpromising appearance of the intensely fluorescent material, optimizing around this condition led to well diffracting crystals (Pusey, 2013 ▶). (c) White-light image of a crystalline precipitate. (d) Image of the same figure taken with SONICC. Images provided by Formulatrix.
6. Conclusions
Since the original paper by Ireton & Stoddard (2004 ▶) defining and describing the microseed matrix seeding method and the subsequent expansion and automation of the technique by D’Arcy et al. (2007 ▶), MMS has been successfully applied to many different families of proteins and protein complexes and has improved hit rates and crystal quality. Its application in the pharmaceutical industry to produce suitable and reproducible crystallization systems for inhibitor studies has had a positive impact on numerous drug-discovery projects. There are many reasons (time, cost, efficiency, simplicity) to favour MMS over the classical approach of fine-screening around initial hits. In our experience, many people dogmatically reject MMS (and seeding in general) on the basis of their attempts on a single project. While MMS cannot solve every problem that prevents crystallization, it can be one of the most straightforward and inexpensive ways to rejuvenate a failing project. We hope that the information in this review will encourage and equip more investigators to use MMS successfully.
Acknowledgments
The authors thank the following people for their contributions to this article: Alexey Rak for data and images of an example of using MMS in an ongoing drug-discovery project; Vaheh Oganeysan, Alexey Rak, Janet Newman and Tom Peat for providing experimental data on crystal forms and space-group changes induced by MMS; Galina Obmolova for data and figures for examples of successful MMS with Fabs and Fab–antigen complexes; Frederic Villard for images and crystallization conditions of protease crystals obtained with MMS; Nikolaus Schiering for comparing the sequence identity and crystal packing in bovine trypsin and porcine pancreatic elastase; Stefan Gerhardt and Oliver Einsele for providing examples of MMS with membrane proteins; Christian Oefner and Fritz Winkler for helpful discussions on cross-seeding and space-group changes; Marc Pusey for providing images of trace-fluorescent labelled proteins; Ellen Gualtieri, Formulatrix for providing SONICC images of crystallization drops; and Tim Sharpe for critical reading of the manuscript.
References
- Abuhammad, A., Lowe, E. D., McDonough, M. A., Shaw Stewart, P. D., Kolek, S. A., Sim, E. & Garman, E. F. (2013). Structure of arylamine N-acetyltransferase from Mycobacterium tuberculosis determined by cross-seeding with the homologous protein from M. marinum: triumph over adversity. Acta Cryst. D69, 1433–1446. [DOI] [PubMed]
- Bergfors, T. (2003). Seeds to crystals. J. Struct. Biol. 142, 66–76. [DOI] [PubMed]
- Björkelid, C., Bergfors, T., Henriksson, L. M., Stern, A. L., Unge, T., Mowbray, S. L. & Jones, T. A. (2011). Structural and functional studies of mycobacterial IspD enzymes. Acta Cryst. D67, 403–414. [DOI] [PubMed]
- Chayen, N. E., Saridakis, E., El-Bahar, R. & Nemirovsky, Y. (2001). Porous silicon: an effective nucleation-inducing material for protein crystallization. J. Mol. Biol. 312, 591–595. [DOI] [PubMed]
- Cowan-Jacob, S. (2007). Personal communication.
- Cowan-Jacob, S. (2014). Personal communication.
- D’Arcy, A., Mac Sweeney, A. & Haber, A. (2003). Using natural seeding material to generate nucleation in protein crystallization experiments. Acta Cryst. D59, 1343–1346. [DOI] [PubMed]
- D’Arcy, A., Villard, F. & Marsh, M. (2007). An automated microseed matrix-screening method for protein crystallization. Acta Cryst. D63, 550–554. [DOI] [PubMed]
- Fermani, S., Falini, G., Minnucci, M. & Ripamonti, A. (2001). Protein crystallization on polymeric film surfaces J. Cryst. Growth, 224, 327–334.
- Gerhardt, S. (2014). Personal communication.
- Gorrec, F. (2009). The MORPHEUS protein crystallization screen. J. Appl. Cryst. 46, 1035–1042. [DOI] [PMC free article] [PubMed]
- Gruss, F., Zähringer, F., Jakob, R. P., Burmann, B. M., Hiller, S. & Maier, T. (2013). The structural basis of autotransporter translocation by TamA. Nature Struct. Mol. Biol. 20, 1318–1320. [DOI] [PubMed]
- Ireton, G. C. & Stoddard, B. L. (2004). Microseed matrix screening to improve crystals of yeast cytosine deaminase. Acta Cryst. D60, 601–605. [DOI] [PubMed]
- Luft, J. R. & DeTitta, G. T. (1999). A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Cryst. D55, 988–993. [DOI] [PubMed]
- Lusty, C. J. (1999). A gentle vapor-diffusion technique for cross-linking of protein crystals for cryocrystallography. J. Appl. Cryst. 32, 106–112.
- Marsh, M. (2008). Structural studies on the enzymes of the gentisate biodegradation pathway. PhD thesis, University of Bristol, England.
- McPherson, A. & Shlichta, P. J. (1987). Facilitation of the growth of protein crystals by heterogeneous/epitaxial nucleation. J. Cryst. Growth, 85, 206–214.
- Newman, J. (2013). Personal communication.
- Newman, J., Pearce, L., Lesburg, C. A., Strickland, C. & Peat, T. S. (2011). Crystallization of an apo form of human arginase: using all the tools in the toolbox simultaneously. Acta Cryst. F67, 90–93. [DOI] [PMC free article] [PubMed]
- Obmolova, G. (2011). Microseed matrix screening crystallization of antibody fragments and antibody-antigen complexes. Recent Advances in Macromolecular Crystallization Presentation Abstracts https://hamptonresearch.com/documents/ramc/RAMC2011_T11_Obmolova.pdf.
- Obmolova, G. (2013). Maximizing crystallization success with microseed matrix screening. Recent Advances in Macromolecular Crystallization Presentation Abstracts. https://hamptonresearch.com/documents/ramc/RAMC2013T_Obmolova.pdf.
- Obmolova, G., Malia, T. J., Teplyakov, A., Sweet, R. & Gilliland, G. L. (2010). Promoting crystallization of antibody-antigen complexes via microseed matrix screening. Acta Cryst. D66, 927–933. [DOI] [PMC free article] [PubMed]
- Obmolova, G., Malia, T. J., Teplyakov, A., Sweet, R. & Gilliland, G. L. (2014). Protein crystallization with microseed matrix screening: application to human germline antibody Fabs. Acta Cryst. F70, 1107–1115. [DOI] [PMC free article] [PubMed]
- Oganesyan, V. (2013). Personal communication.
- Pechkova, E. & Nicolini, C. (2001). Accelerated protein crystal growth by protein thin film template. J. Cryst. Growth, 231, 599–602.
- Pechkova, E. & Nicolini, C. (2002). Protein nucleation and crystallization by homologous protein thin film template. J. Cell. Biochem. 85, 243–251. [DOI] [PubMed]
- Persson, B. D., Schmitz, N. B., Santiago, C., Zocher, G., Larvie, M., Scheu, U., Casasnovas, J. M. & Stehle, T. (2010). Structure of the extracellular portion of CD46 provides insights into its interactions with complement proteins and pathogens. PLoS Pathog. 6, e1001122. [DOI] [PMC free article] [PubMed]
- Punzi, J. S., Luft, J. & Cody, V. (1991). Protein crystal growth in the presence of poly(vinylidene difluoride) membrane. J. Appl. Cryst. 24, 406–408.
- Pusey, M. L. (2011). Developing a fluorescence-based approach to screening for macromolecule crystallization conditions. Cryst. Growth Des. 11, 1135–1142. [DOI] [PMC free article] [PubMed]
- Pusey, M. L. (2013). Crystallization screening using trace fluorescence labeling. Recent Advances in Macromolecular Crystallization Presentation Abstracts. https://hamptonresearch.com/documents/ramc/RAMC2013T_Pusey.pdf.
- Rak, A. (2013a). Personal communication.
- Rak, A. (2013b). The benefits gained from a redesigned crystallisation strategy focused on a high throughput seeding technique. Recent Advances in Macromolecular Crystallization Presentation Abstracts. https://hamptonresearch.com/documents/ramc/RAMC2013T_Rak.pdf.
- Rong, L., Komatsu, H., Yoshizaki, I., Kadowaki, A. & Yoda, S. (2004). Protein crystallization by using porous glass substrate. J. Synchrotron Rad. 11, 27–29. [DOI] [PubMed]
- Saridakis, E., Khurshid, S., Govada, L., Phan, Q., Hawkins, D., Crichlow, G. V., Lolis, E., Reddy, S. M. & Chayen, N. E. (2011). Protein crystallization facilitated by molecularly imprinted polymers. Proc. Natl Acad. Sci. USA, 108, 11081–11086. [DOI] [PMC free article] [PubMed]
- Schiering, N. (2014). Personal communication.
- Shaw Stewart, P. D., Kolek, S. A., Briggs, R. A., Chayen, N. E. & Baldock, P. F. M. (2011). Random microseeding: A theoretical and practical exploration of seed stability and seeding techniques for successful protein crystallization. Cryst. Growth Des. 11, 3432–3441.
- Stieglitz, B., Rittinger, K. & Haire, L. F. (2012). Crystallization of SHARPIN using an automated two-dimensional grid screen for optimization. Acta Cryst. F68, 816–819. [DOI] [PMC free article] [PubMed]
- St John, F. J., Feng, B. & Pozharski, E. (2008). The role of bias in crystallization conditions in automated microseeding. Acta Cryst. D64, 1222–1227. [DOI] [PubMed]
- Thakur, A. S., Newman, J., Martin, J. L. & Kobe, B. (2008). Increasing protein crystallization screening success with heterogeneous nucleating agents. Methods Mol. Biol. 426, 403–409. [DOI] [PubMed]
- Valkov, E., Sharpe, T., Marsh, M., Greive, S. & Hyvönen, M. (2012). Targeting protein-protein interactions and fragment-based drug discovery. Top. Curr. Chem. 317, 145–179. [DOI] [PubMed]
- Walter, T. S., Mancini, E. J., Kadlec, J., Graham, S. C., Assenberg, R., Ren, J., Sainsbury, S., Owens, R. J., Stuart, D. I., Grimes, J. M. & Harlos, K. (2008). Semi-automated microseeding of nanolitre crystallization experiments. Acta Cryst. F64, 14–18. [DOI] [PMC free article] [PubMed]
- Wampler, R. D., Kissick, D. J., Dehen, C. J., Gualtieri, E. J., Grey, J. L., Wang, H.-F., Thompson, D. H., Cheng, J.-X. & Simpson, G. J. (2008). Selective detection of protein crystals by second harmonic microscopy. J. Am. Chem. Soc. 130, 14076–14077. [DOI] [PMC free article] [PubMed]