

Abstract
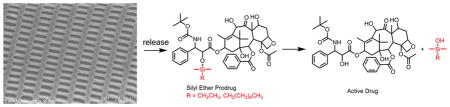
Delivery systems designed to have triggered release after passively targeting the tumor may improve small molecule chemotherapeutic delivery. Particle replication in nonwetting templates was used to prepare nanoparticles to passively target solid tumors in an A549 subcutaneous xenograft model. An acid labile prodrug was delivered to minimize systemic free docetaxel concentrations and improve tolerability without compromising efficacy.
Keywords: Soft-lithography, docetaxel, polylactic acid, silyl ether
The synthesis of prodrugs is a common approach to overcome drug delivery issues, including poor aqueous solubility1 or permeability,2 and to provide site-specific release.3 Nanotechnology can be a powerful tool to improve drug delivery, but does so by altering the biodistribution of the encapsulated small molecule.4,5 In this report, we combined the merits of both approaches to improve the pharmacokinetics and toxicity of the chemotherapeutic docetaxel by passively targeting an encapsulated docetaxel prodrug to solid tumors, where it could selectively release and convert to active docetaxel.
Many chemistries have been utilized to prepare prodrugs of taxanes6–9 or camptothecins.10,11 Hydrazone bonds cleave quickly at acidic conditions, but derivatives of taxanes must first be synthesized to accommodate this chemistry.8 Thus, free paclitaxel or docetaxel is not released immediately after the hydrazone bond cleaves. Many of these other linkers were too stable in vivo and did not release the active therapeutic at high concentrations.6,11 Insufficient prodrug conversion may hinder therapeutic efficacy because the active compound never reaches therapeutically relevant concentrations. Ester prodrugs, though more common in usage for prodrugs, do not offer fast conversion within the tumor microenvironment.9 Amino acid linkers were found to cleave more rapidly at higher pH values, but the tumor microenvironment is thought to be acidic.10,11 Thus, a linker with triggered release at basic conditions may not be preferable for chemotherapeutic delivery. Utilization of the endosomal enzyme cathepsin B has been used with success for targeted delivery systems12 but has yielded mixed results for non targeted polymer drug conjugates. Retrospective analysis of phase III clinical trial results of a cathepsin B sensitive polymer drug conjugate suggested that different levels of cathepsin B activity may lead to differences in survival.13 For this work, we selected a prodrug chemistry with established tunability to ensure active drug release at the target site.14 There are no known enzymes that degrade silyl ethers, thus the use of this chemistry may ensure that the drug is only released within more acidic environments. Chlorosilanes with different combinations of steric bulk and alkyl chain lengths are commercially available to synthesize prodrugs with varying rates of hydrolysis, water solubility, and affinity to the particle matrix in order to control drug release rates. Alkyl silyl ether prodrugs of docetaxel were synthesized for incorporation into poly(lactide-co-glycolide) nanoparticles prepared by the particle replication in nonwetting templates (PRINT) process. The increased lipophilicity of the prodrug improved drug retention in the particles, which delayed the release of the prodrug until the particles passively targeted the solid tumor.
Prodrug Synthesis and Particle Fabrication
The silyl ether docetaxel prodrugs, C2 (ethyldimethylsilyl ether docetaxel) and C8 (octyldimethylsilyl ether docetaxel), were prepared by a single step reaction of docetaxel with chlorodimethyle-thylsilane or chloro(dimethyl) octylsilane, respectfully (Scheme 1). It has been well documented that the C2’ alcohol of taxanes preferentially react with electrophiles, such as acid chlorides and anhydrides.15,16 As expected, the C2’ monosubstituted silyl ether prodrugs of docetaxel formed and were isolated in good yield.
Scheme 1.
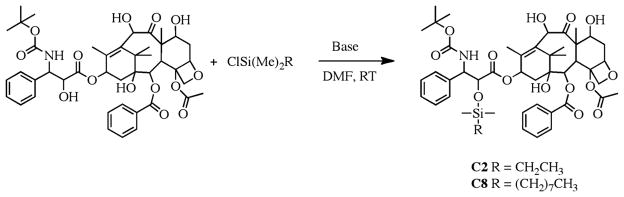
Synthesis of Alkyl Silyl Ether Docetaxel Prodrugs
The rate of conversion of the prodrug to docetaxel is the consequence of simple hydrolysis and can be tuned by altering the substituents on the silicon atom. To achieve rapid prodrug hydrolysis upon release from the NP, alkyl dimethyl silyl chlorides were selected (Figure 1A,B). Hydrolysis of the prodrugs was evaluated in aqueous solutions at different pH conditions (pH 5 and pH 7) and 37 °C in the presence of human serum albumin and measured by LC-MS/MS. At pH 5, the C2 prodrug was quickly degraded; full conversion was achieved at 6 h (Figure 1A), where as the C8 prodrug was not fully converted until 24 h. The conversion of the prodrugs to docetaxel was also studied in mouse plasma at physiological conditions. The t1/2 of C2 and C8 in mouse plasma were similar, 8 h for the C2 prodrug and 10 h for the C8 prodrug. The majority of the C2 and C8 prodrugs are converted within the first 24 h. The toxicity of C2 and C8 were compared to free docetaxel in vitro in A549 cells (Figure 1C). With a 24 h incubation time, the toxicity of the C2 and C8 prodrugs were less than that of free docetaxel. Modification of the C2’ alcohol of taxanes reduces its activity and requires conversion of the prodrug to achieve efficacy.7
Figure 1.
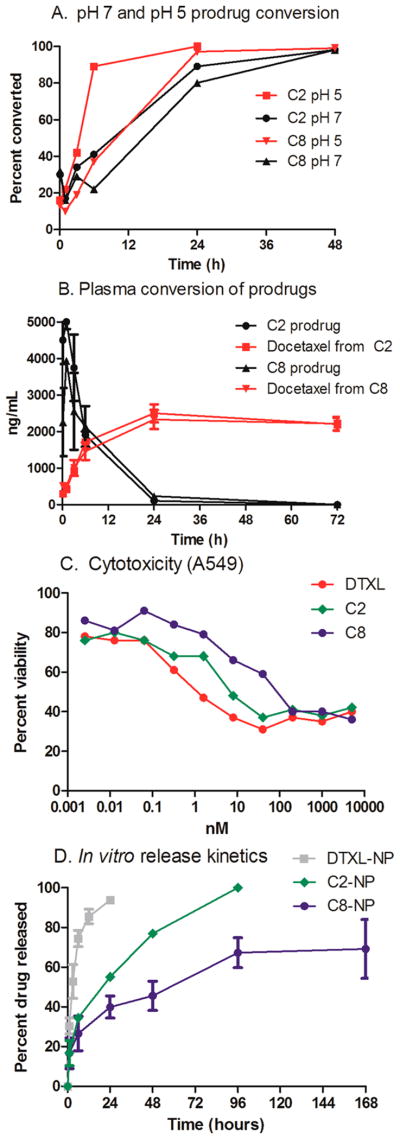
(A) Hydrolysis of prodrugs in pH 5 and pH 7 buffer at 37 °C. (B) Hydrolysis of prodrugs in mouse plasma at physiological conditions. (C) Cytotoxicity of docetaxel, C2 and C8 on A549 cells in vitro. (D) Release kinetics of docetaxel, C2 and C8 from PLGA NPs at pH 7.4 and 37 °C.
Cylindrical particles with diameter (d) = 80 nm and height (h) = 320 nm were prepared using a poly(lactide-co-glycolide) polymer. By dynamic light scattering (DLS), the hydrodynamic radius was measured as ~200 nm. The particle samples were monodisperse with a polydispersity index (PDI) of less than 0.1 and as low as 0.05. PRINT NPs were loaded with drugs at weight percents of 20–22%. NP formulations of docetaxel and two docetaxel prodrugs, C2 and C8, were prepared. All formulations were similar in particle size and drug loading (Table 1). The release kinetics of the three formulations, DTXL-NP (docetaxel nanoparticle), C2-NP, and C8-NP, were evaluated at 37 °C in phosphate-buffered saline (Figure 1D). Release kinetics were dependent upon the length of the alkyl chain of the docetaxel prodrug; prodrugs with the longer alkyl chain lengths had slower release from the particles. Unmodified docetaxel was fully released after 24 h where as the C2 prodrug was fully released after 4 days and C8 prodrug was not fully released after 7 days.
Table 1.
NP Characterization of DTXL-NP, C2-NP and C8-NP Formulations
| formulation | size (nm) | PDI | zeta potential (mV) | weight percent loading |
|---|---|---|---|---|
| DTXL-NP | 213 ± 1 | 0.07 ± 0.01 | −2.81 ± 0.23 | 21.2 ± 0.5 |
| C2-NP | 205 ± 2 | 0.05 ± 0.02 | −2.65 ± 0.52 | 20.7 ± 0.4 |
| C8-NP | 208 ± 7 | 0.09 ± 0.02 | −3.78 ± 0.36 | 22.3 ± 1.9 |
In Vivo Pharmacokinetics of Prodrug NPs
The pharmacokinetic parameters and profiles of DTXL-NP and C2-NP, the two faster-releasing formulations, compared to free docetaxel at equal-molar dosing are shown in Figure 2 and Table 2. Mice with A549 flank tumors were administered one dose at 10 mg/kg docetaxel via vein injection and subsequent drug concentrations were measured in plasma, tumor and tissue by LC-MS/MS. All NP formulations had greater sum total plasma exposures (encapsulated docetaxel or prodrug + released docetaxel) compared to free docetaxel when measured by the area under the concentration − time curve (AUC). A 60-fold increase in AUC was realized for the DTXL-NP where as the C2-NP formulations displayed a 182-fold in AUC. The improved sum total plasma AUC of the C2-NP relative to the DTXL-NP may be attributed to the slower release kinetics of the C2 prodrug. The improved retention of the C2 prodrug in the NPs most likely reduced the tissue distribution of C2 as evidenced by the reduced Vd of sum total C2 compared to sum total docetaxel (237 vs 4513 mL/kg). Comparison of the sum total Cmax also suggests that improved NP retention of drug reduces immediate tissue distribution. The order of Cmax from least to highest for the NP formulations emulated the in vitro release kinetics, docetaxel and C2 (23 359 ± 4528 vs 78 952 ± 6589, P = 0.0002). Furthermore, minimal conversion of C2 docetaxel was observed in plasma of the total drug (Figure 1A). The percentage of released docetaxel from C2-NP was 2.3%. This percentage was calculated by AUCC2/(AUCC2+AUCDTXL) × 100.
Figure 2.

Pharmacokinetic profiles of the following: red circle, free docetaxel; gray square, DTXL-NP; and green diamond, C2-NP formulations (dotted line indicates C2 prodrug and solid line indicates converted DTXL). Each replicate is shown and lines are connected by the means of three replicates at each time point. Mice bearing A549 flank tumors received one iv injection via tail vein at 10 mg/kg docetaxel or docetaxel molar equivalents.
Table 2.
Pharmacokinetic Parameters of Free Docetaxel, DTXL-NP, and C2-NP Formulations
| formulation
|
|||||
|---|---|---|---|---|---|
| taxotere
|
DTXL-NP
|
C2-NP
|
|||
| specimen | parameter | DTXL | DTXL | C2 | DTXL |
| plasma | AUC (ng/mL h) | 1227 | 79 192 | 227 735 | 5381 |
| Cmax (ng/mL) | 2314 ± 362 | 23 359 ± 4 528 | 78 952 ± 6 589 | 1994 ± 107 | |
| CL (mL/h/kg) | 8150 | 126 | 49 | 1858 | |
| Vd (mL/kg) | 10 508 | 4513 | 237 | 9535 | |
| tumor | AUC (ng/mL h) | 73 222 | 60 858 | 26 799 | 12 897 |
| Cmax (ng/mL) | 453 ± 225 | 476 ± 73 | 946 ± 743 | 288 ± 139 | |
In vitro, the C2 had a short conversion half-life of 8 h; the NP likely protects the prodrug in plasma to prevent conversion before the prodrug reaches the target site of the tumor. This design attribute has the potential to decrease systemic toxicity of chemotherapeutics.
Though low C2 plasma conversion is preferred, the prodrug must convert at its target site to be effective. 32.5% of C2 was determined to be converted to docetaxel in the tumor, much higher than the 2.3% observed within the plasma. This percentage compares favorably to other polymeric prodrug strategies that have entered clinical development. For a camptothecin polymer drug conjugate, only 1.3% unconjugated camptothecin was observed in xenograft tumors over 48 h.11 Only 4–17% of unconjugated paclitaxel from a paclitaxel polymer drug conjugate was observed in a xenograft tumors over 144 h.6 In other tissues where the NPs distribute, the liver and spleen (Supporting Information Figure 1 and Table 1) only had 13.2 and 2.7% free docetaxel from C2. The silyl ether prodrug has selective conversion at the target site of the tumor. Silyl ethers are commonly used as protecting groups that are acid-labile.17 The hypothesized acidic tumor microenvironment is thought to contribute to the site selective conversion.
In Vivo Efficacy and Tolerability
The efficacy of the C2-NP formulation was compared to free docetaxel in an A549 subcutaneous xenograft mouse model. Mice were administered weekly doses via tail vein injection over 6 weeks. Figure 3A shows the tumor growth curves for free docetaxel at its MTD18(20 mg/kg), C2-NP at 20 mg/kg, and C2-NP at its MTD of 50 mg/kg. The final relative tumor volume of mice receiving C2-NP was equal to free docetaxel when administered at 20 mg/kg. However, at 50 mg/kg, mice receiving the C2-NP had statistically lower mean relative tumor volume than mice receiving free docetaxel at 20 mg/kg starting at day 10. Body weights (Figure 3B) of mice receiving saline and equimolar doses of free docetaxel and C2-NP remained stable over the course of treatment, while some body weight loss was observed in mice receiving the 50 mg/kg dose of C2-NP over the course of 6 doses. On the basis of the pharmacokinetic profile of the tumor, mice receiving C2-NP at equal molar dosing to free docetaxel did not have increased sum total docetaxel concentrations (Figure 1B). The similar tumor docetaxel levels may explain the similar tumor growth rates for mice receiving C2-NP and free docetaxel at equal molar doses.
Figure 3.
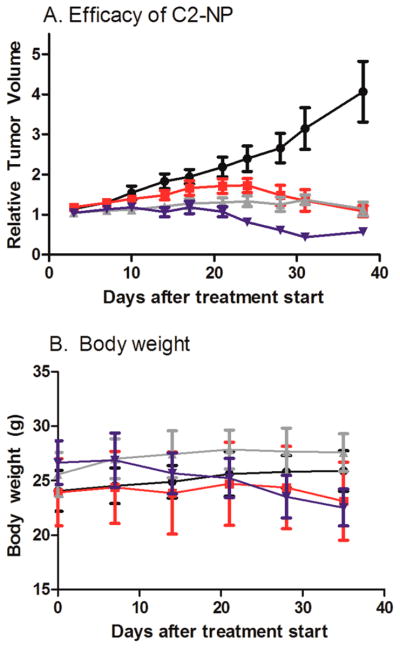
(A) Tumor growth inhibition curve (mean ± sem). Mice bearing A549 flank tumors received 6 weekly doses via IV tail vein injection. Black circle, saline; red square, free docetaxel (20 mg/kg); gray triangle up, C2-NP (20 mg/kg dtxl equivalents); and blue triangle down, C2 NP (50 mg/kg dtxl equivalents) Mice receiving C2 NP at 50 mg/kg had tumor volumes lower than mice receiving free docetaxel (P < 0.05 starting on day 10). (B) Body weights (mean ± std dev). Twenty mg/kg weekly × 6 is the maximum tolerated dose for docetaxel in this model.18
However, the C2-NP formulation improved the tolerability of docetaxel in mice. Neutropenia, characterized by reduced white blood cell counts (WBC), is a common side effect of docetaxel (Taxotere). Patients that experience dose limiting toxicity due to neutropenia may benefit from different dosing schedules, such as receiving doses every 2 weeks versus 3 weeks, but this may be inconvenient to the patient.19 Thus, a formulation change to improve the toxicity profile of docetaxel is an attractive option. Table 3 shows the mean white blood cell counts of mice receiving the C2-NP (20 mg/kg or 50 mg/kg) compared to free docetaxel (20 mg/kg). Blood was collected 4 days after injection and measured for complete blood counts. Mice receiving the C2-NP formulation at an equal molar dose to free docetaxel trended toward statistically higher WBC 4 days after the first dose (P = 0.12) and almost double the WBC 4 days after the sixth dose (P = 0.008). The pharmacokinetic data demonstrates that only a fraction of C2 is converted to docetaxel within the plasma, liver and spleen. The minimized free docetaxel concentrations likely account for the improved tolerability of the C2-NP. With improved tolerability, mice could receive a 2.5 times higher docetaxel dose with the C2-NP than free docetaxel, resulting tumor reduction in the efficacy study.
Table 3.
White Blood Cell Counts Measured 4 Days after Injection with Saline, Free Docetaxel or C2-NP at Two Dose Levels
| time point | saline | free docetaxel (20 mg/kg) | C2-NP (20 mg/kg) | C2-NP (50 mg/kg) |
|---|---|---|---|---|
| WBC (103 cells/μL) | ||||
| day of dose 1 | 2.07 ± 0.21 | 1.34 ± 0.36 | 1.67 ± 0.54 | 1.54 ± 0.35 |
| 4 days after dose 1 | 2.81 ± 0.91 | c0.84 ± 0.65 | a,c1.41 ± 0.65 | c0.49 ± 0.38 |
| 4 days after dose 6 | 3.43 ± 1.13 | c1.53 ± 0.52 | b3.00 ± 1.10 | c0.56 ± 0.28 |
Indicates trend toward statistically significance compared to free docetaxel (P = 0.12).
Indicates statistical significance compared to free docetaxel (P = 0.008).
Indicates statistical significance compared to saline (P < 0.006).
Compared to poly(lactide) docetaxel NPs in clinical development, release of docetaxel directly from NPs has not shown improvement in tolerability relative to the clinical control Taxotere.20,21 Nanoxel-PM is a micelle formulation of docetaxel, consisting of PDLLA-mPEG. In preclinical models, Nanoxel-PM had similar pharmacokinetics to Taxotere and a similar hematological toxicity profile.20 Instability of the micelle may be a potential reason why an improved toxicity profile was not observed. However, BIND-014 is a stable targeted PLA-PEG nanoparticle with controlled release of docetaxel and differentiated pharmacokinetics and efficacy.21 Even with these improved attributes, BIND-014 did not achieve a higher maximum tolerated dose than Taxotere in a phase I clinical trial.21 Thus, unlike reformulations of paclitaxel, where removing cremophor EL as an excipient contributed to increased tolerability,22 there has yet to be a formulation change that improves the hematological toxicity of docetaxel in a clinical setting. Our prior data that evaluated PLGA docetaxel particles in vivo also did not improve the hematological toxicity of docetaxel compared to Taxotere, even though the maximum tolerated dose was increased by 50%.18 Thus, the use of a prodrug strategy appears to be key in improving the toxicity profile of docetaxel by reducing the amount of docetaxel in systemic circulation.
We have demonstrated that combining a labile prodrug of docetaxel with NP delivery improves the tolerability of docetaxel without decreasing efficacy by minimizing systemic conversion of the prodrug but preferential converting within the tumor. Further work is being conducted to more thoroughly identify the appropriate dose level to balance efficacy and toxicity.
Supplementary Material
Acknowledgments
Funding
Joseph DeSimone is a founder and maintains a financial interest in Liquidia Technologies.
The authors acknowledge Charlene Ross of the Animal Studies Core at the University of North Carolina at Chapel Hill. This work was supported by the Carolina Center for Nano-technology Excellence (U54-CA151652 and U54-CA119343), University Cancer Research Fund, and Liquidia Technologies, Inc.
ABBREVIATIONS
- C2
ethyldimethylsilyl ether docetaxel
- C8
octyldimethylsilyl ether docetaxel
- DTXL-NP
docetaxel nanoparticle
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare the following competing financial interest(s): Joseph DeSimone is a founder and maintains a financial interest in Liquidia Technologies. PRINT is a registered trademark of Liquidia Technologies, Inc.
Liver and spleen pharmacokinetics. Additional hematological data. Materials and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Myer-Losic F, Nicolazzi C, Quinonero J, Ribes F, Michel M, Dubois V, de Coupade C, Boukaissi M, Chéné AS, Tranchant I, Arranz V, Zoubaa I, Fruchart JS, Ravel D, Kearsey J. Clin Cancer Res. 2008;14:2145–2153. doi: 10.1158/1078-0432.CCR-07-4580. [DOI] [PubMed] [Google Scholar]
- 2.Gupta D, Varghese Gupta S, Dahan A, Tsume Y, Hilfinger J, Lee KD, Amidon GL. Mol Pharm. 2013;10:512–522. doi: 10.1021/mp300564v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee Y, Kim H, Kim W, Yoon JH, Jeong SH, Jung Y. J Drug Target. 2012;20:524–534. doi: 10.3109/1061186X.2012.693498. [DOI] [PubMed] [Google Scholar]
- 4.Chu KS, Hasan W, Rawal S, Walsh MD, Enlow EM, Luft JC, Bridges AS, Kuijer JL, Napier ME, Zamboni WC, DeSimone JM. Nanomedicine. 2013;9:686–693. doi: 10.1016/j.nano.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang L, Fan TM, Borst LB, Cheng J. ACS Nano. 2012;6:3954–3966. doi: 10.1021/nn300149c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li C, Newman RA, Wu QP, Ke S, Chen W, Hutto T, Kan Z, Brannan MD, Charnsangavej C, Wallace S. Cancer Chemother Pharmacol. 2000;46:416–422. doi: 10.1007/s002800000168. [DOI] [PubMed] [Google Scholar]
- 7.Feng L, Wu H, Ma P, Mumper RJ, Benhabbour SR. Int J Nanomed. 2011;6:2545–2556. doi: 10.2147/IJN.S24954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etrych T, Sirova M, Starovoytova L, Rihova B, Ulbrich K. Mol Pharm. 2010;7:1015–1026. doi: 10.1021/mp100119f. [DOI] [PubMed] [Google Scholar]
- 9.Ma P, Benhabbour SR, Feng L, Mumer RJ. Cancer Lett. 2013;334:253–262. doi: 10.1016/j.canlet.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C, Greenberger LM, Horak ID. Bioconjugate Chem. 2008;19:849–859. doi: 10.1021/bc700333s. [DOI] [PubMed] [Google Scholar]
- 11.Schluep T, Cheng J, Khin KT, Davis ME. Cancer Chemother Pharmacol. 2006;57:654–662. doi: 10.1007/s00280-005-0091-7. [DOI] [PubMed] [Google Scholar]
- 12.Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, Ramchandren R, Bartlett NL, et al. J Clin Oncol. 2012;30:2183–2189. doi: 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chipman SD, Oldham FB, Pezzoni G, Singer JW. Int J Nanomed. 2006;1:375–383. doi: 10.2147/nano.2006.1.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parrott MC, Finniss M, Luft JC, Pandya A, Gullapalli A, Napier ME, DeSimone JM. J Am Chem Soc. 2012;134:7978–7982. doi: 10.1021/ja301710z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ali S, Ahmad I, Peters A, Masters G, Minchey S, Janoff A, Mayhew E. Anticancer Drugs. 2001;12:117–128. doi: 10.1097/00001813-200102000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Huynh L, Leroux JC, Allen C. Org Biomol Chem. 2009;7:3437–3446. doi: 10.1039/b906862g. [DOI] [PubMed] [Google Scholar]
- 17.Wuts PGM, Greene TW. Greene’s Protective Groups in Organic Synthesis. 4. Wiley; New York: 2007. [Google Scholar]
- 18.Chu KS, Schorzman AN, Finniss MC, Bowerman CJ, Peng L, Luft JC, Madden AJ, Wang AZ, et al. Biomaterials. 2013;34:8424–8429. doi: 10.1016/j.biomaterials.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kellokumpu-Lehtinen PL, Harmenberg U, Joensuu T, McDermott R, Hervonen P, Ginman C, Luukkaa M, Nyandoto P, et al. Lancet Oncol. 2013;14:117–124. doi: 10.1016/S1470-2045(12)70537-5. [DOI] [PubMed] [Google Scholar]
- 20.Lee SW, Yun MH, Jeong SW, In CH, Kim JY, Seo MH, Pai CM, Kim SO. J Controlled Release. 2011;155:262–271. doi: 10.1016/j.jconrel.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 21.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, et al. Sci Transl Med. 2012;4:128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 22.Gradishar WJ, Tiulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.