

Comparison of mutant and wild-type hammerhead ribozyme structures reveals active-site structural perturbations consistent with either a general base catalytic mechanism, as previously assumed to be operative in the hammerhead ribozyme, or a specific base catalytic mechanism, which may possess additional explanatory power.
Keywords: hammerhead ribozyme, acid–base catalysis, RNA cleavage
Abstract
The hammerhead ribozyme catalyzes RNA cleavage via acid–base catalysis. Whether it does so by general acid–base catalysis, in which the RNA itself donates and abstracts protons in the transition state, as is typically assumed, or by specific acid–base catalysis, in which the RNA plays a structural role and proton transfer is mediated by active-site water molecules, is unknown. Previous biochemical and crystallographic experiments implicate an invariant purine in the active site, G12, as the general base. However, G12 may play a structural role consistent with specific base catalysis. To better understand the role of G12 in the mechanism of hammerhead catalysis, a 2.2 Å resolution crystal structure of a hammerhead ribozyme from Schistosoma mansoni with a purine substituted for G12 in the active site of the ribozyme was obtained. Comparison of this structure (PDB entry 3zd4), in which A12 is substituted for G, with three previously determined structures that now serve as important experimental controls, allows the identification of structural perturbations that are owing to the purine substitution itself. Kinetic measurements for G12 purine-substituted schistosomal hammerheads confirm a previously observed dependence of rate on the pK a of the substituted purine; in both cases inosine, which is similar to G in pK a and hydrogen-bonding properties, is unexpectedly inactive. Structural comparisons indicate that this may primarily be owing to the lack of the exocyclic 2-amino group in the G12A and G12I substitutions and its structural effect upon both the nucleotide base and phosphate of A9. The latter involves the perturbation of a previously identified and well characterized metal ion-binding site known to be catalytically important in both minimal and full-length hammerhead ribozyme sequences. The results permit it to be suggested that G12 plays an important role in stabilizing the active-site structure. This result, although not inconsistent with the potential role of G12 as a general base, indicates that an alternative hammerhead cleavage mechanism involving specific base catalysis may instead explain the observed rate dependence upon purine substitutions at G12. The crystallographic results, contrary to previous assumptions, therefore cannot be interpreted to favor the general base catalysis mecahnism over the specific base catalysis mechanism. Instead, both of these mutually exclusive mechanistic alternatives must be considered in light of the current structural and biochemical data.
1. Introduction
The hammerhead ribozyme (Prody et al., 1986 ▶; Forster & Symons, 1987 ▶; Uhlenbeck, 1987 ▶), like other nucleolytic RNA enzymes, whether these enzymes are comprised of amino acids or of ribonucleotides, catalyzes RNA cleavage via acid–base catalysis. The full-length hammerhead ribozyme structure, which appears to be the active conformation of the ribozyme, has an invariant nucleotide in the core catalytic sequence, G12, within hydrogen-bonding distance of the nucleophile of the self-cleavage reaction (the 2′OH of C17; Martick & Scott, 2006 ▶).
G12 is hypothesized to be the general base in the hammerhead self-cleavage reaction. When deprotonated at N1, G12 is thought to initiate the cleavage reaction by abstracting a proton from the 2′OH of the cleavage-site nucleotide C17. Proton transfer is thus thought to restore G12 to the proton-bound form. Two sets of experimental results are typically invoked in support of this general base mechanism. The first is the proximity of G12 to the nucleophile in the full-length hammerhead structures. Specifically, N1 of G12 is positioned within hydrogen-bonding distance (∼3 Å) of the 2′O of C17, and thus when ionized is ideally positioned to initiate the cleavage reaction by abstracting the 2′H+ from C17. The second is the observed rate dependence upon the pK a values of G12-substituted purines. Although G12 is an invariant nucleotide in the natural hammerhead sequence, synthetic constructs with purine substitutions at position 12 show a general trend of decreasing catalytic prowess, with a decrease in the pK a of the substituted purine corresponding to protonation or deprotonation at position N1 (Han & Burke, 2005 ▶). Because N1 of G12 is positioned within hydrogen-bonding distance of the 2′O nucleophile of C17, the pK a trend of the G12 purine substitutions is considered in this context to be strong (but circumstantial) evidence in support of the hypothesis that a deprotonated G12 functions as the general base in hammerhead ribozyme catalysis (Martick & Scott, 2006 ▶), in the same manner that a deprotonated His12 is thought to function in general base catalysis in the analogous RNase A reaction (Cuchillo et al., 2011 ▶).
However, substitution of G12 with other purine-nucleotide analogs with different functionality may, at least potentially, also attenuate the reaction rate by perturbing the structure of the hammerhead ribozyme, so in principle some or all of the observed reduction in the rate of catalysis may therefore simply be owing to structural changes. These two effects will nonetheless give rise to the same rate law; the principle of kinetic equivalence entails that we cannot even distinguish between the extremes of a mechanism of pure general base catalysis, in which G12 functions as a Brønsted base, and a mechanism of specific base catalysis, in which the role of G12 is purely structural and water (or hydroxide ion) functions as a Brønsted base, on the basis of rate measurements alone (Fedor, 2009 ▶).
To better understand the contributions of structural perturbations that may potentially disrupt catalysis, we have obtained the 2.2 Å resolution crystal structure of a G12A substitution in a previously well characterized full-length Schistosoma mansoni (Smα1) hammerhead ribozyme crystal form. The new G12A hammerhead structure (PDB entry 3zd4) is compared with three previously determined crystal structures that now serve as crucial experimental controls for the analysis presented here. The new crystal structure and the roles of the three structural controls are summarized in Table 1 ▶.
Table 1. Hammerhead crystal structures.
The first of these controls is the corresponding wild-type sequence and structure (PDB entry 3zp8) with G12 at the active site. The crystal form (space group and unit cell) and crystallization conditions are otherwise identical to this 1.55 Å resolution structure (Anderson et al., 2013 ▶).
The second structural control is the structure of the same unsubstituted S. mansoni hammerhead sequence crystallized under rather different experimental conditions (a high concentration of ammonium sulfate in which bound monovalent and divalent metal ions are not observed). This structure was originally obtained in 2006 (Martick & Scott, 2006 ▶), but has been re-refined using the same, more modern protocols used to refine the newer structures and has been updated in the PDB (PDB entry 3zd5). The re-refinement eliminates the possibility that the observed structural differences may be owing to differing refinement protocols and also permitted verification of the earlier observation that no metal ions are observed to be specifically bound to the ribozyme.
The third structural control (PDB entry 2qus) is the structure of a hammerhead ribozyme sequence from satellite Tobacco ringspot virus (sTRSV), which possesses a distinctly different tertiary contact, in addition to many sequence differences in the nonconserved regions of the ribozyme. The crystallization conditions, sequence, crystal form (including the P1 space group) and tertiary contact are all completely different from the other structures. This ribozyme is also an active G12A-substituted hammerhead in which the 2′OH has not been substituted with a methyl group.
Our results, and comparison to the three structural controls listed in Table 1 ▶, permit us to estimate the contribution of a structural perturbation observed in the G12A-substituted hammerhead owing to the lack of the exocyclic 2-amino group, and thus the relative contributions of the ΔpK a and structural changes to the reaction rate.
2. Experimental methods
2.1. Crystallography
Full-length G12A hammerhead ribozyme crystals were obtained via vapor diffusion as described previously (Martick & Scott, 2006 ▶; Martick et al., 2008 ▶) except that the crystallization conditions were modified as follows. The reservoir consisted of 1.7 M sodium malonate buffered to pH 7.5, 1 mM MgCl2. The hanging drop contained a 1/2 concentration of the reservoir solution mixed with the RNA solution prepared as described previously (Martick & Scott, 2006 ▶). Crystals were stabilized in a mother liquor consisting of 1.7 M sodium malonate pH 7.5, 10 mM MgCl2 and flash-cooled using the sodium malonate as a cryoprotectant. The data collection is summarized in Table 2 ▶. The data were processed using iMosflm (Battye et al., 2011 ▶) and CCP4 (Winn et al., 2011 ▶) and were refined using PHENIX (Afonine et al., 2012 ▶), beginning with rigid-body refinement using 3zp8 after substituting A12 for G12 in the model. This was followed by simulated-annealing and TLS refinement using the default phenix.refine protocols. Model building and adjustment was performed within Coot (Emsley et al., 2010 ▶). Superposition calculations for structural comparisons were performed using least-squares superposition within Coot (Emsley et al., 2010 ▶). Figures were created with PyMOL (DeLano, 2002 ▶). The refined structural coordinates and accompanying F obs are available in the Protein Data Bank as entry 3zd4.
Table 2. Crystallographic data and refinement statistics for 3zd4 .
| Data collection | |
| Data-processing software | iMosflm, CCP4 suite, phenix.xtriage |
| Space group | Monoclinic, C2 |
| Unit-cell parameters (Å, °) | a = 50.14, b = 68.45, c = 60.22, β = 112.56 |
| Solvent content V s (%) | 49.2 |
| Matthews coefficient V M (Å3 Da−1) | 2.42 |
| Resolution range (Å) | 20.22–2.20 (2.32–2.20) |
| No. of unique reflections | 9162 (1556) |
| Multiplicity | 3.9 (4.0) |
| 〈I/σ(I)〉 | 16.80 (3.80) |
| Completeness (%) | 95.42 (98.20) |
| R merge † | 0.03 (0.31) |
| Structure refinement | |
| Model-building software‡ | Coot |
| Refinement software§ | phenix.refine |
| Target | Maximum likelihood (ML) |
| R factors | |
| R cryst ¶ | 0.1820 (0.2845) |
| R free †† | 0.2360 (0.3744) |
| Test-set size (%) | 9.9 (10.3) |
| Geometry | |
| R.m.s.d., bond lengths (Å) | 0.005 |
| R.m.s.d., bond angles (°) | 0.938 |
| R.m.s.d., planarity (°) | 0.007 |
| R.m.s.d., torsion angles (°) | 14.735 |
| ML coordinate error (Å) | 0.33 |
| ML phase error (°) | 30.80 |
| B factor from Wilson plot (Å2) | 53.23 |
| No. of TLS groups‡‡ | 10 |
R
free = 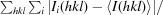
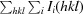 , where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean intensity of all measurements of reflection hkl.
, where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean intensity of all measurements of reflection hkl.
Model building, validation and identification of Na+ was performed using Coot (Emsley et al., 2010 ▶).
Refinement and analysis was carried out using PHENIX (phenix.refine and phenix.xtriage; Afonine et al., 2012 ▶).
R
cryst = 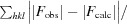
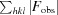 , where F
obs and F
calc are observed and calculated structure factors, respectively.
, where F
obs and F
calc are observed and calculated structure factors, respectively.
R free was calculated in the same way as R cryst but using a test set of about 7.5% of the total unique reflections that were randomly chosen and set aside prior to refinement.
2.2. G12R kinetics
The catalytic activity for all of the hammerhead ribozyme constructs described here was obtained by radio-labelled PAGE analysis using an unmodified RNA substrate corresponding to the sequence crystallized in PDB entry 3zd4, using the procedure that we employed previously for measuring the catalytic effect of the G12A substitution in PDB entry 2qus (Chi et al., 2008 ▶). Reactions were performed under single-turnover conditions at pH 7.5 in which 32P-labeled RNA substrate was mixed with a 100-fold excess of hammerhead ribozyme enzyme strand and then annealed. Owing to the high activity of the G12 wild-type ribozyme, the assay was instead performed at pH 6.5 and the results were extrapolated to obtain an approximate value of ∼50 turnovers per minute at pH 7.5 (Table 3 ▶). The hammerhead ribozyme single-turnover cleavage reactions were initiated by adding 10 mM MgCl2 and reaction aliquots were sampled for 24 h. Reactions in each aliquot were terminated at various time points using a stop dye containing 47.5% formamide and 10 mM EDTA. Each of the reaction time-point aliquots, as well as a corresponding uninitiated reaction sample, was assayed using a 15% polyacrylamide sequencing gel. Radiolabeled gels were subsequently exposed to a radiograph and quantitated using a phosphoimager and the ImageQuant software. Background, zero-time and infinite-time normalization were performed to ensure the accuracy of the catalytic activity (Stage-Zimmermann & Uhlenbeck, 1998 ▶).
Table 3. Measured and predicted rates for G12R substitutions at pH 7.5.
k c is the experimentally observed single-turnover rate of cleavage for each hammerhead ribozyme construct. The value of ∼50 in the k c column is extrapolated from the measured value at pH 6.5 as described in §2.2. The final two entries in the k c column are upper bounds to the measured cleavage rate using the 24 h time point as described in §2. The relative rate trends reported here reproduce those reported previously (Han & Burke, 2005 ▶) in the context of the minimal hammerhead ribozyme (c.f. Fig. 2d of Han & Burke, 2005 ▶).
| Relative rate | ||||
|---|---|---|---|---|
| pK a | k c (min−1) | Predicted | Measured | |
| G12 | 9.6 | ∼50 | 1 | 1 |
| G12I | 8.7 | 0.02 | 1.3 × 10−1 | 4 × 10−4 |
| G12diAP | 5.1 | 0.04 | 3.2 × 10−5 | 8 × 10−4 |
| G12A | 3.8 | ∼10−6 | 1.6 × 10−6 | <10−7 |
| G12 2AP | 3.5 | ∼10−7 | 8 × 10−7 | <10−7 |
3. Results
Using a hammerhead ribozyme sequence derived from the naturally occurring satellite RNA hammerhead discovered in S. mansoni that has previously been optimized for crystallization (Martick & Scott, 2006 ▶), we measured the effects on the single-turnover rate of purine substitutions at G12 (the presumed general base in the cleavage reaction) previously shown to exhibit a pK a-dependent effect upon cleavage rates in the context of the minimal hammerhead ribozyme (Han & Burke, 2005 ▶). We have also attempted to crystallize each of these purine-substituted hammerhead sequences, but we only obtained well diffracting crystals for the G12A substitution.
3.1. Kinetic measurements of G12R substitutions
Table 3 ▶ summarizes the results that we obtained for rate measurements for the wild-type (G12) sequence corresponding to the structures with PDB codes 3zp8 and 3zd4, as well as inosine (G12I), diaminopurine (G12diAP), adenine (G12A, corresponding to the structure 3zd4 reported here) and 2-aminopurine (G12 2AP) substitutions at the general base position in the active site. Whereas most of the substitutions follow a trend that correlates with decreasing pK a, inosine is the clear outlier, with a catalytic rate that is suppressed more than 1000-fold in excess of what would be predicted simply on the basis of the ΔpK a.
3.2. Crystal structure of a G12A substitution: PDB entry 3zd4
Fig. 1 ▶(a) illustrates the overall crystal structure of the G12A-modified hammerhead ribozyme, with G12A and its hydrogen-bonding partners (A9 and C17) highlighted as atomic color-coded stick models. The A9 phosphate, which participates in the coordination of a catalytically important (Wang et al., 1999 ▶) metal ion in both the minimal (Scott, 1999 ▶) and full-length (Martick et al., 2008 ▶) hammerhead structures, is highlighted with a magenta P atom. Fig. 1 ▶(b) shows a close-up of the same region with the same color-coding and the corresponding 2.2 Å resolution σA-weighted 2F o − F c electron-density map shown as a blue mesh. Details of the crystallographic data collection and refinement are reported in Table 2 ▶. The data and coordinates for the structure are available in the Protein Data Bank as entry 3zd4. The G12A structure was obtained using the same crystallization conditions, and in the same crystal form, as 3zp8 using these coordinates.
Figure 1.

The crystal structure of a G12A-substituted hammerhead ribozyme. (a) An overall view of the full-length G12A hammerhead ribozyme crystal structure (PDB entry 3zd4), with the G12A purine substitution and its two hydrogen-bonding partners, A9 and C17, shown with atom color-coding. The A9 P atom is highlighted in magenta. The substrate strand, which contains C17, is depicted as a light blue ribbon and the enzyme strand as a gray ribbon. The remaining nucleotides are shown as stylized representations. (b) Wall-eyed stereoview of the G12A hammerhead ribozyme (PDB entry 3zd4) active site. The 2F o − F c electron-density map contoured at 1.0 r.m.s.d. is shown as a blue mesh. A12, A9 and C17 are highlighted as before, including the A9 P atom, which is highlighted in magenta.
3.3. Comparison to G12 structures: PDB entries 3zd4 versus 3zp8 and 3zd5
The two crystal structures of the G12 hammerhead obtained in the same crystal form but under different crystallization conditions are very similar. The 1.55 Å resolution structure (PDB entry 3zp8) was obtained in the presence of a high concentration of Na+, and Na+ is observed to coordinate the N7 of G10.1 and the pro-R O atom of the A9 phosphate in a manner similar to that previously observed for various divalent cations (Anderson et al., 2013 ▶). The 2.2 Å resolution structure (3zd5, formerly 2goz) was obtained in the presence of NH4 +, and no metal ions are observed to coordinate at the A9 phosphate site (Martick & Scott, 2006 ▶). Both structures contain a modified substrate in which C17 is replaced by 2′OMe-C17, a modification that prevents the cleavage reaction from occurring without introducing any obvious structural perturbations. The N1 of G12 donates a hydrogen bond to the 2′O of C17 in both structures. (Presumably in an active ribozyme the 2′O is a hydrogen-bond donor and a deprotonated N1 will be a hydrogen-bond acceptor. Abstraction of the 2′H+ by G12 presumably initiates the cleavage reaction.)
Based on this comparison, the presence versus absence of a metal ion bound to the A9 phosphate and the N7 of G10.1 in itself does not significantly perturb what appears to be a pre-formed metal-binding site in the wild-type G12 hammerhead. (This binding site in fact persists in all G12 hammerhead structures, including those of the minimal hammerhead.) It is only when the G12A substitution is made (in 3zd4 as well as in 2qus) that this pre-formed metal-binding site becomes significantly perturbed.
3.4. The G12A–C17 interface
The new crystal structure of the corresponding G12A modification, PDB entry 3zd4, reveals several structural perturbations. Because of the absence of a hydrogen-bond donor, the N1 atom of A12 cannot make a hydrogen bond to the 2′O of C17; instead, this distance is 4.3 Å, corresponding to a van der Waals contact (Fig. 2 ▶ a), rather than ∼3 Å as observed in the G12 structures (Figs. 2 ▶ b and 2 ▶ d). The exocyclic 6-amine instead donates a hydrogen bond to the 2′O of C17. This interaction is almost certainly an artifact created by the presence of 2′OMe-C17. In the case of the previously solved G12A sTRSV hammerhead structure (PDB entry 2qus; Chi et al., 2008 ▶), in which an unmodified C17 is present, the N1 of A12 receives a nearly ideal hydrogen bond from the 2′OH of C17 (Fig. 2 ▶ c).
Figure 2.

G12 and A12 hydrogen-bonding networks. Hydrogen-bonding networks involving G12 and A12 are shown for four structures as light blue dotted lines; other distances of relevance are shown as light yellow dotted lines. The distances indicated are in Å. Those for the two G12 structures (PDB entries 3zp8 and 3zd5) are essentially identical. The hydrogen-bonding network involving A12 in the presence of 2′OMe-C17 (PDB entry 3zd4) differs from that in the presence of the unmodified substrate (PDB entry 2qus). The latter makes a good hydrogen bond between N1 of A12 and the 2′OH of C17, whereas the N1 atom of 3zd4 is 4.3 Å from the 2′O of the methylated substrate owing to the fact that no hydrogen bond can form between these atoms. Instead, the 6-amino group appears to make a hydrogen-bond donation to the 2′O of the methylated substrate in 3zd4 and A12 hydrogen bonds to A9 in a manner similar to that observed in the G12 structures, apart from the absence of the 2-amino group.
3.5. The G12A–A9 interface
In the 3zd4 structure, the N3 on the minor-groove face of A12, along with the 2′OH of A12, forms hydrogen bonds to the exocyclic 6-amine of A9 (Fig. 2 ▶ a), similar to the hydrogen bonds formed between the corresponding G12 and A9 (Figs. 2 ▶ b and 2 ▶ d). Because A12 lacks a 2-amine group, the hydrogen bond between this group in G12 and the N7 of A9 does not exist. Hence, it appears that A12 in 3zd4 interfaces with A9 in a manner as analogous as possible to G12 in 3zp8 and 3zd5. In contrast, the G12A structure in 2qus reveals a single 3.3 Å hydrogen bond between A12 and A9 that more tenuously mediates a weakened interface between these purines (Chi et al., 2008 ▶).
3.6. The A9 phosphate potential metal ion-binding site
A set of three least-squares superposition calculations were performed using the high-resolution G12 structure (3zp8) as the reference structure in each case. An all-atom superposition of consecutive residues (G8, A9 and G10.1) from the new G12A structure (3zd4), the previous G12A structure (2qus) and the G12 structure in the absence of metal ions (3zd5) was calculated in each case (Fig. 3 ▶). These calculations reveal that the two G12 structures are nearly identical, despite the presence of metal ions in one structure and their absence in the other. In addition, the two G12A-substituted structures, despite being from two very different sequences and crystal forms and despite the presence (2qus) versus absence (3zd4) of an active substrate, reveal rather similar distortions at the A9 phosphate, demonstrating that it is the G12A substitution itself that perturbs the A9 phosphate, and thus metal ion binding. Indeed, unlike the G12 reference structure (3zp8) in which a partially hydrated, octahedrally coordinated Na+ ion is bound to the N7 of G10.1 and to the pro-R O atom of the A9 phosphate, the new G12A structure (3zd4) obtained under identical crystallization conditions does not reveal the presence of a bound Na+ ion at the A9 phosphate (Fig. 3 ▶). Hence, it appears that this structural perturbation is not only a consequence of the G12A substitution itself, but also results in interference with the most prominent metal ion-binding site in the hammerhead ribozyme, one that is known to have structural and catalytic importance.
Figure 3.
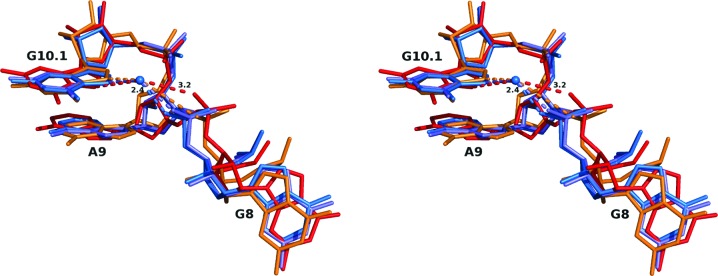
Conformations of the A9 phosphate. A wall-eyed stereo superposition of G12 and G12A hammerhead ribozyme nucleotides including and adjacent to A9. The superpositions were performed in Coot (Emsley et al., 2010 ▶) using PDB entry 3zp8, the highest-resolution structure, as a reference. The color-coding shows two G12A structures (3zd4, red; 2qus, orange) and two G12 structures (3zd5, light blue; 3zp8, blue). The blue sphere represents the position of Na+ in 3zp8. The distance between this ion and the A9 pro-R phosphate O atom in 3zp8 is 2.36 Å, that in 3zd4 is 3.22 Å and that in 2qus is 3.81 Å (see Table 4 ▶).
4. Discussion
There are two primary structural perturbations observed in 3zd4. The first perturbation, found only in the 3zd4 G12A structure, is the misalignment of the N1 of the purine at position 12 with the 2′O of C17, the nucleophile in the cleavage reaction. This perturbation is almost certainly an artifact owing to the presence of the 2′OCH3 on C17 in the 3zd4 G12A structure. In the 2qus structure, which has an unaltered 2′OH at C17, the N1 of A12 is aligned properly to accept a hydrogen bond from the 2′OH of C17. In the G12 wild-type structures, the N1 of G12 is aligned properly with the 2′OCH3 on C17 to donate a hydrogen bond to the 2′O at C17; because it is a hydrogen-bond acceptor in the G12 case, the methyl group does not perturb the geometry of the interaction.
The second perturbation is the movement of the A9 phosphate relative to those of the 3zp8 and 3zd5 controls. We suggest in this case the perturbation is not simply a consequence of the presence of the 2′OCH3 on C17.
The G12 wild-type structures (3zp8 and 3zd5) were obtained from the same sequence in two different crystallization conditions, but belong to the same crystal form as 3zd4. The 3zp8 structure was obtained at a high concentration of Na+ ions and reveals an Na+ ion bound to the N7 of G10.1 and the pro-R O atom of the A9 phosphate, binding in a mode previously observed for divalent metal-ion binding at this same site. The 3zd5 structure was obtained at a high concentration of NH4 + and does not appear to have a metal ion bound to the N7 of G10.1 and the pro-R O atom of the A9 phosphate. Nevertheless, the two structures are otherwise almost identical; the positional difference of the A9 phosphate pro-R O atom between the two structures upon least-squares superposition is about 0.7 Å, which is little more than the sum of the positional errors associated with each of the two structures. The absence of a metal ion binding to this site is thus of minimal structural consequence in the context of the wild-type structure.
The positional difference between the A9 phosphate pro-R O atom of the new G12A-substituted structure 3zd4 and that of 3zp8 is 2.1 Å. The conformation of the phosphate in 3zd4 is much less amenable to cation binding because of the distance between the N7 of G10.1 and the pro-R O atom of the A9 phosphate. Similarly, the corresponding distance in the case of the previous G12A-substituted structure 2qus is 1.7 Å, with the phosphate conformation similarly distorted (Chi et al., 2008 ▶). Based upon these two comparisons, the structural perturbation of the A9 phosphate appears to primarily be a consequence of the G12A substitution itself rather than the presence versus absence of the unmodified C17 2′OH. It is noteworthy that 3zd4 and 2qus are two completely different hammerhead ribozyme sequences belonging to two different tertiary-structural contact classes, and that the crystal forms and crystallization conditions have little in common. Nevertheless, despite all of these differences, the observed structural perturbation is apparent in both G12A structures. Fig. 3 ▶ shows a superposition of all four structures, with 3zp8 and its accompanying Na+ ion and electron density as the reference. Because there is little change in the position of the N7 of G10.1 in the superposed structures, potential coordination distances and angles between the Na+ ion and the other pro-R A9 phosphate O atoms can be estimated (see Table 4 ▶).
Table 4. A9 phosphate geometry.
The penultimate column lists the distances between the N7 atom of G10.1 and the ORP nonbridging A9 phosphate O atom for each of the four structures discussed in the text. In each case, the distance between the ORP nonbridging A9 phosphate O atom of the named structure and the position occupied by the Na+ in 3zp8 (the reference structure) is given in the final column. Each of the last three structures was superposed upon the first (or reference) structure as described in the text.
Comparison of the two G12A structures reveals that the presence versus absence of the unmodified C17 2′OH has a pronounced effect upon the disposition of the purine base. The N1 of A12 is deprotonated near neutral pH, so a hydrogen bond can only form if the 2′O of C17 donates a hydrogen. Thus, the 3zd4 structure is inherently incapable of forming the hydrogen bond relevant to catalysis owing to the presence of the methyl group, and instead the exocyclic 6-amine of A12 forms a potentially catalytically inhibitory hydrogen bond to the 2′O of the 2′OCH3 modification. In the 2qus structure the relevant hydrogen bond is able to form owing to the presence of a 2′OH in the active unmodified substrate. (The G12 structures can also form a hydrogen bond to the 2′O of the methylated substrate because the N1 atom is protonated near neutrality.) Comparison of the A12/A9 nucleotide base hydrogen-bonding interface in the two G12A structures, however, reveals that the 3zd4 structure forms a more stable interaction with two hydrogen bonds present, mimicking the G12 structures, whereas the 2qus structure reveals a less stable interaction with only one hydrogen bond between A12 and A9 present. This suggests that two competing A12 active-site conformational states may exist, in which the stabilizing interface between A12 and A9 must be disrupted in a way that allows the spatial positioning found in the G12/A9 interface to be reproduced for catalysis to take place. In other words, the hydrogen-bonding interface that helps to stabilize G12 and position it favorably for catalysis may instead have an inhibitory effect upon other purines, especially those lacking the 2-amino group present in G12. This would explain the pK a trend anomaly observed for G12I substitutions. Inosine is similar to guanosine, lacking only the 2-amino group. The absence of the 2-amino group lowers the pK a by only one unit, which, assuming a general base catalytic mechanism, should result in an approzimately tenfold reduction in catalytic activity. Instead, the observed reduction is ∼104-fold, implying a rather more dramatic effect arising from the absence of the 2-amino group. This implies a ∼103-fold reduction in catalytic activity owing to structural perturbations alone (assuming an approximately additive energetic effect). A similar violation of the general base-predicted pK a trend, with a pronounced inhibitory effect for the G12I substitution, has previously been observed in the context of a minimal hammerhead sequence (Han & Burke, 2005 ▶), so this observation is very unlikely to be an artifact arising from the peculiarities of our particular full-length hammerhead sequences.
The pronounced inhibitory effect of the G12A substitution in the hammerhead ribozyme is therefore likely to arise from the combination of two contributions, one of which is structural (primarily the absence of the exocyclic 2-amino group) and one of which is a manifestation of the ΔpK a. The catalytic effect of the G12I substitution allows us to separate the contribution of the two effects to at least within an order-of-magnitude approximation, i.e. a ∼103-fold effect upon the rate owing to ablation of the 2-amino group; I is in essence G with the 2-amino group deleted. The pK a is very similar, and the hydrogen-bonding face presented to C17 is the same. A9, however, sees the minor-groove edge of the purine that lacks the 2-amino group, and therefore presents the same hydrogen-bonding potential as a G12A substitution. As a consequence, the observed ∼103-fold excess reduction in catalytic activity must be attributed to structural perturbations similar to those induced by the G12A substitution, and a similar in order-of-magnitude effect upon catalytic activity in the G12A hammerhead may therefore be attributed to perturbing structural effects.
In summary, these results permit us to suggest the following.
(i) The G12A substitution in both cases (3zd4 and 2qus) signficantly perturbs the metal ion-binding site at the A9 phosphate that is preformed in all of the G12 hammerhead structures.
(ii) The 2′OCH3 modification of C17 in 3zd4, by itself, is not responsible for this structural perturbation.
(iii) The absence of the 2-amine on G12A-substituted purine, relative to G12 itself, is most likely to be responsible for the structural perturbation of the A9 phosphate-binding site.
(iv) The structural perturbation of the A9 phosphate-binding site is likely to have a pronounced deleterious effect upon the catalytic prowess of the hammerhead ribozyme, and also explains why the G12I substitution has such a profound effect upon the catalytic rate. This is discussed in the next section, along with the implications for competing mechanisms of base catalysis.
5. Conclusions
The hammerhead ribozyme has traditionally been assumed to catalyze RNA self-cleavage via a general acid–base catalysis mechanism. Based on a comparison of four crystal structures, including the new structure (PDB entry 3zd4) reported here, and based on the anomalously low cleavage rate that we observe for the G12I-substituted hammerhead ribozyme, it appears that the 2-amine of G12, although not thought to participate directly in the chemistry of catalysis, is nonetheless critical to the function of the hammerhead ribozyme. The G12I substitution results in deletion of the 2-amine, but does not significantly perturb the pK a of the purine. Nonetheless, the cleavage rate is attenuated three orders of magnitude beyond what one might expect based upon the difference in pK a between G and I, assuming pure general base catalysis. Similarly, the G12A substitution results in a much greater attenuation of catalytic activity than what one would predict based upon the difference in pK a between G and A, again assuming pure general base catalysis.
The 2-amine of G12 is involved in noncanonical base-pairing hydrogen-bond interactions with A9 in the wild-type hammerhead. Ablation of the 2-amine disrupts the G12/A9 interface. Disruption of the noncanonical base-pairing interaction involving A9 in turn perturbs the phosphate of A9 and therefore the metal-binding site that is formed by the collaboration of the pro-R O atom of the A9 phosphate and the N7 of G10.1.
Genral base catalysis is typically assumed to be a component of the hammerhead ribozyme cleavage mechanism. This assumption is based on the trend observed for purine substitutions at G12, in which the cleavage rate is observed to decrease with decreasing purine pK a. It is also based on the observed hydrogen-bonding interaction between the N1 of G12 and the 2′O of C17. The assumption is that this hydrogen bond observed in the G12 crystal structures, in which the normally protonated N1 of G12 is the donor and the 2′O of C17 is the acceptor, is replaced by a catalytically active hydrogen bond approaching the transition state, in which a transiently deprotonated N1 is now the hydrogen-bond acceptor and the 2′OH of C17 is the hydrogen-bond donor. If G12 successfully abstracts the proton from the 2′OH of C17, it generates the nucleophile for the self-cleavage reaction.
Specific base catalysis and general base catalysis are mutually exclusive reaction mechanisms, despite the fact that both are described by phenomenologically identical kinetics equations. In the context of the hammerhead ribozyme reaction, specific base catalysis would involve the assumption that the hydrogen bond between the N1 of G12 and the 2′O of C17 is purely structural and is not a catalytically active hydrogen bond that directly participates in the chemistry of the reaction. Instead, if specific base catalysis pertains, a water molecule or hydroxide ion abstracts the proton from the 2′OH of C17, an event that is presumably facilitated by this structural hydrogen bond. If the structural hydrogen bond is perturbed, this will result in an attenuation of catalytic activity. Specific base catalysis, in contrast to general base catalysis, does not require the structural hydrogen bond between the N1 of G12 and the 2′O of C17 to be replaced by an active hydrogen bond of the opposite directionality as a consequence of a transient deprotonation of G12.
The biochemical data and the crystal structures are each consistent with both general base catalysis and specific base catalysis. Although general base catalysis is typically assumed in the hammerhead ribozyme mechanism, specific base catalysis also must be considered in the absence of a definitive experimental result that distinguishes between the two incompatible mechanims. To explain the anomolies in the pK a trend of purine-substituted G12 hammerheads observed for substitutions involving ablation of the 2-amine, a general base catalysis mechanism must involve an additional ad hoc assumption involving the effects of structural perturbations. If instead one considers a mechanism employing specific base catalysis, no additional ad hoc assumption involving the effects of structural perturbations need be made; such potential structural effects are an intrinsic part of specific base catalysis. In addition, the assumption that the crystallographically observed hydrogen bond between the N1 of G12 and the 2′O of C17 is purely structural, and does not need to be replaced by an active hydrogen bond of opposite directionality concomitant with deprotonation of the N1 of G12, enjoys considerable parsimony relative to what is required for general base catalysis.
For these reasons, we conclude that specific base catalysis cannot be dismissed as a component of the hammerhead ribozyme catalytic mechanism, and that in some senses it may in fact possess greater explanatory power.
Supplementary Material
PDB reference: schistosomal hammerhead ribozyme, G12A mutant, 3zd4
WebGL html document to accompany Fig. 3.. DOI: 10.1107/S1399004714010608/cb5054sup1.html
Acknowledgments
This work was supported by National Institutes of Health grant R01GM087721 to WGS. We thank Harry Noller and the other members of the Center for Molecular Biology of RNA for helpful discussions, advice and shared facilities.
References
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Anderson, M., Schultz, E. P., Martick, M. & Scott, W. G. (2013). J. Mol. Biol. 425, 3790–3798. [DOI] [PMC free article] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Chi, Y.-I., Martick, M., Lares, M., Kim, R., Scott, W. G. & Kim, S.-H. (2008). PLoS Biol. 6, e234. [DOI] [PMC free article] [PubMed]
- Cuchillo, C. M., Nogués, M. V. & Raines, R. T. (2011). Biochemistry, 50, 7835–7841. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fedor, M. J. (2009). Annu. Rev. Biophys. 38, 271–299. [DOI] [PubMed]
- Forster, A. C. & Symons, R. H. (1987). Cell, 50, 9–16. [DOI] [PubMed]
- Han, J. & Burke, J. M. (2005). Biochemistry, 44, 7864–7870. [DOI] [PubMed]
- Martick, M., Lee, T.-S., York, D. M. & Scott, W. G. (2008). Chem. Biol. 15, 332–342. [DOI] [PMC free article] [PubMed]
- Martick, M. & Scott, W. G. (2006). Cell, 126, 309–320. [DOI] [PMC free article] [PubMed]
- Painter, J. & Merritt, E. A. (2006a). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006b). J. Appl. Cryst. 39, 109–111.
- Prody, G. A., Bakos, J. T., Buzayan, J. M., Schneider, I. R. & Bruening, G. (1986). Science, 231, 1577–1580. [DOI] [PubMed]
- Scott, W. G. (1999). Curr. Opin. Chem. Biol. 3, 705–709.
- Stage-Zimmermann, T. K. & Uhlenbeck, O. C. (1998). RNA, 4, 875–889. [DOI] [PMC free article] [PubMed]
- Uhlenbeck, O. C. (1987). Nature (London), 328, 596–600. [DOI] [PubMed]
- Wang, S., Karbstein, K., Peracchi, A., Beigelman, L. & Herschlag, D. (1999). Biochemistry, 38, 14363–14378. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: schistosomal hammerhead ribozyme, G12A mutant, 3zd4
WebGL html document to accompany Fig. 3.. DOI: 10.1107/S1399004714010608/cb5054sup1.html