

The structure of Xsa43E possesses the characteristic five-bladed β-propeller domain seen in all GH43 enzymes. The functional characterization of Xsa43E shows it to be an arabinofuranosidase capable of cleaving arabinose side chains from short segments of xylan.
Keywords: Xsa43E, GH43, Butyrivibrio proteoclasticus
Abstract
The rumen of dairy cattle can be thought of as a large, stable fermentation vat and as such it houses a large and diverse community of microorganisms. The bacterium Butyrivibrio proteoclasticus is a representative of a significant component of this microbial community. It is a xylan-degrading organism whose genome encodes a large number of open reading frames annotated as fibre-degrading enzymes. This suite of enzymes is essential for the organism to utilize the plant material within the rumen as a fuel source, facilitating its survival in this competitive environment. Xsa43E, a GH43 enzyme from B. proteoclasticus, has been structurally and functionally characterized. Here, the structure of selenomethionine-derived Xsa43E determined to 1.3 Å resolution using single-wavelength anomalous diffraction is reported. Xsa43E possesses the characteristic five-bladed β-propeller domain seen in all GH43 enzymes. GH43 enzymes can have a range of functions, and the functional characterization of Xsa43E shows it to be an arabinofuranosidase capable of cleaving arabinose side chains from short segments of xylan. Full functional and structural characterization of xylan-degrading enzymes will aid in creating an enzyme cocktail that can be used to completely degrade plant material into simple sugars. These molecules have a range of applications as starting materials for many industrial processes, including renewable alternatives to fossil fuels.
1. Introduction
Lignocellulosic plant material is seen as an increasingly important energy resource as it is abundant and renewable. The plant cell wall makes up the majority of plant material and is predominantly composed of polysaccharides, with the most important of these being cellulose, hemicellulose and pectin. Complete degradation of a plant cell wall into its component sugars requires a suite of enzymes with specific activities for hydrolysing the various chemical linkages between the sugars present within these polymers. As well as being an attractive source of renewable carbon, plant material is also the basis of pastoral grazing systems around the world. Ruminants that ingest this material depend on specialized microorganisms in their rumen to release and ferment the component sugars to volatile fatty acids, providing a source of energy for the animal. The enzymes that are used to accomplish this task have the potential to form an ideal toolkit for the industrial depolymerization of plant material.
Butyrivibrio proteoclasticus was first isolated from the rumen contents of a cow and is capable of degrading the hemicellulose xylan (Attwood et al., 1996 ▶; Moon et al., 2008 ▶). Xylan consists of β-1,4-linked xylose units with a variety of decorating side groups including acetyl, arabinofuranosyl and feruloyl moieties. The genome of B. proteoclasticus has been sequenced, revealing a suite of genes annotated as enzymes involved in fibre degradation and which encode part of the glycobiome of the organism (Kelly et al., 2010 ▶). These enzymes cover a broad range of glycoside hydrolase (GH) families, presumably to accommodate the broad range of activities required for the complete degradation of xylan into simple sugars.
A structural genomics approach has been taken to characterize the suite of fibre-degrading enzymes produced by B. proteoclasticus. Enzymes that have been cloned and expressed and for which three-dimensional structures have been determined are then also functionally characterized. Here, we present the structure of Xsa43E (EC 3.2.1.37), a GH43 family enzyme. According to the CAZy database, GH43 family enzymes have a number of known activities including β-xylosidase, β-1,3-xylosidase, α-l-arabinofuranosidase, arabinanase, xylanase and galactan 1,3-β-galactosidase activities (Lombard et al., 2014 ▶; Cantarel et al., 2009 ▶). All structures of GH43 enzymes have a common five-bladed β-propeller domain. This fold has been seen in four different GH families: GH families 32 and 68 of the GH-J clan and GH families 43 and 62 of the GH-F clan. GH43 enzymes have an inverting mechanism with three residues implicated as being involved in catalysis: a general acid, a general base and a third acid residue proposed to modulate the pK a of the general acid as well as to orientate the substrate and general acid. Here, we highlight some of the interesting structural features of Xsa43E; we also present biochemical evidence for its substrate specificity and use point mutations to probe its catalytic mechanism.
2. Experimental procedures
2.1. Cloning, expression and purification
The Xsa43E gene was amplified from B. proteoclasticus B316 genomic DNA by nested PCR using two sets of primers. Gene-specific primers (forward, 5′-GGCAGCGGCGCGATGACAGAAACTATGACAGAAGA-3′; reverse, 5′-GAAAGCTGGGTGTCAGTTTATCTTTTCAACCCCAT-3′) are used in the initial round of PCR. These contain a 12 base-pair overlap with the generic primers that are used in the second round of PCR (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTC-3′ and 5′-GTGGGTCGAAAGAACATGTTTCACCAGGGG-3′). This two-stage strategy adds the attB recombination sequences and also an rTEV cleavage site between the attB sequence and the original ORF translational start, allowing cloning into the Gateway vectors and facilitating cleavage of any purification tag post expression of protein, as previously described by Moreland et al. (2005 ▶). The PCR product was cloned into the pDONR221 Gateway entry vector and then into the pDEST17 Gateway vector for the expression of recombinant protein with an N-terminal His6 affinity tag. The correct insert was confirmed by DNA sequencing. The plasmid containing the appropriate gene was transformed into Escherichia coli BL21 (DE3) cells for recombinant expression. Expression cultures were grown in 1 l LB broth at 37°C to an OD of ∼0.6 at 600 nm and were then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and transferred to 28°C for a further 16–20 h. Cells were harvested and lysed using sonication. Filtered lysate was purified by immobilized metal-affinity chromatography using a 5 ml HisTrap HP column (GE Healthcare). Protein was eluted from the column with 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 300 mM imidazole. The target protein was identified using SDS–PAGE and was further purified by size-exclusion chromatography using an S-200 16/60 column (Amersham Biosciences) in 20 mM Tris–HCl pH 8.0, 150 mM NaCl. The purity of Xsa43E was analysed by SDS–PAGE. Protein concentration was estimated by UV absorbance using a theoretical extinction coefficient of 77 865 M −1 cm−1. Selenomethionine-incorporated Xsa43E (SeMet-Xsa43) was expressed using PASM-5052 medium containing selenomethionine and E. coli DL41 cells and was purified as described above; the buffers contained the reducing agent β-mercaptoethanol (1 mM) to prevent oxidation of the Se atoms.
2.2. Crystallization, data collection and structure determination
Crystallization was carried out using hanging-drop vapour diffusion in 24-well VDX plates (Hampton Research, USA). Each well contained 500 µl mother liquor. 2 µl mother liquor from the well was mixed with 2 µl protein solution (50 mg ml−1 in buffer consisting of 20 mM Tris–HCl pH 8.0, 150 mM NaCl) on a 22 mm square unsiliconized glass cover slip. The cover slip was then inverted and placed on top of the well; a seal was formed using a layer of grease to exclude air. Crystals of Xsa43E were grown with mother liquor consisting of 18–20% PEG 8000, 0.2 M NaCl, 0.1 M phosphate–citrate buffer pH 4.6. Crystals of SeMet-Xsa43E were grown in buffer that also contained 1 mM β-mercaptoethanol and were grown with mother liquor consisting of 20% PEG 8000, 0.2 M NaCl, 0.1 M phosphate–citrate buffer pH 3.4. All crystals were grown at 18°C. Crystals were transferred to a cryoprotectant consisting of mother liquor containing 20%(v/v) glycerol before being flash-cooled in liquid nitrogen. Diffraction data for native protein and selenomethionine-derivatized protein were collected on beamline 11-1 of the Stanford Synchrotron Radiation Laboratory (SSRL), USA equipped with a MarMosaic 325 CCD detector (MAR USA).
SAD (single-wavelength anomalous diffraction) data were collected at a wavelength of 0.9795 Å. The images were integrated using iMosflm (Battye et al., 2011 ▶) and scaled using the CCP4 program SCALA (Evans, 2006 ▶). The structure was then solved using AutoSol in PHENIX (Adams et al., 2010 ▶) to a resolution of 1.33 Å. AutoSol located nine sites with a figure of merit (FOM) of 0.50. Following density modification with RESOLVE (Terwilliger, 2003 ▶) the FOM improved to 0.73; this was followed by AutoBuild (Terwilliger et al., 2008 ▶). The output model was put through an initial round of refinement in the program REFMAC5 from CCP4 (Winn et al., 2011 ▶). The output was used as a starting point for manual building. Manual model building was performed in Coot (Emsley & Cowtan, 2004 ▶) and progress was checked and final refinement was performed with restrained refinement in REFMAC5. Electron-density maps used for model building were σA-weighted 2|F o| − |F c| (contoured at 1.0σ) and |F o| − |F c| (contoured at ±3.0σ). Model building required inserting alternative conformations for some residues. Waters were added in Coot (Emsley & Cowtan, 2004 ▶) and checked manually. Structure validation was performed with PROCHECK (Laskowski et al., 1993 ▶) from the CCP4 suite. Data-collection and refinement statistics are shown in Table 1 ▶. Difference-map peak heights were generated using PHENIX. Coordinates and structure factors have been deposited in the PDB as entry 4nov.
Table 1. Data and refinement statistics for Xsa43E.
Values in parentheses are for the outer shell.
| Data statistics | |
| Space group | P212121 |
| Wavelength (Å) | 0.9795 |
| Unit-cell parameters | |
| a (Å) | 59.13 |
| b (Å) | 76.54 |
| c (Å) | 82.56 |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 90 |
| Resolution range (Å) | 56.2–1.33 (1.4–1.33) |
| R merge (%) | 7.7 (51.0) |
| No. of measured reflections | 463689 (44441) |
| No. of unique reflections | 82628 (11406) |
| Mean I/σ(I) | 14.9 (2.3) |
| Completeness (%) | 95.9 (91.7) |
| Anomalous completeness (%) | 88 (78.3) |
| Multiplicity | 5.6 (3.9) |
| Anomalous multiplicity | 3.0 (2.2) |
| Refinement statistics | |
| R work/R free (%) | 16.9/18.6 |
| Total No. of atoms | 2827 |
| No. of protein atoms | 2499 |
| Other molecules/ions | 2 [1 Tris, 1 Ca] |
| No. of waters | 319 |
| R.m.s. deviation from standard geometry | |
| Bond lengths (Å) | 0.031 |
| Bond angles (°) | 2.343 |
| Average B factors (Å2) | |
| Protein | 15.653 |
| Water | 26.678 |
2.3. Inductively coupled plasma mass spectrometry
Inductively coupled plasma mass spectrometry (ICP-MS) and the resulting analysis was performed as a service provided by the Chemistry Department at the University of Waikato, Hamilton, New Zealand.
2.4. Site-directed mutagenesis
Site-directed mutagenesis was performed by overlap extension, incorporating custom oligonucleotides containing the desired mutation into the gene by PCR. Mutated genes were then cloned back into the vector (pDEST17) as described for the wild type.
2.5. Enzyme assays
2.5.1. Substrates
p-Nitrophenyl substrates were purchased from Sigma; all other substrates were purchased from Megazymes (Ireland) unless otherwise stated.
2.5.2. Substrate specificity assays
The activity of Xsa43E was tested on the following range of substrates: arabinobiose, arabinohexaose, xylose oligosaccharides with a degree of polymerization from 2 to 6 and arabinoxylan. Xsa43E was incubated with the substrate for a set time at 37°C in 20 mM sodium phosphate buffer pH 7.2. The mixture was analysed by thin-layer chromatography (TLC) for a qualitative result, which was run in a solvent system of chloroform:acetic acid:water (6:7:1) and visualized by the addition of 5% sulfuric acid in ethanol and incubation at 110°C for 10 min. Quantitative assays were carried out as above to determine the specific activity for release of arabinose from arabinobiose, arabinoxylan and arabinoxylan in conjunction with a xylanase from Trichoderma viride. The specific activity for release of arabinose was determined using the Lactose/Galactose Rapid Assay Kit (Megazymes International Ltd) following the manufacturer’s instructions. The kit contains β-galactose dehydrogenase, which oxidizes β-d-galactose (using NAD+) to d-galactonic acid; the amount of NADH formed in this reaction is measured, which is stoichiometric with the amount of d-galactonic acid. The NADH is measured by the increase in absorbance at 340 nm. The kit can also be used to determine the amount of l-arabinose released in a system through the same chemistry.
2.6. Model substrate assays
Kinetic data were obtained using 50 mM p-nitrophenyl-α-l-arabinofuranoside or 50 mM p-nitrophenyl-α-l-xylanopyranose. Reactions were performed in 20 mM sodium phosphate buffer pH 7.2. All assays were performed in triplicate and with a blank using purification buffer alone (20 mM Tris–HCl, 150 mM NaCl) without enzyme. The reactions were initiated by addition of enzyme and the absorbance was monitored at 1 min intervals at 405 nm for 30 min in a BMG FLUOstar Optima F plate reader at 21°C. The data were corrected for non-enzymatic decay of the substrate. Michaelis–Menten kinetics were modelled using the following formulae and were processed using GraphPad Prism 5 or Microsoft Excel:
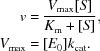 |
Enzyme concentration, pH, temperature, buffer and volume were fixed while the substrate concentration was varied. Rates were measured at a range of substrate concentrations and plotted using GraphPad Prism 5.
3. Results and discussion
3.1. Overall structure of Xsa43E
One monomer, consisting of one catalytic domain, is present in the asymmetric unit. The majority of the protein is visible in the electron density from residue 9 through to the terminal residue 313. The final structure also contains 319 water molecules, a molecule of tris(hydroxymethyl)aminomethane (Tris) and a calcium ion (Fig. 1 ▶). The presence of calcium in a solution of Xsa43E protein was confirmed using ICP-MS. The highest resolution (1.33 Å) data were obtained from a crystal of selenomethionine-derivatized protein; the final R factor and R free for this data set were 16.9% and 18.6%, respectively. Full data statistics are given in Table 1 ▶. The structure of Xsa43E has 96.4% of the residues within the most favoured region and 3.6% in allowed regions of the Ramachandran plot (structure validation using PROCHECK in CCP4; Laskowski et al., 1993 ▶).
Figure 1.

Cartoon representation of Xsa43E showing the five blades of the β-propeller, the Tris molecule and the calcium ion.
Xsa43E has one domain, with the overall structure consisting of a five-bladed β-propeller. This domain is common to all reported three-dimensional structures of GH43 enzymes. The first reported five-bladed β-propeller structure was that of tachylectin (Beisel et al., 1999 ▶). Since then this fold has been seen in four different GH families, GH families 32 and 68 of the GH-J clan and GH families 43 and 62 of the GH-F clan, as recorded in the CAZy database (Lombard et al., 2014 ▶; Cantarel et al., 2009 ▶). The five blades of the propeller form a cylindrical shape and are organized around a central axial cavity. This central cavity is approximately 30 Å long and contains the active site and a calcium ion. The calcium-ion position corresponds to a peak in the difference map of 56σ compared with an average water peak of 19.6σ. Each blade of the propeller consists of four antiparallel β-strands connected by loops of varying sizes. The loops between the first and second strand of each blade are short hairpins two to three residues in length; the loops between the third and fourth strands are also relatively short at between seven and 12 residues long. The loops between the second and third strands (six to 18 residues in length) and the fourth strand of one blade and the first strand of the next blade are longer (12–23 residues long), and they form the substrate-binding cavity and the active site. The inner strands of the five blades form the base of the tapered end of the cup-like propeller structure (Fig. 1 ▶). Intriguingly, the inner strands are all preceded by a proline residue. The four strands that make up each blade twist through approximately 90° from the inside to the outside of the structure. The N-terminal and C-terminal ends of the protein have short β-sheets that are at hydrogen-bonding distance and effectively seal the propeller structure. The C-terminal strand is the fourth strand of the fifth blade of the propeller and the N-terminal end acts almost as a fifth strand in that blade, in a ‘molecular Velcro’ fashion.
The active site accommodates a molecule of Tris, most likely from the purification buffer. The three catalytic residues previously observed in GH43 family structures (Brüx et al., 2006 ▶; Vandermarliere et al., 2009 ▶; Cartmell et al., 2011 ▶) were identified by superimposing the structure of the arabinanase Arb43A (PDB entry 1gyd; sequence identity = 26%; Nurizzo et al., 2002 ▶) from Cellvibrio japonicus with that of Xsa43E. Asp24 is the general base, Glu202 is the general acid and Asp141 is the third catalytic residue that is believed to modulate the pK a of the catalytic acid as well as orientating the catalytic acid and the substrate (Nurizzo et al., 2002 ▶). These residues are located on the innermost strands of blades 1, 4 and 3, respectively. As the catalytic base (Asp24) activates a water molecule, which attacks the anomeric C atom from one side, the catalytic acid (Glu202) donates a proton to the leaving group on the other side, breaking the glycosidic bond between the two sugar moieties and in the process inverting the configuration of the anomeric C atom. In GH43 enzymes the active site lies in a groove that is shown to accommodate multiple sugar moieties. The substrate-binding groove has defined subsites which accommodate these sugars. The subsites are labelled based on their proximity to the active site. Subsites −1 and +1 are the sites either side of the glycosidic bond being cleaved. In Xsa43E subsite −1 accommodates the arabinose side chain while subsite +1 accommodates the xylose sugar of the xylan backbone that the arabinose branches from. It is the glycosidic bond between these two sugar moieties that is hydrolysed (Brüx et al., 2006 ▶; Nurizzo et al., 2002 ▶; Pons et al., 2004 ▶; Vandermarliere et al., 2009 ▶).
Metal ions are common in protein structures and are often important for structural integrity and/or activity (Dokmanić et al., 2008 ▶). The structure of Xsa43E reveals a calcium ion in close proximity to the active site and within ligand-bonding distance of His258, located on the innermost strand of blade 5. It has previously been shown in other GH43 family enzymes with the same fold that this conserved histidine is important for catalysis (de Sanctis et al., 2010 ▶; Cartmell et al., 2011 ▶). We propose the mechanism by which this histidine is involved in catalysis is via an electron-withdrawing pathway between the divalent calcium ion and the catalytic acid Glu202, helping to modulate the pK a of that acidic residue. The N∊2 atom in the imidazole ring of His258 acts as a ligand for the central Ca2+ ion. His258 is hydrogen-bonded to a water molecule via the ND1 atom in the imidazole ring. This water molecule forms a second hydrogen bond to the OE2 atom of Glu202 as shown in Fig. 2 ▶. This electron-withdrawing pathway shows that the calcium ion and His258 are positioned in a way that would allow them to exert an effect on the pK a of Glu202. We have performed site-directed mutagenesis to help support this proposal, mutating the histidine to alanine. This effectively removes all activity from Xsa43E against the model substrate p-nitrophenyl-α-l-arabinofuranoside, whereas mutating the histidine to glutamine allows the enzyme to retain some activity (k cat = 0.20 s−1), although it is significantly less than the wild-type enzyme (k cat = 0.70 s−1) (Table 2 ▶). Modelling a glutamine in place of His258 positions the amide in a way that is analogous to the two N atoms of the histidine ring. In theory, this would allow the ‘electron-withdrawing pathway’ to be maintained.
Figure 2.
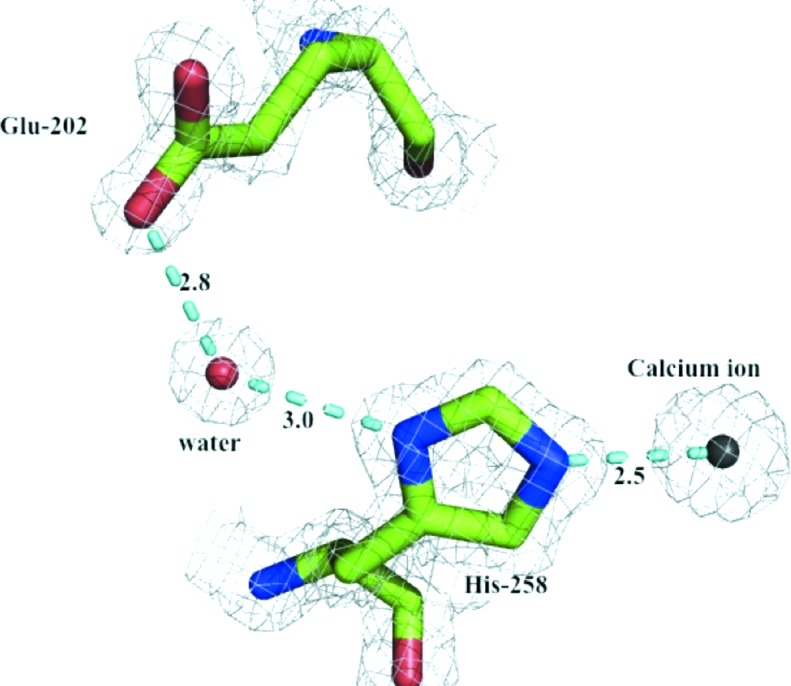
Proposed pathway by which the calcium ion and His258 can affect the environment of the catalytic acid Glu202. Electron density is shown as a 2|F o| − |F c| map contoured at 1.0σ.
Table 2. Kinetic values for activity of Xsa43E and mutants of Xsa43E with model substrate p-nitrophenyl-α-L-arabinofuranoside (PNPA) and p-nitrophenyl-α-L-xylopyranose (PNPX) at pH 7.2 and specific activity of Xsa43E with natural substrates.
(a).
Activity of Xsa43E and mutants with the model substrates PNPA and PNPX. ND, not determined.
| Substrate | Enzyme | K m (mM) | k cat (s−1) | k cat/K m (mM −1 s−1) |
|---|---|---|---|---|
| PNPX | Xsa43E | 8 ± 2 | 1.25 ± 0.10 | 0.15 ± 0.05 |
| PNPA | Xsa43E | 3.6 ± 0.5 | 0.70 ± 0.03 | 0.20 ± 0.04 |
| PNPA | Xsa43E D24A | ND | ND | ND |
| PNPA | Xsa43E D141A | ND | ND | ND |
| PNPA | Xsa43E E202A | ND | ND | ND |
| PNPA | Xsa43E H258A | ND | ND | ND |
| PNPA | Xsa43E H258Q | 16.6 ± 3.2 | 0.20 ± 0.02 | 0.012 ± 0.003 |
(b).
Specific activity of Xsa43E with natural substrates.
| Substrate | Enzyme | Specific activity (nmol min−1 mg−1) |
|---|---|---|
| Arabinobiose | Xsa43E | 2 ± 0.5 |
| Arabinoxylan | Xsa43E | 10 ± 4 |
| Arabinoxylan | Xsa43E with xylanase from T. viride | 25 ± 2 |
3.2. Calcium-ion environment
The calcium ion shows pentagonal bipyramidal seven-coordinate geometry (Fig. 3 ▶ a). It has five equatorial ligands, which are all water molecules. It is also coordinated to one water molecule in an axial position, while the other axial position is occupied by N∊2 of the His258 ring. All bonding distances for the calcium are between 2.4 and 2.5 Å.
Figure 3.

The calcium-ion environment (a) with the seven ligands in a pentagonal bipyramidal coordination and (b) with the five equatorial waters and their interactions with either a proline or the preceding residue from adjacent blades of the β-propeller. Electron density is shown as a 2|F o| − |F c| map contoured at 1.0σ.
The calcium environment corresponds with the symmetry of the overall structure. Each of the five equatorial water molecules are positioned midway between two of the blades of the propeller. This allows each water molecule to hydrogen-bond to two adjacent blades of the structure. Each water molecule hydrogen-bonds to either a proline residue or the preceding residue located at the inner loops of two consecutive blades (Fig. 3 ▶ b). These prolines are located where the loops terminate and the innermost β-strands of each blade start. They are also located near the catalytic residues in the sequence of Xsa43E. Pro25 is the proline at the end of the loop that terminates at the first β-strand of the first blade positioned next to Asp24, the general base. Pro88 is located at the end of the loop that runs from the final β-strand of blade 1 and terminates at the first β-strand of the second blade; it participates in hydrogen bonding to one of the equatorial waters. This is part of the highly conserved WAP region of GH43 domains, of which the tryptophan is thought to form hydrophobic stacking interactions with the substrate (Brüx et al., 2006 ▶; Vandermarliere et al., 2009 ▶). Pro142 is located at the end of the loop that runs from the last β-strand of the second blade and the innermost β-strand of the third blade, adjacent to Asp141, which is part of the previously reported catalytic triad. Pro204 and Pro259 are the remaining two prolines involved in coordinating the equatorial waters surrounding the central calcium ion. Pro204 is two residues away from the general acid Glu202 and is located in the loop that terminates at the innermost β-strand of the fourth blade, and Pro259 is next to His258, again located just prior to the end of the loop that runs into the innermost β-strand of the fifth blade of the propeller. In Xsa43E we propose that this may help to position the catalytic residues in the correct spatial configuration and generally assist in formation of the active-site pocket. Structural alignments of 14 different GH43 enzyme structures from the PDB show that although some of these proline residues are highly conserved, it is not common to see all five in the GH43 structures.
3.3. Substrate specificity of Xsa43E and mechanistic investigation
GH43 family enzymes listed in the CAZy database have a number of known activities, including β-xylosidase, β-1,3-xylosidase, α-l-arabinofuranosidase, arabinanase, xylanase and galactan 1,3-β-galactosidase activities (Lombard et al., 2014 ▶; Cantarel et al., 2009 ▶).
To characterize the function of Xsa43E, we have undertaken a range of enzymatic assays with both natural and synthetic model substrates. The specific activity of Xsa43E on natural substrates was determined using the Lactose/Galactose Rapid Assay Kit (Megazymes International Ltd). The results indicate that Xsa43E can release arabinose units from arabinobiose with a specific activity of 2 ± 0.5 nmol min−1 mg−1 but is unable to cleave arabinohexaose. It cleaves arabinose from arabinoxylan with a specific activity of 10 ± 4 nmol min−1 mg−1. This activity is greatly enhanced in the presence of a xylanase from T. viride to 25 ± 2 nmol min−1 mg−1 (Table 2 ▶).
Xsa43E shows no activity on xylose oligosaccharides with a degree of polymerization from 2 to 6 (i.e. xylobiose to xylohexaose). It also shows no activity towards the azo-wheat arabinoxylan substrate (Megazymes). Xsa43E can cleave the glycosidic bond in p-nitrophenyl-α-l-arabinofuranoside and p-nitrophenyl-α-l-xylopyranose, but cannot cleave p-nitrophenyl-α-d-xylopyranose. The kinetic parameters for Xsa43E with these two model substrates are given in Table 2 ▶. Structural analysis of the substrate-binding cleft and the positioning of the catalytic residues indicate that Xsa43E would cleave arabinose side chains from the xylose backbone of arabinoxylan. The experimental results support this hypothesis and show that Xsa43E works more efficiently on short xylose segments, as indicated by its increased activity on arabinoxylan with the addition of a xylanase. Xsa43E is a nonsecreted enzyme, and it is known that B. proteoclasticus can only transport short oligosaccharides into the cell (Kelly et al., 2010 ▶). This is also consistent with Xsa43E acting on short arabinoxylan substrates.
4. Conclusions
Members of the GH43 family of enzymes have an inverting mechanism; the configuration of the anomeric C atom of the sugar substrate is inverted as opposed to being retained. This is achieved by having the catalytic acid and the catalytic base on opposite sides of the substrate in the binding pocket of the enzyme. As the catalytic base activates a water molecule which attacks the anomeric C atom from one side, a catalytic acid donates a proton to the leaving group on the other side, breaking the glycosidic bond between the two sugar moieties and in the process inverting the configuration of the anomeric C atom. GH43 enzymes have three residues implicated in catalysis: a general acid, a general base and a third acidic residue proposed to modulate the pK a of the general acid as well as orientate the substrate and general acid. The central calcium ion and a histidine residue in a GH43 family enzyme have also been suggested to be important for activity. de Sanctis and coworkers have postulated that this histidine may be the true pK a modulator in GH43 family enzymes (de Sanctis et al., 2010 ▶). We have used site-directed mutagenesis to mutate each active-site residue to alanine, including His258. In addition, we have also mutated His258 to glutamine. The resulting enzymes all fail to show detectable levels of activity with model substrates (Table 2 ▶) with the exception of H258Q, which showed modest activity (although the k cat was only 29% of the wild-type activity). These experiments lend support to the proposed mechanism and indicate that His258 is important in catalysis.
This characterization of Xsa43E shows it to be an arabinofuranosidase capable of cleaving arabinose side chains from short segments of xylan. GH43 enzymes all have a five-bladed β-propeller structure but can have a range of functions. The structural and functional characterization of Xsa43E presented here supports its inclusion in the GH43 family of enzymes.
B. proteoclasticus survives in a very crowded microbial environment; it has a large suite of xylan-degrading enzymes that allow it to harvest the complex polysaccharide xylan as a food source. Through understanding how these enzymes facilitate the depolymerization of xylan, we can develop the potential to exploit these systems for industrial processes.
Supplementary Material
PDB reference: Xsa43E, 4nov
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Attwood, G. T., Reilly, K. & Patel, B. K. (1996). Int. J. Syst. Bacteriol. 46, 753–758. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Beisel, H. G., Kawabata, S., Iwanaga, S., Huber, R. & Bode, W. (1999). EMBO J. 18, 2313–2322. [DOI] [PMC free article] [PubMed]
- Brüx, C., Ben-David, A., Shallom-Shezifi, D., Leon, M., Niefind, K., Shoham, G., Shoham, Y. & Schomburg, D. (2006). J. Mol. Biol. 359, 97–109. [DOI] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res. 37, D233–D238. [DOI] [PMC free article] [PubMed]
- Cartmell, A., McKee, L. S., Peña, M. J., Larsbrink, J., Brumer, H., Kaneko, S., Ichinose, H., Lewis, R. J., Viksø-Nielsen, A., Gilbert, H. J. & Marles-Wright, J. (2011). J. Biol. Chem. 286, 15483–15495. [DOI] [PMC free article] [PubMed]
- Dokmanić, I., Šikić, M. & Tomić, S. (2008). Acta Cryst. D64, 257–263. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Kelly, W. J., Leahy, S. C., Altermann, E., Yeoman, C. J., Dunne, J. C., Kong, Z., Pacheco, D. M., Li, D., Noel, S. J., Moon, C. D., Cookson, A. L. & Attwood, G. T. (2010). PLoS One, 5, e11942. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. (2014). Nucleic Acids Res. 42, D490–D495. [DOI] [PMC free article] [PubMed]
- Moon, C. D., Pacheco, D. M., Kelly, W. J., Leahy, S. C., Li, D., Kopecny, J. & Attwood, G. T. (2008). Int. J. Syst. Evol. Micriobiol. 58, 2041–2045. [DOI] [PubMed]
- Moreland, N., Ashton, R., Baker, H. M., Ivanovic, I., Patterson, S., Arcus, V. L., Baker, E. N. & Lott, J. S. (2005). Acta Cryst. D61, 1378–1385. [DOI] [PubMed]
- Nurizzo, D., Turkenburg, J. P., Charnock, S. J., Roberts, S. M., Dodson, E. J., McKie, V. A., Taylor, E. J., Gilbert, H. J. & Davies, G. J. (2002). Nature Struct. Biol. 9, 665–668. [DOI] [PubMed]
- Pons, T., Naumoff, D. G., Martinez-Fleited, C. & Hernandez, L. (2004). Proteins, 54, 424–432. [DOI] [PubMed]
- Sanctis, D. de, Inácio, J. M., Lindley, P. F., de Sá-Nogueira, I. & Bento, I. (2010). FEBS J. 277, 4562–4574. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Methods Enzymol. 374, 22–37. [DOI] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L.-W., Read, R. J. & Adams, P. D. (2008). Acta Cryst. D64, 61–69. [DOI] [PMC free article] [PubMed]
- Vandermarliere, E., Bourgois, T. M., Winn, M. D., van Campenhout, S., Volckaert, G., Delcour, J. A., Strelkov, S. V., Rabijns, A. & Courtin, C. M. (2009). Biochem. J. 418, 39–47. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Xsa43E, 4nov