

Abstract
Particle replication in nonwetting templates (PRINT) is a continuous, roll-to-roll, high-resolution molding technology which allows the design and synthesis of precisely defined micro- and nanoparticles. This technology adapts the lithographic techniques from the microelectronics industry and marries these with the roll-to-roll processes from the photographic film industry to enable researchers to have unprecedented control over particle size, shape, chemical composition, cargo, modulus, and surface properties. In addition, PRINT is a GMP-compliant (GMP = good manufacturing practice) platform amenable for particle fabrication on a large scale. Herein, we describe some of our most recent work involving the PRINT technology for application in the biomedical and material sciences.
Keywords: colloids, drug delivery, nanomedicine, nano-particles, soft lithography
1. Introduction
The major approaches to fabricate colloidal particles can be divided into two categories: bottom-up and top-down. Bottom-up methods rely upon weak and noncovalent interactions between molecules to self-assemble into particles such as micelles,[1] vesicles,[2] liposomes,[3] and polymersomes.[4] The bottom-up methods have been intensively explored in the field of nano-medicine and have achieved significant success for drug and gene delivery. For example, approximately 12 liposome-based drugs have been approved for clinical use with more at different stages of clinical trials.[5] Despite many advantages for the bottom-up methods, limitations such as formulation instability, large size distribution, and limited particle shape still exist.[6] In contrast to bottom-up methods, top-down methods process macroscopic materials to create particles of micron-or nanoscale sizes. Milling, grinding, and emulsion are examples of top-down fabrication methods.[7] However, these techniques also have limited control over particles size and shape. Photolithography,[8] microfludic synthesis,[9] and molding technology[10] represent the more advanced forms of top-down fabrication techniques and have been used to fabricate particles for various applications. Herein we highlight the particle replication in nonwetting templates (PRINT) technology, a unique soft lithography-based molding technology which allows the engineering of precisely defined particles.
The PRINT technology was conceived after the development of a novel class of fluoropolymers, photocurable perfluoropolyethers (PFPE). The photocurable fluoropoly-mers were created to address the issue of polydimethylsiloxane (PDMS) microfluidic channel swelling by certain organic solvents and the challenges associated with molding of features from low-molar-mass curable compounds which would tend to swell PDMS-based molds. PFPEs have excellent resistance to swelling by low-molecular-weight organic molecules,[11] and enables the fabrication of less than 100 nm high-resolution features with high fidelity.[12] Of particular interest are the unique attributes of these fluoropolymers, including an extremely low surface energy, low modulus, high gas permeability, and low toxicity, which made the system amenable to the creation of isolated micro- and nanosized particles.[10c] Based upon this discovery, PRINT process was developed for the fabrication of monodisperse particles. Unlike other micro- and nanoparticle fabrication techniques, PRINT allows complete control over the physicochemical characteristics of the particle. This platform technology is demonstrating the potential to impact both the biomedical and material sciences for applications in drug delivery, electronics, optics, sensing, and imaging.[13]
2. The PRINT Process
A typical PRINT process is schematically illustrated in Figure 1a. Silicon wafers patterned with different features (grey) are fabricated using standard photolithographic techniques and are used as master templates in the PRINT process. In the past few years, the PRINT technology has evolved from small batch work on the lab bench to an automated continuous roll-to-roll system, which allows for particle production suitable for pre-clinical studies and clinical trials (Figure 1b).
Figure 1.

a) Preparation of PRINT mold and fabrication of PRINT particles. A liquid PFPE precursor can completely wet the silicon wafer with micro- and nanosized patterns because of its positive spreading coefficient on almost all surfaces and can be photocured to generate an elastomeric PRINT mold with negative micro/nanofeatures derived from the patterns on the silicon wafer. A liquid pre-particle material (red) is filled into the cavities without wetting the land area surrounding the cavities using a roll-to-roll process which involves a film-split technique against a high-surface-energy polyethylene terephthalate (PET) counter sheet when passed through a nip of a roller which applies pressure to the mold. The liquid in the mold cavities is then converted into a solid through a number of different processes including photocuring, or perhaps through vitrification by filling at an elevated temperature and cooling down, or by solvent evaporation. Once the liquid in the mold cavities is solidified, the array of particles (red) can be removed from the mold by bringing the mold in contact with an adhesive layer (yellow) which can pull the particles from the low-surface-energy mold. The particles can now be easily freed from the surface by dissolving the adhesive layer. b) Technical innovations in the PRINT technology: A) a roll of low-cost thin mold for PRINT particle fabrication and B) a roll-to-roll equipment for continuous PRINT particle manufacturing. Adapted from Ref. [14] with permission from Future Medicine Ltd.
The PRINT process enables the design and synthesis of well-defined micro- and nanoparticles without the challenges that plague other particle fabrication strategies including molecular self-assembly, emulsification, precipitation, and coacervation techniques.[14,15] To date, precise template fabrication has been achieved in the electronics industry as a result of the advancements in standard lithography, and the PRINT process has taken full advantage of these advancements to make uniform particles of specific shapes and sizes down to 50 nm. The composition of particles fabricated from the PRINT process can be readily tuned, and particles have been made by PRINT from materials which include: 1) hydrogels such as cross-linked poly(ethylene glycol)s (PEG) and poly(silyl ether)s;[10c,16] 2) thermoplastic polymers such as poly(lactic acid) (PLLA) and poly(lactic-co-glycolic acid) (PLGA);[10c,17] 3) biologics such as insulin and albumin, and pure small-molecule compounds including sugars and small molecular drugs.[18] Multiphasic and regiospecifically functionalized particles can also be fabricated using PRINT (Figure 2). Our group has developed a strategy to prepare end-labeled particles, biphasic Janus particles, and multiphasic shape-specific particles by integrating two compositionally different chemistries into a single particle.[19] In addition, the surface properties of the PRINT particles can be easily modified, and the porosity, texture, and modulus of the particles can be altered through careful design of matrix formulation. This review will cover the most recent progress of PRINT technology in the life sciences and material sciences, and directions for future research.
Figure 2.

Scanning-electron and light micrographs showcasing the ability of the PRINT process to synthesize particles of different shapes, sizes, and compositions. (A)[20], (B)[21], (C)[22], (D)[16b], (E)[23], (F)[24], (G)[25], (H)[26], and (I)[19] reprinted with permissions.
3. Life Sciences
Over the past few decades, there has been a large focus on the design and synthesis of micro- or nanoscale particle-based drug carriers to address the problems associated with conventional forms of drugs, such as low efficacy or harmful side effects.[3c,27] Several critical factors, including the size and shape of the particle, particle matrix, surface chemistry, and drug transport through the matrix, must be considered in the rational design of many nanoparticle drug delivery carriers. PRINT has independent control over all aspects of particle design, and the use of this technology is playing a significant role in elucidating the impact of particle design on particle biodistribution and pharmacokinetics,[25,28,29] differential lung deposition, and cellular internalization.[24,30,31] In addition, PRINT particles can carry a diverse array of cargos such as chemotherapeutics, small interfering RNAs (siRNA), RNA replicons, and contrast agents through straightforward incorporation in the liquid pre-particle material.
3.1. Delivery of Small Molecules and Prodrugs
To increase the therapeutic efficacy of highly toxic small-molecule drugs, one approach is to encapsulate the drugs in nanocarriers.[27,32] One such clinically relevant example is Doxil, a liposomal formulation of doxorubicin, which shows less cardiotoxicity compared to doxorubicin.[3b] Nanocarriers have the potential to improve the therapeutic efficacy by decreasing drug exposure to healthy cells, thus increasing drug concentration at diseased tissues and providing sustained drug release.[3c,33] Using the PRINT process, we have evaluated two different particle matrixes for the delivery of docetaxel, camptothecin, desatinib, and gemcitabine: PLGA, which is a lipophilic, biodegradable thermoplastic, and hydrogel materials based on cross-linked PEG.
PLGA has been used to physically entrap and release therapeutics in a sustained manner by a number of different researchers.[7a,34] Docetaxel-loaded PLGA particles have been mainly fabricated using emulsion,[35] microfludics,[36] film rehydration,[37] and ultrasonication techniques.[38] Docetaxel loading in PLGA particles has been reported in the literature to be in the range of 0.5 to 15%, with the drug encapsulation efficiencies ranging from 11 to 95%. Using the PRINT process, up to 40% of docetaxel can be loaded into cylindrical PLGA particles (diameter [d] = 200 nm; height [h] = 200 nm) at 90% drug encapsulation efficiency.[17] The in vitro efficacy of PRINT PLGA particles, containing varying concentrations of docetaxel (0–40%), against the ovarian cancer cell line, SKOV3, was compared to the clinically administered form of docetaxel, Taxotere. The nondrug-loaded particles were shown to be nontoxic, and particles loaded with docetaxel exhibited dose-dependent cytotoxicity. PRINT PLGA particles containing 10 and 20% docetaxel were slightly less toxic than Taxotere, while particles with 30 and 40% docetaxel were more toxic at equivalent doses. The PRINT PLGA particles loaded with 40% docetaxel had an IC50 value almost an order of magnitude greater than Taxotere and 30 times greater than the particles loaded with 10% docetaxel. This study demonstrated that docetaxel can be encapsulated in PRINT PLGA particles at exceptionally high loadings and released in its active form. The results also suggest that higher drug loadings have the potential to increase drug potency at lower doses. Furthermore, in vivo studies demonstrated that docetaxel released from PLGA PRINT particles achieved an elevated plasma exposure and tumor delivery compared to free docetaxel.[39] Future work will focus on optimizing the system for in vivo efficacy to reduce tumor burden.
The biocompatible hydrogel PEG is amenable to the incorporation of prodrugs covalently linked into the PEG matrix.[40] The decrease in pH value at tumor sites and inside tumor cells can be utilized for the release of the drug in its active form at the site of the tumor. The group of DeSimone has developed a new class of prodrug linker chemistry based on bis(dialkyl)silyl ethers, which offers the ability to tune drug release in acidic environments by varying the alkyl substituents on the silicon.[16a,23] Silyl ether functionalized camptothecin, desatinib, and gemcitabine were synthesized and incorporated into PRINT PEG hydrogel particles (Figure 3) by free radical polymerization methods. The release rates of the drug from cylindrical particles (d = 200 nm; h = 200 nm) were found to increase as the size of the substituent on the silicon atom decreased (ethyl > isopropyl > tert-butyl), with the prodrug carrying the largest substituent showing no appreciable degradation over time. Cell viability experiments demonstrated that the particles containing the ethyl- and isopropyl-based prodrug gemcitabine showed toxicity against LNCaP cancer cells, whereas the particles containing the tert-butyl-based prodrug showed the same cytotoxicity as that of blank particles, thereby illustrating the ability to incorporate the drug but not releasing it in a relevant timeframe. The substituents on the silicon atom can be changed to achieve desired drug-release kinetics. In addition, the silyl-ether-based prodrug allows the drug to be released in its original form upon bond cleavage with the generation of nontoxic water soluble polymers.
Figure 3.
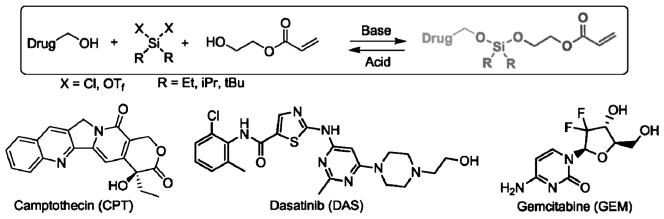
Unsymmetric bifunctional silyl ether (ABS) prodrugs of camptothecin, dasatinib, and gemcitabine. Each ABS prodrug is composed of three parts: 1) a chemotherapeutic, 2) a silyl ether linkage, and 3) a polymerizable monomer for particle incorporation. Adapted from Ref. [23]; copyright 2012 American Chemical Society.
PRINT has enabled a plug-and-play approach for delivery of small molecules, thus allowing us to choose a particle matrix according to the nature of the drug and control drug loading and release. In particular, the use of prodrugs enables a more accurate release of drugs from particles which is triggered by a change in the chemical environment. Investigation of the performance of drug-loaded particles against a series of in vivo tumor models is now underway.
3.2. Protecting and Delivering Biopharmaceuticals
The versatility of the PRINT process has been further demonstrated through the ability to deliver therapeutics based on small interfering RNAs (siRNA) and RNA replicons. Among the RNA therapeutics, siRNA has demonstrated great potential for the treatment of diseases such as breast cancer and prostate cancer by knocking down the gene expression of target proteins, and opens up the possibility of “drugging the undruggable”.[41,42] An RNA replicon can encode therapeutically desirable proteins at high concentrations in the cytosol of cells and thus holds great promise to trigger immune responses against a variety of diseases including influenza and certain cancers.[43] An ideal carrier should be able to protect the RNA, transport the RNA to the site of interest, achieve internalization by the target cells, and release the RNA to the cytosol with minimal toxicity. Using the PRINT process, we have established a siRNA-PLGA-lipid nanoparticle system and a prodrug siRNA-PEG nano-particle system for delivery of siRNA, and a disulfide cross-linked bovine serum albumin (BSA) particle system for delivery of RNA replicon.
The vast majority of the strategies reported in the literature to deliver siRNA involve the formation of polyplexes. Through a melt-solidification methodology, 1–2 wt% of siRNA was incorporated into cylindrical PLGA particles (d = 80 nm; h = 320 nm) with an encapsulation efficiency between 20% to 40%.[21] The particles were harvested from the adhesive layer using a solution containing a mixture of cationic lipids (DOTAP/DOPE = 50:50 wt/wt; DOTAP = 1,2-dioleoyl-3-trimethylammoniumpropane, DOPE = dioleoylphosphatidylethanolamine). Surface modification with cationic lipid facilitates cell internalization and transfection. Coating the particles with the cationic lipids increased particle zeta potentials (−3.45 ±1.9 mV for non-lipid coated versus +5.29 ±1.5 mV for lipid coated), and lipid-coated particles containing dye-labeled siRNA were shown to be readily internalized by cell lines including HeLa/luc, PC3, DU145, and Raw264.7. LNCaP and HepG2 cells revealed a lower efficiency in particle uptake, likely as a result of the long doubling time of these cell lines. Luciferase siRNA knockdown studies demonstrated that the lipid-coated PLGA particles were able to deliver luciferase siRNA and knockdown luciferase expression effectively in HeLa/luc cells with minimal toxicity. Further studies revealed that the siRNA-PLGA-lipid particles were able to deliver a therapeutically relevant KIF11 siRNA to three prostate cancer cell lines (LNCaP, PC3, and DU145). Quantitative real-time PCR revealed a statistically significant reduction in the KIF11 mRNA level in all three prostate cancer cells lines dosed with KIF11-siRNA-PLGA particles, whereas the control siRNA-loaded particles did not show any decrease in the KIF11 mRNA level.
There are very few reported examples of hydrogel-based particles for delivery of siRNA. All of the examples were limited to emulsion fabrication techniques and base-pairing between complimentary siRNA strands.[44] The PRINT process allows direct physical entrapment of siRNA in the PEG hydrogel particles.[22] We were able to achieve significant siRNA knockdown with monodisperse, cylindrical PEG particles (d = 200 nm; h = 200 nm) fabricated with siRNA electrostatically entrapped. However, subsequent efforts to modulate in vivo behavior by conjugation of targeting ligands to the particle surface resulted in extensive premature release of siRNA.
In an effort to reduce premature release of siRNA during the conjugation of targeting ligands, the siRNA was polymerized into the hydrogel matrix using a prodrug strategy. The siRNA was derivatized with a photopolymerizable acrylate bearing a degradable disulfide linkage for reversible covalent incorporation into the PRINT hydrogel nanoparticles (Figure 4). The time-dependent release of the siRNA from the prodrug siRNA PRINT hydrogel particles was evaluated under physiological and reducing conditions, thus revealing that siRNA was retained in the hydrogel particles over 48 h at 37°C in PBS. However, the siRNA was quickly released from the prodrug siRNA hydrogel nanoparticles when incubated in a reducing environment (5 mM glutathione). In addition, prodrug siRNA loaded into PRINT hydrogel particles was protected from degradation by RNases when incubated in serum for over 48 h. Dose-dependent silencing of luciferase expression in HeLa/luc cells was effectively elicited by the prodrug-siRNA-containing hydrogel particles, while the control particles did not elicit significant gene knockdown. The transfection efficiency between siRNA-complexed, PEGylated particles and prodrug siRNA hydrogels was comparable.
Figure 4.

a) Structures of degradable and nondegradable siRNA macromers as well as native siRNA. b) Illustration of pro-siRNA hydrogel behavior under physiological and intracellular conditions. Adapted from Ref. [22]; copyright 2012 American Chemical Society.
The predominant technique for RNA replicon delivery involves viruslike particles (VLP).[45] A nonviral delivery system would be advantageous for controlling size, shape, and surface chemistry. The delivery of RNA replicon, a high-molecular-weight (8–10 kb), single-stranded RNA, was achieved using a BSA matrix.[20] Protein matrices for drug delivery are useful because of their biodegradability, biocompatibility, and amenability to surface modification. A melt-solidification strategy was employed in the fabrication of cylindrical protein particles (d = 1 μm; h = 1 μm) using lactose and glycerol as processing aids. The BSA particles were cross-linked with reductively labile dithiobis(ethyl-1H-imid-azole-1-carboxylate) (DIC), which rendered the protein-based particles transiently insoluble in aqueous solutions until their introduction to a reducing environment. To demonstrate the capability of the particles to deliver biological cargos in a reducing cellular environment, an RNA replicon encoding for the chloramphenicol acetyl transferase (CAT) protein was chosen. Successful delivery of this RNA into the cytosol of Vero cells would yield expression of CAT protein, which can be detected by an enzyme-linked immunosorbent assay (ELISA) using antigen-specific antibodies. TransIT-mRNA transfection reagent (TransIT) was mixed with BSA particles to introduce a positive surface charge on the BSA particle to enhance cell uptake and endosomal escape. The BSA particles coated with TransIT were readily internalized, and the CAT protein was generated (Figure 5). CAT protein production by delivery of PRINT particles was comparable to CAT protein produced using the transfection agent, TransIT.
Figure 5.

CAT protein generated from Vero cells (2 × 104 cells per well) measured by ELISA. Dotted black: CAT RNA standards with TransIT; dotted gray: blank particles with TransIT; striped: CAT RNA containing BSA particles with TransIT (2, 1, and 0.5 μgmL−1 of particles correspond to 30, 15, and 7.5 ng mL−1 of RNA, respectively); checkered: supernatant from particles incubated in PBS for 4 h at 37 °C with TransIT. Adapted from Ref. [20]; copyright 2012 American Chemical Society.
In addition to incorporation of cargo inside the PRINT particles, biologics can also be protected and delivered by absorption on the particle surface. It has long been known that antigens delivered in particulate form increase antibody titers compared to soluble antigens. Enhanced presentation of vaccine antigens to the immune system was achieved by a PRINT particle-based system in which a commercial trivalent injectable influenza vaccine was electrostatically bound to the surface of cationic PLGA particles.[46]
These results have demonstrated that PRINT particles are capable of protecting and delivering the delicate biopharmaceutical cargo. The cargo can be incorporated in a prodrug form, either physically encapsulated or absorbed, depending on the nature of the cargo and the chemical composition of the particles. The next step is to further optimize the particle design for successful RNA delivery in vivo and antigen delivery for vaccine development.
4. Materials Science
In addition to its wide array of applications in the life sciences, the PRINT technology also has significant applicability in material sciences, where it has contributed to the advancement of microlens and solar cell research.[47,48] An area of particular interest is the fabrication of micro- and nanoparticles for studying colloidal assembly. Similar to larger analogues of atoms building up to form molecules, polymers, and crystals, colloidal particles can be arranged into larger structures, such as clusters and chains when the short-and long-range inter-particle interactions are well balanced.[49] This has great implications for understanding atomic-level bonding and is increasingly useful in the development of catalysts, optics, plasmonic devices, and electronics.[50] Unsymmetric and anisotropic particles have garnered much attention in the field of photonics, where phases beyond that of simple symmetry, such as face-centered cubic, hexagonal close-packed, and body-centered cubic, are desired.[51,52] While the fabrication and self-assembly of inorganic anisotropic particles is a well-researched field, the assembly of organic polymeric particles is relatively unexplored. This may be due to the lack of effective and scalable fabrication processes that satisfy the particle monodispersity and shape-control requirements needed for in-depth studies. With the development of PRINT, however, these limitations can be resolved, thus providing the exciting opportunity to study the assembly of multifunctional uniquely shaped particles.
4.1. Assembly by Electric and Magnetic Fields
Dielectrophoresis (DEP), the interaction of liquids or particles with nonuniform alternating electric fields, is a powerful process which allows alignment of particles in situ through directed self-assembly.[53] While most examples of electrophoretic assembly of monodisperse colloidal systems have used spherical particles, the electrophoretic assembly of nonspherical particles has recently garnered attention for the ability to enable alignment in directions which may improve mechanical and electrical performance.[54,55]
Since the PRINT technology allows the preparation of anisotropic particles with a wide range of shapes and sizes, we became interested in using this ability to study the assembly of various organic particles.[56] Thus, rod-, disc-, hexnut-, and boomerang-shaped particles composed of trimethylolpropane ethoxylate triacylate were prepared and suspended in an aqueous solution containing hexadecyltrimethylammonium bromide (CTAB). Upon the application of a nonuniform alternating electric field across two planar electrodes, the particles were polarized and directed towards the region with the highest field intensity. In this process, the particles reoriented and exhibited alignment with their longest axis parallel to the applied field (Figure 6a). Furthermore, particle chaining was observed for all particle shapes with the exception of the boomerang particles. Using the PRINT technology, the study of colloidal assemblies can be made facile using organic particles with unique geometries.
Figure 6.

a) Representative fluorescence images of randomly dispersed (A, C, E, G) and (B, D, F, H) electrically aligned particles. Adapted from Ref. [56]; copyright 2008 American Chemical Society. b) Representative fluorescence images of aligned magnetite–polymer composite particles: A) block-shaped composite particles in the absence of a magnetic field, B) particles without linear magnetite aggregates in an applied magnetic field formed disordered chains, C) particles with linear magnetite aggregates parallel to and D) perpendicular to the length of the composite particle formed somewhat organized chains stacking head-to-tail and side-to-side, respectively. Cartoon insets provided for clarity, and scale bars are 20 microns. Arrows indicate direction of applied magnetic field. Adapted from Ref. [57]; copyright 2010 American Chemical Society.
Building upon this work, we have also successfully fabricated magneto-polymer composite particles by incorporating superparamagnetic and ferromagnetic magnetite nano-particles into PEG hydrogel particles.[57] Particles composed of Fe3O4 nanoparticles embedded in a cross-linked PEG network were prepared in nanorice, nanoworm, micron-sized block, and micron-sized boomerang shapes. The PRINT technology uniquely enables the ability to manipulate the alignment of the Fe3O4 nanoparticles within these particles. Prior to polymerization of the PEG monomer in the fabrication step, the magnetite can be permanently aligned in different directions relative to the composite particle axis, which plays a significant role in dictating particle dynamics under magnetic fields.
Block particles loaded with Fe3O4 nanoparticles were dispersed in water at high concentrations, and based upon the direction of the long axis of the Fe3O4 nanoparticles in the matrix, the chaining of long block particles under an applied magnetic field was observed in three distinct manifestations (Figure 6b). Samples with magnetite aggregates aligned with the long axis of the particle formed chains with all of the particles in line with the applied magnetic field. Samples with the linear Fe3O4 aggregates aligned along the short axis of the particle formed chains of stacked composite particles. Lastly, samples with unaligned magnetite showed chaining with no specific orientation of the composite particles. The PRINT process has allowed us to report the first example of shape-and size-specific magneto-polymer composite nanoparticles fabricated using a top-down method.
4.2. Assembly by van der Waals Interactions
It has been previously reported that diblock and triblock particles can be synthesized through various methods.[58] However, PRINT technology exhibits more flexibility in controlling diblock and triblock particle composition, size, and shape. The synthesis of anisotropic amphiphilic rods from organic materials with tunable multiphases has been achieved using this process.[26] Consisting of a hydrophobic and hydrophilic component of trimethylolpropane ethoxylate triacrylate and poly(ethylene glycol) diacrylate, respectively, amphiphilic rod diblock, triblock, and multiblock particles can be prepared (Figure 7). It is important to note that the hydrophilic/hydrophobic ratio in the triblock and diblock structures can be precisely tuned by changing the concentration of the initial monomer solution.
Figure 7.

a) Schematic illustration of the formation of triblock rods and the corresponding microscopic images of the molds in each step. In brief, the fabrication of triblock particles starts with filling the mold with a dilute solution of the hydrophilic monomer in DMF (i). As the solvent is removed by evaporation, the remaining monomer is drawn by capillary forces to both ends of the rectangular mold cavity (ii), and subsequently exposed to a low intensity UV light source which converts the monomer into a soft gel (iii). The second hydrophobic monomer is added to fill the remaining volume in the middle of the cavity (iv). The final monomer composition is fully cured by intense UV radiation. b) Schematic illustration of the formation of diblock rods and the corresponding microscopic images of the molds: A) Unsymmetric diblock particles were fabricated by introducing mold rotation around the axis perpendicular to the mold plane. B) The mold was first filled with the hydrophilic monomer. C) The centrifugal force draws the hydrophilic monomer composition to the outer end of the mold cavity. The hydrophobic monomer is subsequently used to fill the open space, thus resulting in a range of amphiphilic diblock rods upon complete photocuring. Adapted from Ref. [26]; copyright 2012 American Chemical Society.
Furthermore, the self-assembly of these amphiphilic tri-block and diblock particles was investigated at the water/perfluorodecalin (PFD) interface (Figure 8). The triblock particles preferred a side-to-side assembly, thus forming ordered ribbon structures at the interface and behaving like bolaamphiphiles. Diblock particles, which had an unsymmetric structure similar to molecular surfactants, self-assembled into two-dimensional bilayer structures at the water/PFD interface. This novel work illuminates the relationship between particle architecture and their self-assembly behavior.
Figure 8.

Fluorescence microscopic images of different particles assembled at the water/PFD interface. A) ABA hydrophilic/hydrophobic/hydrophilic triblock particles. B) AB diblock particles fabricated. The scale bar of the inserts: 200 μm. Self-assembly models are illustrated for the particles at a water/oil interface for the C) triblock and D) diblock particles. Adapted from Ref. [26]; copyright 2012 American Chemical Society.
The aforementioned work presents the versatility of PRINT in its ability to study colloidal assembly of unique organic particles. The work discussed herein is only a small selection illustrating PRINT3s potential to make a significant impact in applications as well as in the fundamental understanding of materials science principles. As the preparation of Janus- and magnetic-nanoparticles-incorporated PRINT particles involves a fabrication route that slightly departs from the standard PRINT method, significant changes would have to be made for the fabrication of these particle types to be scalable. Currently, we are developing other strategies for preparing similar anisotropic particles that may be scalable in a continuous roll-to-roll process.
5. Summary and Outlook
It is becoming increasingly evident that the attributes of particle size, shape, chemical composition, and surface properties are critical in biology and materials science. PRINT offers the first GMP-compliant (GMP = good manufacturing practice) platform for the design and fabrication of monodisperse particles from a wide range of matrices with complete control over the physicochemical properties of the particle. We have developed a wide variety of unique micro-or nanoparticles, and we are now investigating their potential for applications in drug delivery and colloidal assembly.
In the life sciences portion of this review, we have demonstrated that the PRINT technology is delicate and versatile enough for the delivery of a wide variety of active therapeutics including small-molecule drugs, siRNA, and RNA replicon. For delivery of biopharmaceuticals, our current work focuses upon using reporter genes, proteins, or peptides to optimize the system for successful delivery in vivo. The focus of future work will be on using the PRINT particles for in vivo delivery of therapeutics or imaging reagents in the fields of disease prevention, detection, diagnosis, and treatment. In this path forward, we envision the development of particle systems based on specific diseases or therapeutic purposes. For a given therapeutic application, PRINT particles can be designed in an effort to achieve cell or tissue selectivity, as well as desired release profiles. Ultimately, the unique attributes of the PRINT technology may enable a transformation in the ways drugs are developed and administered.
Furthermore, in the materials science portion of this review, we have demonstrated strategies used to create particles of unique sizes, shapes, and compositions for assembly induced by an electronic field, magnetic field, and van der Waals interactions. Our future work will continue to elucidate the influence of different chemical compositions, sizes, and shapes on particle manipulation. Besides the fundamental research, we are also interested in application of these particles in the development of catalysts, optics, plasmonic devices, and electronics. Particular attention will focus on applications where excluded volume interactions of shape-specific particles can be used to advantage, especially in the design of new, light-weight, high-strength structural composites.
Acknowledgments
This work was supported by Liquidia Technologies, the Carolina Center for Cancer Nanotechnology Excellence (U54A151652), University Cancer Research Fund, NIH3s Pioneer Award (IDP10D006432), R01 (R01EB009565), and the National Science Foundation (DMR-0923604 and DMR-0906985). We also want to thank Crista Farrell for editing and suggestions.
Biography

Joseph DeSimone is Chancellor’s Eminent Professor of Chemistry at UNC-Chapel Hill, William R. Kenan, Jr. Professor of Chemical Engineering at NC State University and of Chemistry at UNC-CH, and Director of the Frank Hawkins Kenan Institute of Private Enterprise at UNC-CH. He is a member of the National Academy of Sciences and the National Academy of Engineering. He has published over 290 scientific articles and holds over 130 patents. He received his B.S. in Chemistry in 1986 from Ursinus College and his Ph.D. in Chemistry in 1990 from Virginia Tech.
Contributor Information
Dr. Jing Xu, Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC 27599 (USA)
Dominica H. C. Wong, Department of Chemistry, University of North Carolina Chapel Hill, NC 27599 (USA)
James D. Byrne, Eshelman School of Pharmacy, University of North Carolina Chapel Hill, NC 27599 (USA)
Kai Chen, Department of Chemistry, University of North Carolina Chapel Hill, NC 27599 (USA).
Dr. Charles Bowerman, Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC 27599 (USA)
Prof. Joseph M. DeSimone, Email: desimone@email.unc.edu, Department of Chemistry, University of North Carolina Chapel Hill, NC 27599 (USA). Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC 27599 (USA). Eshelman School of Pharmacy, University of North Carolina Chapel Hill, NC 27599 (USA). Department of Pharmacology, Carolina Center of Cancer Nano-technology Excellence, Institute for Advanced Materials, Institute for Nanomedicine, University of North Carolina, Chapel Hill, NC 27599 (USA) and Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695 (USA) and Sloan-Kettering Institute for Cancer Research, Memorial Sloan-Kettering Cancer Center, New York, NY 10021 (USA)
References
- 1.a) Bae Y, Fukushima S, Harada A, Kataoka K. Angew Chem. 2003;115:4788–4791. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:4640–4643. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]; b) Perkin KK, Turner JL, Wooley KL, Mann S. Nano Lett. 2005;5:1457–1461. doi: 10.1021/nl050817w. [DOI] [PubMed] [Google Scholar]
- 2.a) Guo X, Szoka FC. Acc Chem Res. 2003;36:335–341. doi: 10.1021/ar9703241. [DOI] [PubMed] [Google Scholar]; b) Du J, Armes SP. J Am Chem Soc. 2005;127:12800–12801. doi: 10.1021/ja054755n. [DOI] [PubMed] [Google Scholar]
- 3.a) Malone RW, Felgner PL, Verma IM. Proc Natl Acad Sci USA. 1989;86:6077–6081. doi: 10.1073/pnas.86.16.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) O3Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]; c) Farokhzad OC, Langer R. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 4.a) Meng F, Zhong Z, Feijen J. Biomacromolecules. 2009;10:197–209. doi: 10.1021/bm801127d. [DOI] [PubMed] [Google Scholar]; b) Christian DA, Cai S, Bowen DM, Kim Y, Pajerowski JD, Discher DE. Eur J Pharm Biopharm. 2009;71:463–474. doi: 10.1016/j.ejpb.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang HI, Yeh MK. Int J Nanomed. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Dass CR, Choong PFM. J Controlled Release. 2006;113:155–163. doi: 10.1016/j.jconrel.2006.04.009. [DOI] [PubMed] [Google Scholar]; b) Immordino ML, Brusa P, Arpicco S, Stella B, Dosio F, Cattel L. J Controlled Release. 2003;91:417–429. doi: 10.1016/s0168-3659(03)00271-2. [DOI] [PubMed] [Google Scholar]
- 7.a) Senthilkumar M, Mishra P, Jain NK. J Drug Targeting. 2008;16:424–435. doi: 10.1080/10611860802088598. [DOI] [PubMed] [Google Scholar]; b) Shoyele SA. Methods Mol Biol. 2008;437:149–160. doi: 10.1007/978-1-59745-210-6_7. [DOI] [PubMed] [Google Scholar]
- 8.a) Jang JH, Ullal CK, Kooi SE, Koh C, Thomas EL. Nano Lett. 2007;7:647–651. doi: 10.1021/nl0626277. [DOI] [PubMed] [Google Scholar]; b) Moon JH, Kim AJ, Crocker JC, Yang S. Adv Mater. 2007;19:2508–2512. [Google Scholar]
- 9.a) Kenis PJA, Ismagilov RF, Whitesides GM. Science. 1999;285:83–85. doi: 10.1126/science.285.5424.83. [DOI] [PubMed] [Google Scholar]; b) Cohen I, Li H, Hougland JL, Mrksich M, Nagel SR. Science. 2001;292:265–267. doi: 10.1126/science.1059175. [DOI] [PubMed] [Google Scholar]
- 10.a) Glangchai LC, Caldorera-Moore M, Shi L, Roy K. J Controlled Release. 2008;125:263–272. doi: 10.1016/j.jconrel.2007.10.021. [DOI] [PubMed] [Google Scholar]; b) Guan JJ, Ferrell N, Lee LJ, Hansford DJ. Biomaterials. 2006;27:4034–4041. doi: 10.1016/j.biomaterials.2006.03.011. [DOI] [PubMed] [Google Scholar]; c) Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. J Am Chem Soc. 2005;127:10096–10100. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]
- 11.Rolland JP, Van Dam RM, Schorzman DA, Q Sr, DeSimone JM. J Am Chem Soc. 2004;126:8349–8349. doi: 10.1021/ja031657y. [DOI] [PubMed] [Google Scholar]
- 12.Rolland JP, Hagberg EC, Denison GM, Carter KR, DeSimone JM. Angew Chem. 2004;116:5920–5923. [Google Scholar]; Angew Chem Int Ed. 2004;43:5796–5799. doi: 10.1002/anie.200461122. [DOI] [PubMed] [Google Scholar]
- 13.a) Gratton SE, Williams SS, Napier ME, Pohlhaus PD, Zhou Z, Wiles KB, Maynor BW, Shen C, Olafsen T, Samulski ET, Desimone JM. Acc Chem Res. 2008;41:1685–1695. doi: 10.1021/ar8000348. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hampton MJ, Templeton JL, DeSimone JM. Langmuir. 2010;26:3012–3015. doi: 10.1021/la904787k. [DOI] [PubMed] [Google Scholar]; c) Williams SS, Retterer S, Lopez R, Ruiz R, Samulski ET, DeSimone JM. Nano Lett. 2010;10:1421–1428. doi: 10.1021/nl100326q. [DOI] [PubMed] [Google Scholar]
- 14.Jeong W, Napier ME, DeSimone JM. Nanomedicine. 2010;5:633–639. doi: 10.2217/nnm.10.34. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Byrne JD, Napier ME, DeSimone JM. Small. 2011;7:1919–1931. doi: 10.1002/smll.201100442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Parrott MC, Luft JC, Byrne JD, Fain JH, Napier ME, Desimone JM. J Am Chem Soc. 2010;132:17928–17932. doi: 10.1021/ja108568g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kersey FR, Merkel TJ, Perry JL, Napier ME, Desimone JM. Langmuir. 2012;28:8773–8781. doi: 10.1021/la301279v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enlow EM, Luft JC, Napier ME, DeSimone JM. Nano Lett. 2011;11:808–813. doi: 10.1021/nl104117p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly JY, DeSimone JM. J Am Chem Soc. 2008;130:5438–5439. doi: 10.1021/ja8014428. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Nunes JK, Gratton SEA, Herlihy KP, Pohlhaus PD, DeSimone JM. New J Phys. 2009;11:075018. [Google Scholar]
- 20.Xu J, Wang J, Luft JC, Tian S, Owens G, Jr, Pandya AA, Berglund P, Pohlhaus P, Maynor BW, Smith J, Hubby B, Napier ME, DeSimone JM. J Am Chem Soc. 2012;134:8774–8777. doi: 10.1021/ja302363r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasan W, Chu K, Gullapalli A, Dunn SS, Enlow EM, Luft JC, Tian S, Napier ME, Pohlhaus PD, Rolland JP, DeSimone JM. Nano Lett. 2012;12:287–292. doi: 10.1021/nl2035354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn SS, Tian S, Blake S, Wang J, Galloway AL, Murphy A, Pohlhaus PD, Rolland JP, Napier ME, DeSimone JM. J Am Chem Soc. 2012;134:7423–7430. doi: 10.1021/ja300174v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parrott MC, Finniss M, Luft JC, Pandya A, Gullapalli A, Napier ME, DeSimone JM. J Am Chem Soc. 2012;134:7978–7982. doi: 10.1021/ja301710z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia A, Mack P, Williams S, Fromen C, Shen T, Tully J, Pillai J, Kuehl P, Napier M, Desimone JM, Maynor BW. J Drug Delivery. 2012;2012:941243. doi: 10.1155/2012/941243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merkel TJ, Chen K, Jones SW, Pandya AA, Tian S, Napier ME, Zamboni WE, Desimone JM. J Controlled Release. 2012;162:37–44. doi: 10.1016/j.jconrel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JY, Wang Y, Sheiko SS, Betts DE, DeSimone JM. J Am Chem Soc. 2012;134:5801–5806. doi: 10.1021/ja2066187. [DOI] [PubMed] [Google Scholar]
- 27.Langer R. Science. 1990;249:1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 28.Merkel TJ, Jones SW, Herlihy KP, Kersey FR, Shields AR, Napier M, Luft JC, Wu H, Zamboni WC, Wang AZ, Bear JE, DeSimone JM. Proc Natl Acad Sci USA. 2011;108:586–591. doi: 10.1073/pnas.1010013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gratton SEA, Pohlhaus PD, Lee J, Guo J, Cho MJ, DeSimone JM. J Controlled Release. 2007;121:10–18. doi: 10.1016/j.jconrel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci USA. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Tian S, Petros RA, Napier ME, Desimone JM. J Am Chem Soc. 2010;132:11306–11313. doi: 10.1021/ja1043177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green MR, Manikhas GM, Orlov S, Afanasyev B, Makhson AM, Bhar P, Hawkins MJ. Ann Oncol. 2006;17:1263–1268. doi: 10.1093/annonc/mdl104. [DOI] [PubMed] [Google Scholar]
- 33.Merkel TJ, DeSimone JM. Sci Transl Med. 2011;3:73ps8. doi: 10.1126/scitranslmed.3002137. [DOI] [PubMed] [Google Scholar]
- 34.Yan F, Zhang C, Zheng Y, Mei L, Tang L, Song C, Sun H, Huang L. Nanomedicine. 2010;6:170–178. doi: 10.1016/j.nano.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Musumeci T, Ventura CA, Giannone I, Ruozi B, Montenegro L, Pignatello R, Puglisi G. Int J Pharm. 2006;325:172–179. doi: 10.1016/j.ijpharm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 36.Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W, Langer R, Farokhzad OC. Nano Lett. 2008;8:2906–2912. doi: 10.1021/nl801736q. [DOI] [PubMed] [Google Scholar]
- 37.Shin HC, Alani AWG, Rao DA, Rockich NC, Kwon GS. J Controlled Release. 2009;140:294–300. doi: 10.1016/j.jconrel.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Wang YG, Chen XM, Wang JC, Zhang X, Zhang Q. J Controlled Release. 2009;139:56–62. doi: 10.1016/j.jconrel.2009.05.030. [DOI] [PubMed] [Google Scholar]
- 39.Chu KS, Hasan W, Rawal S, Walsh MD, Enlow EM, Luft JC, Bridges AS, Kuijer JL, Napier ME, Zamboni WC, Desimone JM. Nanomedicine. doi: 10.1016/j.nano.2012.11.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee SS, Aher N, Patil R, Khandare J. J Drug Delivery. 2012;2012:103973. doi: 10.1155/2012/103973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbasi S, Paul A, Prakash S. Cell Biochem Biophys. 2011;61:277–287. doi: 10.1007/s12013-011-9201-9. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Chen YC, Tseng YC, Mozumdar S, Huang L. J Controlled Release. 2010;142:416–421. doi: 10.1016/j.jconrel.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.a) Zimmer G. Viruses. 2010;2:413–434. doi: 10.3390/v2020413. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ying H, Zaks TZ, Wang RF, Irvine KR, Kammula US, Marincola FM, Leitner WW, Restifo NP. Nat Med. 1999;5:823–827. doi: 10.1038/10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.a) Lee SH, Chung BH, Park TG, Nam YS, Mok H. Acc Chem Res. 2012;45:1014–1025. doi: 10.1021/ar2002254. [DOI] [PubMed] [Google Scholar]; b) Raemdonck K, Van Thienen TG, Vandenbroucke RE, Sanders NN, Demeester J, De Smedt SC. Adv Funct Mater. 2008;18:993–1001. [Google Scholar]
- 45.Kofler RM. Proc Natl Acad Sci USA. 2004;101:1951–1956. doi: 10.1073/pnas.0307145101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galloway AL, Murphy A, Desimone JM, Di J, Herrmann JP, Hunter ME, Kindig JP, Malinoski FJ, Rumley MA, Stoltz DM, Templeman TS, Hubby B. Nanomedicine. 2012;9:523–531. doi: 10.1016/j.nano.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Ko DH, Tumbleston JR, Henderson KJ, Euliss LE, DeSimone JM, Lopez R, Samulski ET. Soft Matter. 2011;7:6404–6407. [Google Scholar]
- 48.Hampton MJ, Williams SS, Zhou Z, Nunes J, Ko DH, Templeton JL, Samulski ET, DeSimone JM. Adv Mater. 2008;20:2667–2673. doi: 10.1002/adma.200702495. [DOI] [PubMed] [Google Scholar]
- 49.Li F, Josephson DP, Stein A. Angew Chem. 2011;123:378–409. doi: 10.1002/anie.201001451. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:360–388. doi: 10.1002/anie.201001451. [DOI] [PubMed] [Google Scholar]
- 50.Xia YN, Gates B, Yin YD, Lu Y. Adv Mater. 2000;12:693–713. [Google Scholar]
- 51.Velikov KP, van Dillen T, Polman A, van Blaaderen A. Appl Phys Lett. 2002;81:838–840. [Google Scholar]
- 52.Glotzer SC, Solomon MJ. Nat Mater. 2007;6:557–562. doi: 10.1038/nmat1949. [DOI] [PubMed] [Google Scholar]
- 53.van den Ende DA, van Kempen SE, Wu X, Groen WA, Randall CA, van der Zwaag S. J Appl Phys. 2012;111:124107. [Google Scholar]
- 54.a) Park C, Robertson RE. Mater Sci Eng A. 1998;257:295–311. [Google Scholar]; b) Park C, Wilkinson J, Banda S, Ounaies Z, Wise KE, Sauti G, Lillehei PT, Harrison JS. J Polym Sci Part B. 2006;44:1751–1762. [Google Scholar]
- 55.a) Tomer V, Randall CA, Polizos G, Kostelnick J, Manias E. J Appl Phys. 2008;103:034115. [Google Scholar]; b) Wilson SA, Maistros GM, Whatmore RW. J Phys D. 2005;38:175–182. [Google Scholar]
- 56.Herlihy KP, Nunes J, Desimone JM. Langmuir. 2008;24:8421–8426. doi: 10.1021/la801250g. [DOI] [PubMed] [Google Scholar]
- 57.Nunes J, Herlihy KP, Mair L, Superfine R, DeSimone JM. Nano Lett. 2010;10:1113–1119. doi: 10.1021/nl904152e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.a) Jiang S, Chen Q, Tripathy M, Luijten E, Schweizer KS, Granick S. Adv Mater. 2010;22:1060–1071. doi: 10.1002/adma.200904094. [DOI] [PubMed] [Google Scholar]; b) Merkel TJ, Herlihy KP, Nunes J, Orgel RM, Rolland JP, DeSimone JM. Langmuir. 2010;26:13086–13096. doi: 10.1021/la903890h. [DOI] [PMC free article] [PubMed] [Google Scholar]