

The expression, purification and crystallization of acylpeptide hydrolase from D. radiodurans (GenBank accession No. NP_293889) and its preliminary X-ray diffraction analysis to 2.6 Å resolution are reported.
Keywords: acylpeptide hydrolase, Deinococcus radiodurans, S9 serine peptidase
Abstract
Acylpeptide hydrolase (APH; EC 3.4.19.1), which belongs to the S9 family of serine peptidases (MEROPS), catalyzes the removal of an N-acylated amino acid from a blocked peptide. The role of this enzyme in mammalian cells has been suggested to be in the clearance of oxidatively damaged proteins as well as in the degradation of the β-amyloid peptides implicated in Alzheimer’s disease. Detailed structural information for the enzyme has been reported from two thermophilic archaea; both of the archaeal APHs share a similar monomeric structure. However, the mechanisms of substrate selectivity and active-site accessibility are totally different and are determined by inter-domain flexibility or the oligomeric structure. An APH homologue from a bacterium, Deinococcus radiodurans (APHdr), has been crystallized using microbatch-under-oil employing the random microseed matrix screening method. The protein crystallized in space group P21, with unit-cell parameters a = 77.6, b = 189.6, c = 120.4 Å, β = 108.4°. A Matthews coefficient of 2.89 Å3 Da−1 corresponds to four monomers, each with a molecular mass of ∼73 kDa, in the asymmetric unit. The APHdr structure will reveal the mechanisms of substrate selectivity and active-site accessibility in the bacterial enzyme. It will also be helpful in elucidating the functional role of this enzyme in D. radiodurans.
1. Introduction
Acylpeptide hydrolase (APH; EC 3.4.19.1), also referred to as acylaminoacyl peptidase, is a member of the S9 family of serine proteases that use a unique Ser-Asp-His catalytic triad for the hydrolysis of peptides (MEROPS database; Rawlings et al., 2014 ▶). It catalyzes the removal of an N-acylated amino-acid residue from a blocked peptide, producing an acylamino acid and a peptide with a free amino-terminus. It cleaves a variety of peptides with different N-terminal acyl groups such as acetyl, chloroacetyl, formyl and carbamyl, including a bioactive peptide (α-melanocyte-stimulating hormone; Jones et al., 1986 ▶). In addition to exopeptidase activity, APH homologues from mammals and archaea have also been shown to possess low levels of endopeptidase activity (Fujino et al., 2000 ▶; Kiss et al., 2007 ▶). Indirect evidence suggests that APH enzymes play important roles in the cellular physiology of mammals. For example, in human erythrocytes this enzyme preferentially cleaves oxidatively damaged and glycated proteins (Fujino et al., 2000 ▶). Moreover, the enzyme also degrades β-amyloid peptide, a pathogenic fragment associated with Alzheimer’s disease (Yamin et al., 2009 ▶). A truncated form of the APH homologue in mice (55 kDa) cleaves the lens β2-crystallin protein, resulting in the accumulation of low-molecular-weight peptides that promote cataract formation (Santhoshkumar et al., 2014 ▶).
APH homologues are ubiquitous in distribution and have been identified in genomes from all three domains of life: bacteria, archaea and eukaryotes. Biochemical characterizations of APH enzymes have been reported from thermophilic archaea, mammals and plants (Abraham & Nagle, 2013 ▶), while crystal structures have been reported from two genera of archaea (Bartlam et al., 2004 ▶; Menyhárd et al., 2013 ▶). APH and other proteins of the S9 family share a similar monomeric structure consisting of two domains: an N-terminal seven-bladed or eight-bladed β-propeller and a catalytic α/β-hydrolase domain. However, the proteins have evolved different mechanisms to shield the active site to avoid the degradation of nontargeted cellular proteins. The mechanisms for substrate selectivity and access to the active sites vary across the archaeal APHs and the other S9 family enzymes (Fülöp et al., 2000 ▶; Aertgeerts et al., 2004 ▶; Shan et al., 2005 ▶; Harmat et al., 2011 ▶; Menyhárd et al., 2013 ▶).
Deinococcus spp. are extremely radiation-resistant. One of the mechanisms for this exceptional resistance is the protection of the proteome against the oxidative damage caused by ionizing radiation (Krisko & Radman, 2013 ▶). A possible role of APH homologues in eliminating oxidized proteins has been suggested in mammalian cells (COS-7) under oxidative stress (Shimizu et al., 2003 ▶). Thus, from this viewpoint, it is important to study this enzyme from D. radiodurans. Here, we report the crystallization and preliminary X-ray diffraction studies of the APH homologue from the bacterium D. radiodurans (APHdr; GenBank accession No. NP_293889). The structural studies will be useful in elucidating the mechanisms of substrate-size selectivity and active-site accessibility in the bacterial APH enzyme and will also be helpful in understanding the functional role of this enzyme in D. radiodurans.
2. Materials and methods
2.1. Macromolecule production
The coding DNA sequence (CDS) of a gene encoding the APHdr protein (GenBank accession No. NP_293889) was PCR-amplified from the genomic DNA of D. radiodurans R1 (NCBI taxonomic ID 243230). The oligos flanking the CDS, opx175 (CGTGGATCCAACAATTCGGAAACCCCGGCC) and opx176 (CGTTGTACATTACAGCCAGCGTTCCAGCCAGG), containing BamHI and BsrGI restriction-enzyme sites, respectively, were used to PCR-amplify the CDS using Pfu DNA polymerase (Table 1 ▶). The CDS was cloned into pST50Tr, a T7-promoter-based expression plasmid (Tan et al., 2005 ▶), so as to form an in-frame translational fusion of APHdr with a Strep-tag II–hexahistidine–Tobacco etch virus protease site tag (STRHISTEV; 3.8 kDa). The cloning and expression hosts used in the present studies were Escherichia coli strains XL1 Blue and BL21(DE3)pLysS, respectively. The identity and integrity of the construct was verified by restriction-digestion analysis and DNA sequencing using flanking T7 and T7-terminator oligos. The expressing clone was grown in 5 l 2×TY broth at 310 K until the OD600 reached 0.4 and was then shifted to 301 K for further growth. The culture was then induced at an OD600 of ∼0.8 for 18 h by the addition of 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). The cells were harvested by centrifugation at ambient temperature and the pellet was immediately suspended in P300 buffer (50 mM sodium phosphate pH 7.0, 300 mM NaCl). The cells were disrupted by sonication. The soluble fraction of the cell lysate containing the desired protein (STRHISTEV-APHdr) was subjected to metal-ion affinity chromatography by the batch method at 277 K. The lysate was mixed with ∼10 ml of equilibrated Ni Sepharose resin (GE Healthcare). The bound proteins on the resin were eluted with P300 buffer containing 500 mM imidazole. The eluted protein (STRHISTEV-APHdr) was subjected to digestion by Tobacco etch virus protease (TEV; 1:50 molar ratio) at 293 K while dialyzing against T200 buffer (20 mM Tris–HCl pH 8.0, 200 mM NaCl) for 48 h. The desired protein (APHdr) was separated from the STRHISTEV peptide tag and undigested protein using a 5 ml HisTrap column on an ÄKTA system (GE Healthcare) at ambient temperature. The purified protein was concentrated to greater than 16 mg ml−1 using a 30 kDa cutoff ultrafiltration device (Sartorius). Glycerol was added to the protein solution to 20%(v/v) and it was then aliquoted and stored at 203 K until further use. Size-exclusion chromatography was performed with T200 buffer on a Superdex 200 10/300 GL column using an ÄKTA purifier system (GE Healthcare).
Table 1. Macromolecule-production information.
| Source organism | D. radiodurans R1 |
| DNA source | Bacterial cells |
| Forward primer | CGTGGATCCAACAATTCGGAAACCCCGGCC |
| Reverse primer | CGTTGTACATTACAGCCAGCGTTCCAGCCAGG |
| Cloning vector | None |
| Expression vector | pST50Tr |
| Expression host | E. coli BL21(DE3)pLysS |
| Complete amino-acid sequence of the construct produced | MASWSHPQFEKGSSHHHHHHSSGSGGGGGENLYFQGSNNSETPAPGPDSLLALAFPSDPQVSPDGKQVAFVLAQISEEDPAKPDKDFARPRYRSGLWLSEGGAARPLTHAETGRGDSAPRWSPDGQNLAFVRSAGEVKAALMLLPLKGGEARRVTHFKNGVSGPQWSPDGRFIAFTTTADTEDKRDERGEARVLTRPVYRANGADWLPERPAALWLYDVEADKLREWYAPEIGIGALSWWPDSRGVLIVQSEDEWQASQWRQDVYDLPLPTADAPAAPQKLLDWNSAAHGLAPHPDGQRFALIGRPAGKGNTEHAHLYLIENGQHRRLDTGHDHPVGDAVGGDCHVGAFPEGPRWLDGDTLLFSSTVRGSVGLFTAHIGGGVKAYDHDPQGVISAFTANEHGVALIRESATRFPEVELNGQRVTDLHARFPFPVREPQRVTFETELGEGEGWVLLPEGEQKVPALLNIHGGPHTDYGHGFTHEFQLMAARGYGVCYSNPRGSVGYGQAWVDAIYGRWGTVDADDLLNFFDRCLEAVPRLDAAKTAVMGGSYGGFMTNWITGHTTRFQAAITDRCISNLISFGGTSDIGLRFWDDELGLDFSRRADALKLWDLSPLQYVENVKTPTLIVHSVLDHRCPVEQAEQWYAALHKHQVPVRFVRFPEENHELSRSGRPDRRLTRLNEYFAWLERWL |
2.2. Crystallization
For the crystallization experiment, an aliquot of stored protein was subjected to dialysis using T200 buffer. The equilibrated protein was concentrated to ∼15 mg ml−1 for setting up crystallization trials. Initial crystallization trials were set up employing the commercial crystallization screens Index (Hampton Research) and JCSG-plus (Molecular Dimensions). Crystallization was carried out at 293 K employing the microbatch-under-oil method (Chayen et al., 1992 ▶), in which 1 µl protein solution was mixed with 1 µl crystallization solution and overlaid with 50 µl Al’s oil [1:1(v:v) silicone oil:mineral oil] in 96-well U-bottom plates. A single crystal hit was observed in one of the JCSG-plus crystallization conditions [0.1 M bis-tris pH 5.5, 0.2 M ammonium sulfate, 25%(w/v) polyethylene glycol (PEG) 3350]. The thin plate-like crystals were of poor quality and the crystallization condition was not further optimized. Instead, we used these crystals for microseeding employing the random microseed matrix screening method (D’Arcy et al., 2007 ▶). In this method, nuclei formed using one crystallization condition are transferred to other random crystallization conditions to screen for a condition that supports crystal growth. The microseeds were introduced into drops (pre-equilibrated for 5 d) of a factorial screen by streak-seeding. The factorial screen was prepared in-house using combinations of buffers (of different pHs), monovalent and divalent ions and different PEG 3350 concentrations. The microseeding yielded crystals of different sizes and shapes in seven different crystallization conditions from our factorial screen, three of which produced crystals that were suitable for diffraction studies. The crystals exhibiting the best diffraction resolution were grown from the crystallization condition 50 mM sodium acetate pH 4.5, 200 mM NaCl, 10 mM magnesium chloride, 20%(w/v) PEG 3350 (Table 2 ▶). The crystals were cryoprotected using Parabar 10312 oil (Hampton Research). They were picked out from the microbatch drops with a cryoloop, rinsed for 1–2 min in Parabar 10312 oil to remove excess mother liquor and then flash-cooled in liquid nitrogen.
Table 2. Crystallization.
| Method | Microbatch-under-oil |
| Plate type | 96-well U-bottom plate |
| Temperature (K) | 293 |
| Protein concentration (mg ml−1) | 15 |
| Buffer composition of protein solution | 20 mM Tris–HCl pH 8.0, 200 mM NaCl |
| Composition of reservoir solution | 50 mM sodium acetate pH 4.5, 200 mM NaCl, 10 mM magnesium chloride, 20% PEG 3350 |
| Volume and ratio of drop | 2 µl (1:1) |
| Volume of reservoir | Nil (microbatch) |
2.3. Data collection and processing
The single-crystal diffraction experiments on protein crystals were carried out on the recently commissioned protein crystallography (PX-BL21) beamline at the 2.5 GeV Indus-2 synchrotron, India. The diffraction images were collected from a cryocooled crystal (100 K) using 1° oscillation at a wavelength of 0.97947 Å on a MAR225 CCD (Rayonix) detector. The crystal diffracted to 2.6 Å resolution. The data were indexed and integrated using XDS (Kabsch, 2010 ▶) and subsequently scaled using AIMLESS. The intensity data statistics are summarized in Table 3 ▶. Self-rotation function calculations were performed using POLARRFN. A molecular-replacement solution was obtained using Phaser (McCoy et al., 2007 ▶). AIMLESS, POLARRFN and Phaser were used as part of the CCP4 package (Winn et al., 2011 ▶).
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Beamline | PX-BL21, Indus-2, India |
| Wavelength (Å) | 0.97947 |
| Resolution (Å) | 47.96–2.60 (2.64–2.60) |
| Temperature (K) | 100 |
| Space group | P1211 |
| Unit-cell parameters (Å, °) | a = 77.6, b = 189.6, c = 120.4, α = γ = 90, β = 108.4 |
| Total No. of reflections | 240282 (11801) |
| No. of unique reflections | 96084 (4810) |
| Multiplicity | 2.5 (2.5) |
| R meas † (%) | 12.2 (60.4) |
| 〈I/σ(I)〉 | 10.2 (2.6) |
| Completeness (%) | 95.1 (96.8) |
| Matthews coefficient (Å3 Da−1) | 2.89 |
| Solvent content (%) | 58 |
| No. of monomers in the asymmetric unit | 4 |
| Overall B factor from Wilson plot (Å2) | 27.42 |
R
meas = 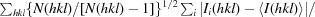
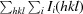 .
.
3. Results and discussion
The typical yield of the purified protein (APHdr) was found to be ∼60 mg per litre of bacterial culture. The purified protein showed a single symmetric peak on Superdex 200 size-exclusion chromatography corresponding to a molecular mass of 265 kDa (Fig. 1 ▶). This observed molecular mass matches reasonably well with the molecular mass calculated for an APHdr tetramer (291 kDa) from the protein sequence. It thus confirms the existence of APHdr in a tetrameric form in the given conditions. The purified APHdr enzyme was found to be active towards N-CBZ-l-Ala-p-nitrophenyl ester, with an enzymatic efficiency (k cat/K m) of about 132 mM −1 s−1 (unpublished results). Initial crystallization screening produced thin plate-like crystals with inferior morphology in the JCSG-plus crystal screen. When these crystals were used as a source of microseeds in 96 different conditions of a factorial screen produced in-house using the random microseed matrix screening method, crystals were yielded in seven different crystallization conditions. The crystals which gave the best diffraction resolution were grown from the crystallization condition 50 mM sodium acetate pH 4.5, 200 mM NaCl, 10 mM magnesium chloride, 20% PEG 3350 (Fig. 2 ▶). Data were collected to a resolution of 2.6 Å at 100 K. The unit-cell parameters were a = 77.6, b = 189.6, c = 120.4 Å, β = 108.4° and the crystals belonged to space group P21. The expected molecular mass of monomeric APHdr deduced from its sequence is 72.64 kDa. The Matthews coefficients (V M; Matthews, 1968 ▶) calculated from the unit-cell parameters were 2.89 and 2.31 Å3 Da−1 for four and five monomers in the asymmetric unit, respectively. The ambiguity of four or five monomers in the asymmetric unit was resolved using a self-rotation function and the size-exclusion chromatography results. A self-rotation function calculated using POLARRFN showed the presence of twofold noncrystallographic symmetry (NCS) axes in the ac plane perpendicular to the crystallographic b axis (Fig. 3 ▶). The twofold NCS axes are most probably owing to the presence of four monomers rather than five monomers in the asymmetric unit, which is consistent with the tetrameric nature of the protein as deduced from size-exclusion chromatography. The closest sequence homologue of APHdr in the Protein Data Bank (PDB) is acylaminoacyl peptidase from Pyrococcus horikoshii (APHph; PDB entry 4hxg; Menyhárd et al., 2013 ▶), with which it shares ∼29% sequence identity. Molecular replacement (MR) in Phaser using a monomer of APHph as a search model gave a unique solution with a final LLG score of 804. The MR solution consists of four monomers in the asymmetric unit of space group P21. This is in complete agreement with the gel-filtration and self-rotation function analysis. Initial refinement of the structure solution using PHENIX (Adams et al., 2010 ▶) resulted in an R work and R free of 31 and 36%, respectively. Further structure refinement and model building are in progress. Structural analysis of APHdr will reveal the mechanism of substrate selectivity, size exclusion and access to the active site in the tetrameric form of APH enzymes. Additionally, structure–function information on APHdr will shed light on its possible role in the degradation of oxidatively damaged proteins in D. radiodurans, which is known to undergo severe oxidative stress on exposure to ionizing radiation.
Figure 1.

Gel-filtration profile of APHdr (marked by an arrow) using a Superdex 200 column with 20 mM Tris–HCl pH 8.0, 200 mM NaCl. The column was calibrated using proteins of standard molecular mass (Sigma; catalogue No. MWGF200-1KT). The observed molecular mass of APHdr is 265 kDa (marked by an arrow in the upper inset). A 15% SDS–PAGE of purified APHdr is shown in the lower inset; lane 1 contains standard molecular-mass markers (labelled in kDa) and lane 2 contains APHdr protein.
Figure 2.

An APHdr protein crystal grown from the crystallization condition 50 mM sodium acetate pH 4.5, 200 mM NaCl, 10 mM magnesium chloride, 20%(w/v) PEG 3350.
Figure 3.

Self-rotation function (κ = 180° section) calculated using POLARRFN in CCP4 (Winn et al., 2011 ▶) in the resolution range 20–3.0 Å with an integration radius of 20 Å; the contour starts at 30% with an interval of 5%. The ω angle varies in the radial direction and the ϕ angle varies along the circumference. Two peaks (marked by arrows) at ω = 18.5°, ϕ = 180° and ω = 71.4°, ϕ = 0°, each with 71% of the magnitude of the origin peak, represent twofold noncrystallographic symmetry (NCS) axes.
Acknowledgments
We thank Dr S. M. Sharma for his constant support and for leading the development and commissioning of the protein crystallography beamline at the Indus-2 synchrotron, RRCAT, India. We sincerely thank Drs P. D. Gupta, S. K. Deb and G. S. Lodha for providing the necessary support and infrastructure at Raja Ramanna Centre for Advanced Technology, Indore to carry out this research. We thank the staff at Indus-2 for providing the synchrotron beam for the present work. We are highly grateful to S. K. Bhattacharjee for his support during the work.
References
- Abraham, C. R. & Nagle, M. W. (2013). Handbook of Proteolytic Enzymes, edited by N. D. Rawlings & G. Salvesen, pp. 3401–3403. New York: Academic Press.
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aertgeerts, K., Ye, S., Tennant, M. G., Kraus, M. L., Rogers, J., Sang, B.-C., Skene, R. J., Webb, D. R. & Prasad, G. S. (2004). Protein Sci. 13, 412–421. [DOI] [PMC free article] [PubMed]
- Bartlam, M., Wang, G., Yang, H., Gao, R., Zhao, X., Xie, G., Cao, S., Feng, Y. & Rao, Z. (2004). Structure, 12, 1481–1488. [DOI] [PubMed]
- Chayen, N. E., Shaw Stewart, P. D. & Blow, D. M. (1992). J. Cryst. Growth, 122, 176–180.
- D’Arcy, A., Villard, F. & Marsh, M. (2007). Acta Cryst. D63, 550–554. [DOI] [PubMed]
- Fujino, T., Watanabe, K., Beppu, M., Kikugawa, K. & Yasuda, H. (2000). Biochim. Biophys. Acta, 1478, 102–112. [DOI] [PubMed]
- Fülöp, V., Szeltner, Z. & Polgár, L. (2000). EMBO Rep. 1, 277–281. [DOI] [PMC free article] [PubMed]
- Harmat, V., Domokos, K., Menyhárd, D. K., Palló, A., Szeltner, Z., Szamosi, I., Beke-Somfai, T., Náray-Szabó, G. & Polgár, L. (2011). J. Biol. Chem. 286, 1987–1998. [DOI] [PMC free article] [PubMed]
- Jones, W. M., Manning, L. R. & Manning, J. M. (1986). Biochem. Biophys. Res. Commun. 139, 244–250. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kiss, A. L., Hornung, B., Rádi, K., Gengeliczki, Z., Sztáray, B., Juhász, T., Szeltner, Z., Harmat, V. & Polgár, L. (2007). J. Mol. Biol. 368, 509–520. [DOI] [PubMed]
- Krisko, A. & Radman, M. (2013). Cold Spring Harb. Perspect. Biol. 5, a012765. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Menyhárd, D. K., Kiss-Szemán, A., Tichy-Rács, É., Hornung, B., Rádi, K., Szeltner, Z., Domokos, K., Szamosi, I., Náray-Szabó, G., Polgár, L. & Harmat, V. (2013). J. Biol. Chem. 288, 17884–17894. [DOI] [PMC free article] [PubMed]
- Rawlings, N. D., Waller, M., Barrett, A. J. & Bateman, A. (2014). Nucleic Acids Res. 42, D503–D509. [DOI] [PMC free article] [PubMed]
- Santhoshkumar, P., Xie, L., Raju, M., Reneker, L. & Sharma, K. K. (2014). J. Biol. Chem. 289, 9039–9052. [DOI] [PMC free article] [PubMed]
- Shan, L., Mathews, I. I. & Khosla, C. (2005). Proc. Natl Acad. Sci. USA, 102, 3599–3604. [DOI] [PMC free article] [PubMed]
- Shimizu, K., Fujino, T., Ando, K., Hayakawa, M., Yasuda, H. & Kikugawa, K. (2003). Biochem. Biophys. Res. Commun. 304, 766–771. [DOI] [PubMed]
- Tan, S., Kern, R. C. & Selleck, W. (2005). Protein Expr. Purif. 40, 385–395. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yamin, R., Zhao, C., O’Connor, P. B., McKee, A. C. & Abraham, C. R. (2009). Mol. Neurodegener. 4, 33. [DOI] [PMC free article] [PubMed]