

Abstract
Objective
To study the efficacy of anti-miRNA-33 therapy on the progression of atherosclerosis.
Approach and Results
Ldlr−/− mice were injected subcutaneously with PBS, control or anti-miR-33 oligonucleotides weekly and fed a Western diet for 12 weeks. At the end of treatment, the expression of miR-33 target genes was increased in the liver and aorta, demonstrating effective inhibition of miR-33 function. Interestingly, plasma HDL cholesterol (HDL-C) was significantly increased in anti-miR-33 treated mice but only when they were fed a chow diet. However, HDL isolated from anti-miR-33 treated mice showed an increase cholesterol efflux capacity compared to HDL isolated from non-targeting oligonucleotide treated mice. Analysis of atherosclerosis revealed a significant reduction of plaque size and macrophage content in mice receiving anti-miR-33. In contrast, no differences in collagen content and necrotic areas were observed between the three groups.
Conclusions
Long-term anti-miR-33 therapy significantly reduces the progression of atherosclerosis and improves HDL functionality. The anti-atherogenic effect is independent of plasma HDL-C levels.
Keywords: miR-33, atherosclerosis, HDL, macrophage, ABCA1
INTRODUCTION
Several groups have recently reported the important role of miR-33a and miR-33b, intronic miRNAs located within the Srebp2 and Srebp1 genes, respectively, in regulating cholesterol and fatty acid metabolism1–5. Hence, miR-33 controls the expression of ABCA1 and ABCG1 in the liver and peripheral tissues, thereby regulating HDL biogenesis and cellular cholesterol efflux. The strong inverse correlation between low circulating HDL-C with coronary heart disease found in human epidemiological studies led Rayner and colleagues to study the potential benefit of anti-miR-33 therapy in promoting atherosclerotic plaque regression in Ldlr−/− mice6. Importantly, in this later study, therapeutic inhibition of miR-33 resulted in an increase in circulating HDL-C and a significant reduction in atherosclerotic plaque size. Moreover, the morphological analysis of the atheromata revealed a significant increase in collagen content and fibrous cap size, which are characteristic features of stable atherosclerotic plaques. Even though this study strongly suggested that anti-miR-33 therapy would be useful for treating the regression of atherosclerosis, no studies have been performed to assess the efficacy of anti-miR-33 on the progression of atherosclerosis. Therefore, we herein we aimed to determine the efficacy of anti-miR-33 therapy in Ldlr−/− mice fed a Western diet (WD).
MATERIALS AND METHODS
Detailed materials and methods appear in the online-only Data Supplemental materials (http://atvb.ahajournals.org). We used Ldlr−/− mice treated with anti-miR-33 or control oligonucleotides and assessed atheroma formation and lipoprotein metabolism.
RESULTS
We first determined the efficacy of the anti-miR-33 treatment by measuring hepatic expression of miR-33 target genes and circulating HDL-C in mice fed a chow diet. Hepatic expression of several miR-33 target genes including ABCA1, CPT1A, IRS2, AMPK and HADHB was increased in mice treated with anti-miR-33 compared with those receiving PBS or control anti-miR (Figure. SIB). Similarly, hepatic ABCA1 and CROT protein expression were also increased in mice treated with anti-miR-33 (Figure. SIC). As reported previously2–5, anti-miR-33 therapy increased plasma HDL-C without affecting the cholesterol distribution in other lipoprotein fractions (Figure. SID and SIE). Plasma triglyceride (TG) levels were the same in the three groups of mice (Figure. SIF).
Next, we analyzed the efficacy of anti-miR-33 therapy during the progression of atherosclerosis by feeding mice a WD. Similar to mice fed a chow diet, ABCA1, ABCG1, CROT, AMPK and HADHB mRNA expression were increased compared to control mice (Figure 1B). However, we did not observe differences in hepatic ABCA1 and CROT protein expression (Figure. 1C). In agreement with this observation, we did not also observe differences in total cholesterol, HDL-C and TG levels among treatments (Figure. 1D–F), suggesting that anti-miR-33 therapy fails to increase hepatic ABCA1 expression and circulating HDL levels in mice fed a WD. The cholesterol distribution in different lipoproteins was also not affected by anti-miR-33 treatment (Figure.1G). These results might be explained by the reduced expression of miR-33 in the liver of mice fed a WD compared with mice fed a chow diet5.
Figure 1. Lipid analysis and gene expression in Ldlr−/− mice injected with PBS, Ctrl ASO or miR-33 ASO during a progression study.
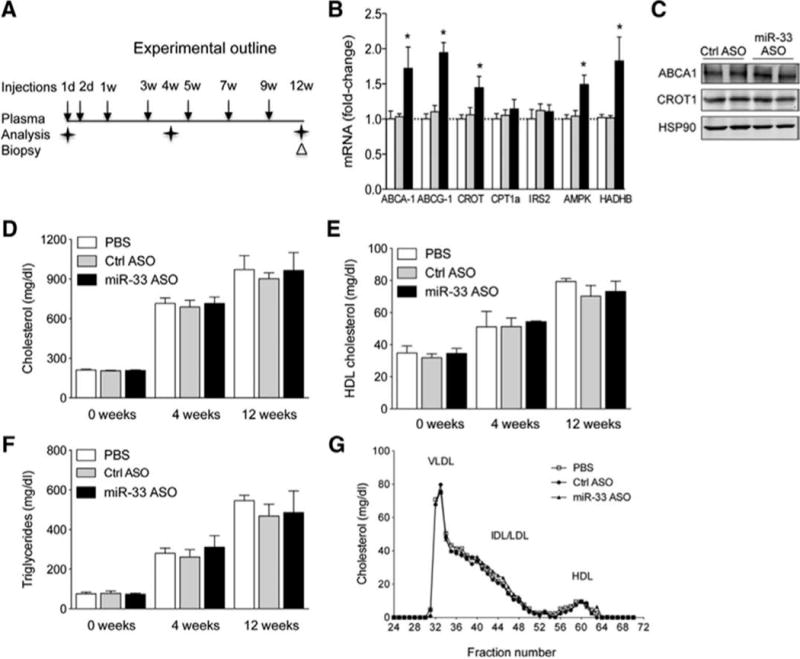
A) Experimental outline of PBS, Ctrl ASO or miR-33 ASO treatment of Ldlr−/− mice fed a WD for 12 weeks. B–C) Hepatic mRNA (left panel) and protein (right panel) expression of miR-33 target genes from mice treated with PBS, Ctrl ASO and miR-33 ASO for 12 weeks and fed a WD. *Indicates p< 0.05 compared with miR-33 ASO with PBS and control ASO group. D–F) Total cholesterol (D), HDL-C (E) and triglyceride (F) levels at starting point (0 week) and at 4 and 12 weeks on WD and PBS, Ctrl ASO and miR-33 ASO treatment. All of the data represent the mean ± SEM; (PBS n=5, control ASO n=9 and miR-33 ASO n=9). G) Cholesterol lipoprotein profile from pooled plasma from Ldlr−/− mice injected with PBS, Ctrl ASO and miR-33 ASO after 12 weeks on WD.
Despite the absence of changes in HDL-C between treatments, we found that atherosclerotic lesion areas were significantly reduced in the aortic roots of mice injected with anti-miR-33, compared to PBS and Ctrl ASO groups (Figure. 2A and 2B). Further analysis of the atheromata revealed no differences in the fibrous cap and necrotic core between treatments (Figure. 2A–C). On the other hand, CD68 staining revealed a decrease in the macrophage content in the proximal aortas of mice injected with anti-miR-33 oligonucleotides compared to the PBS and Ctrl ASO treated mice (Figure. 2D).
Figure 2. Reduction in lesion area and gene expression of inflammatory, adhesion, matrix and macrophage markers from RNA of total aortas in Ldlr−/− mice injected with PBS, Ctrl ASO or miR-33 ASO during a progression study.

A–D) Representative histological analysis of cross-sections from the aortic sinus stained with Oil-red-O (ORO) (A), hematoxylin and eosin (H&E) (B), Masson’s trichrome (MT) (C), and CD68, α-SMC actin (D). Quantification of the lesion area, fibrous cap, necrotic core and macrophage content are represented in the right panels. All of the data represents the mean ± SEM; (PBS n=5, control ASO n=9 and miR-33 ASO n=9). *Indicates p<0.05 compared to miR-33 ASO with PBS and Ctrl ASO group. E) Expression profile of atherosclerotic-related genes assessed by real-time qPCR. Five independent qPCR reactions were carried out for each condition. The fold change for each gene of mice treated with miR-33 ASO compared to mice treated with Ctrl ASO. The data represents the mean ± SEM. *Indicates p< 0.05 compared to the miR-33 ASO with the Ctrl ASO group. Bar scale = 400μm. F) Representative Western blot analysis of ABCA1 and HSP90 expression from aortic lysates of mice treated with Ctrl ASO and miR-33 ASO.
We further analyzed the potential mechanism that mediates atheroprotection in anti-miR-33 treated mice. It is has been previously shown that HDL functionality has a strong inverse association with atherosclerotic vascular disease independent of the total plasma HDL-C7. Therefore, we analyzed the HDL functionality by assessing its ability to promote cellular cholesterol efflux and to protect endothelial cells from cytokine-induce inflammation. As seen in Figure SIIA, HDL isolated from anti-miR-33 treated mice induced greater macrophage cholesterol efflux than did HDL isolated from Ctrl ASO treated mice. In addition to cholesterol efflux, we also assessed the effect of HDL isolated from both groups of mice on TNF-induced inflammation in human aortic endothelial cells (HAECs). As expected, VCAM-1 and ICAM-1 were significantly downregulated when we treated HAECs with HDL, but no differences were noted between HDL isolated from both groups of mice (Figure SIIB). The reasons behind the increased cholesterol efflux capacity of HDL from mice treated with anti-miR-33 remain unclear and will require further investigation.
Finally, we analyzed the expression of inflammatory cytokines, adhesion molecules and matrix remodeling enzymes in the aortas from the three groups of mice. In contrast to the results observed in the liver, anti-miR-33 therapy significantly increased the expression of ABCA1 at mRNA and protein levels (Figure. 2E and 2F). Moreover, we observed an upregulation of MMP-2, COL1A-1 and COL3A-1 suggesting that miR-33 might also be important in regulating plaque remodeling (Figure. 2E). Similar to the results observed in the miR-33−/− Apoe−/− mice8, we found a significant increase in the pro-inflammatory cytokines, TNFα and IL-1β, while IL-6 expression was reduced (Figure. 2E). In agreement with the reduced macrophage content observed in atherosclerotic plaques from mice treated with anti-miR-33 in regression studies, the expression of CD68 and MAC-2 was significantly reduced in this group of mice (Figure. 2E). To analyze whether the macrophage infiltration correlated with reduced levels of adhesion molecules in the aorta, we analyzed the expression of ICAM-1, VCAM-1 and ESEL. As shown in Figure. 2E, anti-miR-33 treatment reduced significantly ICAM-1, VCAM-1 and ESEL expression indicating that miR-33 might regulate the expression of these molecules in endothelial cells (ECs), thereby controlling macrophage infiltration in the artery wall. To elucidate whether the anti-inflammatory effect of anti-miR-33 therapy in the artery wall is mediated by a direct effect of miR-33 on EC activation, we transfected HAECs with anti-miR-33 oligonucleotides and stimulate them with TNF for 6h. The results shown that miR-33 inhibition did not reduced significantly the expression of ICAM-1 and VCAM-1 compared with cells treated with Ctrl ASO, suggesting that the anti-inflammatory effect of anti-miR-33 oligonucleotides is likely mediated by increasing macrophages cholesterol efflux and reducing their accumulation in the artery wall (Figure SIIC).
DISCUSSION
Previous studies by Rayner and colleagues demonstrated that a 2′F/MOE modified anti-miR-33 was effective in an atherosclerosis regression model6. In Ldlr−/− mice with established atherosclerotic plaques, anti-miR-33 treatment resulted in significant reduction in plaque size and elevated circulating HDL-C. In contrast, here we reported that anti-miR-33 failed to increase HDL-C when mice were fed a WD. The mechanism by which the anti-miR-33 did not increase circulating HDL-C in WD-fed animals remains unclear and requires further investigation. Even though the circulating HDL-C levels were the same in all experimental groups, we observed a significant reduction in atherosclerotic plaque size in mice treated with miR-33 ASO. Importantly, the cholesterol efflux capacity of HDL isolated from mice treated with anti-miR-33 was significantly higher compared to HDL isolated from Ctrl ASO mice. In addition to the increased functionality of the HDL particles, these results also suggest that the atheroprotective effects of miR-33 ASO might be in part mediated by their effect in the artery wall. While this paper was under preparation, Horie and colleagues reported that miR-33−/− Apoe−/− have decreased atherosclerosis compared to Apoe−/− mice8. To determine the contribution of miR-33 in monocytes/macrophages, the authors employed bone marrow transplantation experiments. The results demonstrated that miR-33 was important in regulating ABCA1 expression in macrophages and lipid accumulation in the artery wall. However, the deficiency of miR-33 in myeloid cells did not reduce atherosclerotic plaque size as expected. This observation suggests that the anti-atherosclerotic effect of anti-miR-33 might be mediated by another cell type in the artery wall, such as ECs and vascular smooth muscle cells. Indeed, absence of ABCA1 and ABCG1 in ECs leads to endothelial dysfunction in mice fed a high-cholesterol diet9. Therefore, tissue-specific null mice for miR-33 will be important to determine the contribution of miR-33 in hepatocytes, macrophages, endothelial and smooth muscle cells during atherogenesis.
While this study was under revision, another group reported that anti-miR-33 therapy fails to increase circulating HDL-C levels and to slow the progression of atherosclerosis in Ldlr−/− mice10, in contrast to our results. There are several important differences between the two studies, however. First, while the 2’F/MOE anti-miR has been shown in this study and in the previous publication6 to target miR-33 and regulate gene expression in atherosclerotic plaque and whole aorta, the effect of the Locked Nucleic Acid (LNA) modified oligonucleotide used in the Marquart et. al. study was not examined in the artery wall10. It is possible that the LNA anti-miR was not effectively taken up in the plaque macrophages. The second important difference between the studies is the cholesterol concentration in the diets. In our study, the WD contained 0.3% cholesterol, while Marquart et. al. used 1.25%, a more severe model of atherosclerosis. This is an important point because it is expected that miR-33 levels are reduced as cellular cholesterol content increases, and high dietary cholesterol concentration may reduce miR-33 expression to levels where the anti-miR-33 therapy could not have further effect in the liver and peripheral tissues. Finally, Marquart et. al. observed that anti-miR-33 therapy increases plasma TG content while we did not find differences in our study. Altogether, these studies suggest that different oligonucleotide chemistry and cholesterol content in the diet could be relevant to the efficacy of anti-miR-33 therapy. Future studies dissecting the relative importance of these variables to the phenotype will be important to clarify the full therapeutic potential for miR-33 targeting.
Supplementary Material
SIGNIFICANCE.
Alterations in the metabolic control of lipid homeostasis predispose an individual to develop cardiometabolic diseases such as atherosclerosis. Work over the last years has suggested that miRNAs play an important role in regulating cholesterol metabolism. Specifically, miR-33, an intronic miRNA encoded within the Srebp genes, has been shown to play an important role in regulating plasma HDL-C levels. Here, we demonstrate that anti-miR-33 therapy protects against the progression of atherosclerosis but fails to increase circulating HDL-C in mice fed a Western diet. Importantly, HDL isolated from mice treated with anti-miR-33 oligonucleotides has greater cholesterol efflux capacity than HDL isolated from non-targeting oligonucleotide treated mice. These findings provide greater insights into the molecular mechanism by which anti-miR-33 therapy confers atheroprotection.
Acknowledgments
The authors thank Leigh Goedeke for editing and helpful comments on the manuscript
SOURCES OF FUNDING: This work was supported by grants from the National Institutes of Health [R01HL107953 and R01HL106063 (to C.F.-H.)], Ministerio de Educación [Programa Nacional de Movilidad de Recursos Humanos del Plan Nacional de I-D+i 2008–2011 (to N. R.)] and the American Heart Association [(12POST9780016) (to C.M.R)].
Footnotes
DISCLOSURES: C.F.-H has patents on the use of miRNA-33 inhibitors. Christine C. Esau is employee of Regulus Therapeutics
References
- 1.Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, Macdougald OA, Bommer GT. Expression of mir-33 from an srebp2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. 2010;285(44):33652–61. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T. Microrna-33 encoded by an intron of sterol regulatory element-binding protein 2 (srebp2) regulates hdl in vivo. Proc Natl Acad Sci U S A. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marquart TJ, Allen RM, Ory DS, Baldan A. Mir-33 links srebp-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. Microrna-33 and the srebp host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. Mir-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of mir-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. The New England journal of medicine. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horie T, Baba O, Kuwabara Y, et al. Microrna-33 deficiency reduces the progression of atherosclerotic plaque in apoe−/− mice. Journal of the American Heart Association. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terasaka N, Yu S, Yvan-Charvet L, Wang N, Mzhavia N, Langlois R, Pagler T, Li R, Welch CL, Goldberg IJ, Tall AR. Abcg1 and hdl protect against endothelial dysfunction in mice fed a high-cholesterol diet. J Clin Invest. 2008;118:3701–3713. doi: 10.1172/JCI35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marquart TJ, Wu J, Lusis AJ, Baldan A. Anti-mir-33 therapy does not alter the progression of atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2013;33:455–458. doi: 10.1161/ATVBAHA.112.300639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.