

Abstract
Human apolipoprotein (apo) C-II is one of several lipid-binding proteins that self-assemble into fibrils and accumulate in disease-related amyloid deposits. A general characteristic of these amyloid deposits is the presence of lipids, known to modulate individual steps in amyloid fibril formation. ApoC-II fibril formation is activated by sub-micellar phospholipids but inhibited by micellar lipids. We examined the mechanism for the activation by sub-micellar lipids using the fluorescently-labelled, short-chain phospholipid, 1-dodecyl-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]-2-hydroxy-glycero-3-phosphocholine (NBD-lyso-12-PC). Addition of submicellar NBD-lyso-12-PC increased the rate of fibril formation by apoC-II approximately two-fold. Stopped flow kinetic analysis using fluorescence detection and low, non-fibril forming concentrations of apoC-II indicated NBD-Lyso-12-PC binds rapidly, in the millisecond timescale, followed by the slower formation of discrete apoC-II tetramers. Sedimentation velocity analysis showed NBD-Lyso-12-PC binds to both apoC-II monomers and tetramers at approximately 5 sites per monomer with an average dissociation constant of approximately 10 μM. Mature apoC-II fibrils formed in the presence of NBD-Lyso-12-PC were devoid of lipid indicating a purely catalytic role for sub-micellar lipids in the activation of apoC-II fibril formation. These studies demonstrate the catalytic potential of small amphiphilic molecules to control protein folding and fibril assembly pathways.
The aggregation of proteins into amyloid fibrils is associated with a wide variety of diseases, ranging from neurodegenerative Alzheimer’s and Parkinson’s diseases through to systemic amyloidoses (1). The formation of these fibrillar aggregates appears to be a general feature of proteins, as over 20 individual proteins form amyloid in vivo (2), while several other proteins readily form amyloid fibrils in vitro under a variety of solution conditions (1). Amyloid deposits in vivo also contain non-fibrillar material, including the amyloid specific proteins apolipoprotein (apo) E and serum amyloid P, proteoglycans and lipids (2, 3). The importance of lipids in amyloid deposits is underscored by the number of reports of lipid modulation of amyloid fibril formation. Several studies (4–12) have noted that the effect of lipids depends on the lipid-protein ratio and the nature of the interaction between the polypeptide and the lipid surface. Insertion of the protein into the surface inhibits fibril formation (4) while transient electrostatic interactions can enhance the process by increasing the local protein concentration and providing a scaffold for amyloid prone conformations (13). Studies with micellar and sub-micellar lipids provide an alternate approach to the analysis of the effects of lipids on amyloid fibril formation and permit the role of individual lipid molecules to be examined (10, 12, 14).
Apolipoproteins are lipid binding proteins that constitute a high proportion of the proteins which form amyloid in vivo. ApoA-I, apoA-II and apoC-II deposit in atherosclerotic lesions, and may contribute to the progression of cardiovascular diseases (15–18). In addition, apoA-I, apoA-II and apoA-IV amyloid formation is associated with several hepatic, systemic and renal amyloid diseases (19–24). Human apoC-II is an 8914 Da exchangeable apolipoprotein that associates with VLDL and chylomicrons, where it acts as a co-factor for lipoprotein lipase. In the presence of micellar lipid mimetics apoC-II adopts a predominantly α-helical structure (25, 26). Conversely, lipid-free apoC-II rapidly self-assembles into homogenous fibrils with increased β-structure and all of the hallmarks of amyloid (27). A structural model for apoC-II fibrils composed of a linear assembly of monomers in a “letter G-like” conformation has recently been described (28).
ApoC-II amyloid fibril formation is inhibited by micellar concentrations of phospholipids such as dihexanoylphosphatidylcholine (DHPC) whereas sub-micellar DHPC enhances fibril formation via the induction of a tetrameric intermediate which acts as a nucleus for fibril elongation (29–31). Screening a large number of lipids and related amphiphiles at sub-micellar concentrations identified a range of activators and inhibitors of apoC-II fibril formation (32). Biophysical studies showed that activators promoted the formation of a tetrameric intermediate enriched in β-structure while inhibitors induced dimeric species with increased α-structure. To further investigate the mechanism for the effects of lipid modulators on amyloid fibril formation pathways we have used the fluorescently-labelled, short-chain phospholipid, 1-dodecyl-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]-2-hydroxy-glycero-3-phosphocholine (NBD-lyso-12-PC). Our results show that apoC-II monomers and tetramers bind several molecules of lipid while mature fibrils are essentially lipid-free. The observation that apoC-II fibrils formed in the presence of NBD-lyso-12-PC lack bound fluorescence indicates that activation by NBD-lyso-12-PC is catalytic with the release of monomer and tetramer bound lipid accompanying fibril elongation and growth.
EXPERIMENTAL PROCEDURES
Alexa 594 C5 maleimide was obtained from Invitrogen-Molecular Probes (Eugene, Oregon) and 1-(dodecyl-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]lauroyl)-2-hydroxy-sn-glycero-3-phosphocholine (NBD-Lyso-12-PC) was obtained from Avanti Polar Lipids, Inc. (Alabaster, Alabama). ApoC-II was expressed and purified as described previously (12). Purified apoC-II stock solutions were stored in 5M guanidine hydrochloride, 10 mM Tris.HCl, pH 8.0 at a concentration of approximately 45 mg/ml. ApoC-IIS61C was provided by Dr. Chi Pham (University of Melbourne) and was conjugated with Alexa 594 as described previously (29). ApoC-II lipid interactions and fibril formation were performed by dilution of the stock solution apoC-II solution into refolding buffer (100mM sodium phosphate, 0.1% sodium azide, pH 7.4).
Fluorescence measurements
The time course of fibril formation was determined using a previously described centrifugal pelleting assay (29), where the proportion of non-sedimenting material in the supernatant was measured using tryptophan fluorescence (excitation 295nm, emission 350 nm). Fluorescence resonance energy transfer (FRET) between NBD-Lyso-12-PC and Alexa 594-labelled apoC-II was measured using a Cary Eclipse (Varian, Palo Alto, California), with excitation at 430nm and emission spectra collected from 450–750 nm. Stopped flow measurements were conducted using an RX-6200 portable stopped flow device (Applied Photophysics, Leatherhead, Surrey, UK) equipped with a pneumatic drive and 2 × 2 mL syringes for the ligand and acceptor solutions. In all cases stopped flow was conducted using a stopping volume of 150 μL. A pelleting assay, described previously to measure the interaction of HDL with apoC-II amyloid fibrils (33), was used to test whether NBD-Lyso-12-PC binds to apoC-II fibrils. Briefly, apoC-II fibrils (0.3 mg/ml) in the presence and absence of NBD-Lyso-12-PC were layered onto a 20% sucrose cushion and centrifuged for 30 min at 100,000 rpm (436,000g) in an OptimaMax centrifuge using a TLA-100 rotor (Beckman Coulter Instruments, Inc., Fullarton, California). The original sample, pellet and supernatant fractions were assayed for NBD-fluorescence.
Sedimentation velocity analysis
Sedimentation velocity experiments were conducted using an AnTi50 rotor and analytical ultracentrifuge cells equipped with quartz windows and double sector charcoal epon centerpieces. Sedimentation velocity experiments for apoC-II alone were conducted using optical density measurements at 280 nm. Fluorescence detection sedimentation experiments were conducted using an XL-A analytical ultracentrifuge (Beckman Coulter Instruments, Inc., Fullarton, California) equipped with a fluorescence detection system (FDS; Aviv Associates Inc). Sedimentation velocity data for NBD-Lyso-12-PC in the absence and presence of unlabelled apoC-II or apoC-II fibrils were obtained at 20°C using a rotor speed of 50,000 rpm (180,000 g). Fluorescence data were collected at 1 minute intervals from 6.0–7.25 cm with the excitation laser focused to a spot 20 μm in diameter, 31 μm below the surface of the sapphire window. Sedimentation velocity data obtained from these experiments were analysed using the c(s) model in SEDFIT9.4 (34, 35). Data on the concentration dependence of the weight-average sedimentation coefficient of apoC-II in the presence of NBD-Lyso-12-PC were analysed according to a reversible monomer-tetramer equilibrium model assuming a sedimentation coefficient of 0.93 and 2 S for the sedimentation coefficient of apoC-II monomer and tetramer, respectively (29). For these calculations data collected at apoC-II concentrations less than 1 μM were omitted due to significant overlap of fluorescence from non-sedimenting NBD-Lyso-12-PC.
RESULTS
Sub-micellar NBD-lyso-12-PC activates apoC-II fibril formation
The critical micelle concentration (CMC) for NBD-lyso-12-PC was determined from the intensity of scattered 600 nm light as a function of NBD-lyso-12-PC concentration (Figure 1A). The data shows a shallow linear gradient at low concentrations of NBD-lyso-12-PC followed by a much steeper gradient at higher concentrations. The breakpoint between these two lines indicates a CMC of approximately 0.2mM for NBD-lyso-12-PC. While previous studies have demonstrated the activation of apoC-II fibril formation by sub-micellar lipids (29, 30) this has not been shown for sub-micellar NBD-lyso-12-PC. The effect of sub-micellar NBD-lyso-12-PC on apoC-II amyloid fibril formation was measured using a centrifugal pelleting assay (Figure 1B). ApoC-II (0.3 mg/ml) in the absence of lipid showed a time-dependent increase in the amount sedimenting material, with time to half maximum (t50) of approximately 28 h. Addition of 60 μM NBD-lyso-12-PC reduced the t50 to 11.8 h indicating significant activation, comparable to the effects previously observed with other sub-micellar phospholipids (29, 30).
Figure 1.

Effect of submicellar NBD-Lyso-12-PC on apoC-II fibril formation. A. Determination of CMC for NBD-Lyso-12-PC using light scattering. B. ApoC-II fibril formation (0.3 mg/mL) monitored using a centrifugal pelleting assay in the absence (squares) and presence (circles) of NBD-Lyso-12-PC (60 μM) and control data where the apoC-II sample with NBD-Lyso-12-PC was not centrifuged (triangles).
NBD-Lyso-12-PC binds rapidly to apoC-II monomers
The association of Alexa 594-labelled apoC-II with NBD-Lyso-12-PC was examined using FRET measurements. Figure 2A shows the spectra of NBD-Lyso-12-PC alone, Alexa 594 labelled apoC-II alone and a mixture at the same concentrations of NBD-Lyso-12-PC and Alexa 594 labelled apoC-II. The decrease in intensity at 540 nm and the increase in intensity at 620 nm indicate significant transfer of excitation from the NBD fluorophore to the Alexa 594 fluorophore, suggesting an interaction between non-fibrillar apoC-II and NBD-Lyso-12-PC. This FRET signal was exploited to examine the millisecond time-scale kinetics of the lipid-protein interactions using stopped flow analysis. Figure 2B shows the intensity of NBD-Lyso-12-PC at 540 nm upon excitation at 430 nm over time in the presence and absence of Alexa 594 apoC-II. In the absence of Alexa 594-labelled apoC-II, the NBD-Lyso-12-PC fluorescence was constant over the time course of the experiment. Addition of Alexa 594-labelled apoC-II induced a rapid decay in the fluorescence intensity which reached completion over 100 milliseconds. The decay in fluorescence intensity due to FRET was dependent on the concentration of labelled apoC-II and indicates a very rapid association of protein with the lipid. These fluorescence decays were globally fitted to provide a pseudo first order rate constant of 156,000 s−1.
Figure 2.

FRET analysis of the interaction between NBD-Lyso-12-PC and freshly refolded Alexa 594 apoC-II. A. Fluorescence spectra using excitation at 430 nm for NBD-Lyso-12-PC (60 μM) alone (solid line), the same concentration of NBD-Lyso-12-PC in the presence of Alexa 594 apoC-II (5 μM) (dashed line) and Alexa 594 apoC-II (5 μM) alone (dashed dotted line). B. Stopped flow analysis of the interaction of NBD-Lyso-12-PC with apoC-II monitored using FRET (excitation 430 nm, emission 540 nm). Data obtained using NBD-Lyso-12-PC (60 μM) and Alexa 594 labelled apoC-II concentrations of 0 (diamonds), 1.25 (circles), 2.5 (squares) and 5 (triangles) μM Alexa 594 labelled apoC-II. The solid lines are global best-fits to a pseudo first order rate constant.
NBD-Lyso-12-PC binds to multiple sites on apoC-II monomers and tetramers
The fluorescence properties of NBD-lyso-12-PC permitted fluorescence detection using the analytical ultracentrifuge to measure the interaction of this lipid with apoC-II, at low non-fibril-forming concentrations of apoC-II. Representative sedimentation velocity data sets (Figures 3A and B) show an increased amount of fluorescence in the sedimenting boundary compared to the non-sedimenting fluorescence as the concentration of apoC-II is increased. Approximate values for the molecular mass and partial specific volume of NBD-Lyso-12-PC (0.93 ml/g and 600 respectively) yield a sedimentation coefficient for NBD-lyso-12-PC of less than 0.06S, consistent with the observation that this molecule does not sediment appreciably at 50,000 rpm (180,000 g). With the small sedimentation coefficient of NBD-lyso-12-PC and the similarity of the sedimentation coefficient of free and NBD-lyso-12-PC bound apoC-II (see below), the effective particle theory of sedimentation of reacting systems (36) shows, supported by simulations, that the non-sedimenting fluorescence in the supernatant provides a reliable estimate of the free NBD-lyso-12-PC in the original solution. Accordingly, the dependence of the non-sedimenting fluorescence on the concentration of added apoC-II (over the range 0.45 to 28 μM) was used to obtain the proportion of bound NBD-lyso-12-PC per apoC-II molecule as a function of free lipid (Figure 3C). Analysis of this data, assuming a multiple sites model, yielded a stoichiometry of approximately 5 lipid molecules/molecule of apoC-II and an intrinsic dissociation constant of approximately 10 μM (Figure 3C). This binding constant may be considered as an average of the affinities of NBD-lyso-12-PC for apoC-II monomers and tetramers, since under the condition studied the protein existed as an equilibrium mixture of these two species (see Figure 4).
Figure 3.

Sedimentation velocity data using fluorescence detection of NBD-Lyso-12-PC (60 μM) in the presence of apoC-II at 3.5 and 14 μM (panels A and B, respectively). C. The molar ratio of bound NBD-Lyso-12-PC per apoC-II was estimated from the proportion of non-sedimenting fluorescence material for NBD-Lyso-12-PC samples (60 μM) containing a range of apoC-II concentrations (0.45 μM to 28 μM). The solid line is the best fit to the data assuming a simple multiple site binding model.
Figure 4.

Sedimentation velocity analysis of the interaction between NBD-Lyso-12-PC and apoC-II. Sedimentation velocity data using fluorescence detection was obtained for NBD-Lyso-12-PC samples (60 μM) containing a range of apoC-II concentrations (0.45 μM to 28 μM). A. Analysis of the data assuming a continuous sedimentation coefficient distribution c(s) model. B. The weight average sedimentation coefficients (closed circles), obtained by integration of sedimentation coefficient distributions in panel A. The solid line is a fit to the data assuming a monomer-tetramer model. The weight average sedimentation coefficients for apoC-II samples alone, monitored by optical density measurements at 280 nm, are shown for comparison (open squares).
Sedimentation velocity data using fluorescence detection of NBD-lyso-12-PC over a range of apoC-II concentrations was analysed using a c(s) model to obtain coefficient distributions (Figure 4A). The distributions at the lower apoC-II concentrations show two distinct peaks at approximately 1 S and 2 S, corresponding to the sedimentation coefficients observed for monomeric and tetrameric apoC-II formed in the presence of DHPC (29). Increasing the concentration of apoC-II shifted the distributions towards a single broad peak with a modal sedimentation coefficient of 1.9 S. Since the measured fluorescence signal arises from the lipid component, these distributions indicate that NBD-lyso-12-PC associates with apoC-II and binds to both apoC-II monomers and tetramers.
The c(s) distributions in Figure 4 were integrated to obtain signal weighted average sedimentation coefficients, sw,NBD. Based on the observation that the fluorescence emission of NBD-lyso-12-PC does not change in the presence of apoC-II (data not shown), this will correspond to the standard weight-average sedimentation coefficient of the ligand-bound species. The sedimentation coefficient for apoC-II alone, detected using optical density measurements, did not display any significant change over the course of the experiment. However, sw,NBD of apoC-II in the presence of NBD-lyso-12-PC, displayed an increase that was dependent on apoC-II concentration and consistent with lipid-induced tetramerisation. Due to the low sedimentation coefficient of NBD-lyso-12-PC alone, the increasing sw,NBD value cannot be attributed to multiple bound ligands. Analysis of the dependence of the weight average sedimentation coefficient on apoC-II concentration yielded a value of 3.5 × 10−3 μM−3 for the monomer-tetramer equilibrium constant (Figure 4B). This value is similar in magnitude to estimates provided from previous kinetic analyses of DHPC-induced apoC-II tetramerisation (29).
ApoC-II fibrils do not bind NBD-Lyso-12-PC
Our studies of apoC-II fibril formation show that sub-micellar phospholipids activate fibril nucleation rather than elongation (37). However, these observations do not preclude interactions between lipid and fibrillar material, which could also play a role in promoting fibril aggregation. It was therefore of interest to determine if phospholipid remained associated with the fibrillar end product. The binding of lipids to apoC-II amyloid fibrils was initially investigated with a FRET assay (Figure 5). The time course for the change in FRET signal observed for NBD-Lyso-12-PC and monomer and tetramers of Alexa 594-labelled apoC-II (Figure 2) decreased exponentially during incubation under fibril forming conditions indicative of a loss of bound lipid. The lack of binding of NBD-lyso-12-PC to apoC-II fibrils was supported by fluorescence emission spectra which showed no significant increase in intensity at 620nm indicating that there is little or no FRET between the NBD-Lyso-12-PC and Alexa 594-labelled fibrillar apoC-II (Figure 5). This result was confirmed by sedimentation velocity analysis using the FDS, where no significant sedimentation was observed at 50000 rpm for either NBD-Lyso-12-PC alone (60 μM) or in presence of apoC-II fibrils (33 μM) which were either preformed or formed in the presence of NBD-Lyso-12-PC (Figure 6). Further confirmation of the lack of an interaction between NBD-Lyso-12-PC and apoC-II amyloid fibrils was provided by a centrifugal pelleting assay using a 20 % sucrose cushion to separate free lipid from bound lipid (Table 1). This assay has previously been used to identify the binding of HDL particles to apoC-II fibrils (33). NBD-Lyso-12-PC alone (60 μM) did not pellet through the sucrose solution, as the control without any fibrillar material contained <0.0003% of the fluorescence contained in the original solution and supernatant fractions. A similar low level of fluorescence in the pellet fraction was observed for NBD-Lyso-12-PC solutions containing preformed apoC-II amyloid fibrils or apoC-II fibrils formed in the presence of NBD-Lyso-12-PC. Control experiments using apoC-II in the absence of NBD-Lyso-12-PC displayed no fluorescence either prior to centrifugation or after centrifugation.
Figure 5.
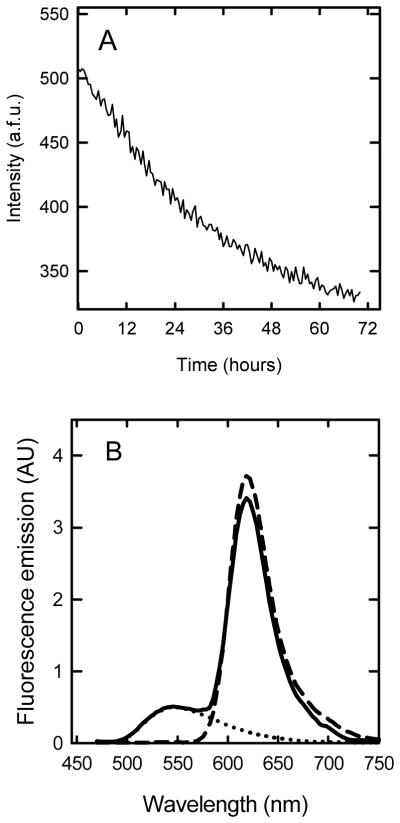
FRET analysis of the interaction between Alexa 594 apoC-II fibrils and NBD-Lyso-12-PC. A. Time course for the change in FRET during apoC-II fibril formation. Alexa 594-labelled apoC-II (33 μM) was incubated with NBD-Lyso-12-PC (60 μM) at 20 °C and the fluorescence emission at 620 nm measured with excitation at 430 nm. B. Fluorescence emission spectra (in arbitrary units) for Alexa 594 apoC-II fibrils alone (5 μM, dashed line), Alexa 594 apoC-II fibrils (5 μM) in the presence of 60 μM NBD-Lyso-12-PC (solid line) and 60 μM NBD-Lyso-12-PC alone (dotted line) were acquired using excitation at 430nm.
Figure 6.

Sedimentation velocity analysis using fluorescence detection of NBD-Lyso-12-PC in the presence and absence of apoC-II fibrils. Samples of NBD-Lyso-12-PC alone (A) and in the presence of apoC-II fibrils (33 μM) which were either preformed (B) or formed in the presence of NBD-Lyso-12-PC (60 μM) by incubation at 20°C for 5 days (C) were analyzed using fluorescence detection in the analytical ultracentrifuge. The radial scans are for data collected after 20 min (solid circles) and 5 h (open triangles) using a rotor speed of 50000 rpm. For the sake of clarity only every 40th data point is presented.
Table 1.
Sedimentation analysis of NBD-Lyso-12-PC in the presence and absence of apoC-II fibrilsa
| sample | Original sample | Supernatant | Pellet fraction |
|---|---|---|---|
| NBD-Lyso-12-PC alone | 1 | 1 | 2.16 × 10−6 |
| NBD-Lyso-12-PC plus preformed apoC-II fibrils | 1 | 1 | 2.26 × 10−6 |
| ApoC-II fibrils formed in presence of NBD-Lyso-12-PC | 1 | 1 | 2.23 × 10−6 |
| ApoC-II fibrils alone | 2.20 × 10−6 | 2.20 × 10−6 | 2.19 × 10−6 |
Samples of NBD-Lyso-12-PC (60 μM) were prepared in the absence and presence of apoC-II fibrils (33 μM) which were either preformed or formed in the presence of NBD-Lyso-12-PC by incubation at 20°C for 5 days. The samples were centrifuged at 100,000 rpm (436,000g) in a preparative ultracentrifuge for 30 min and the fluorescence emission of NBD-Lyso-12-PC (60 μM) determined for the original solution, supernatant and pellet fractions made up to the original volume. Fluorescence emission is expressed relative to the initial NBD-Lyso-12-PC solution.
DISCUSSION
Amphipathic lipids and lipid mimetics affect several amyloid forming systems (4–12) with no obvious pattern concerning the magnitude and specificities of the effects observed. In the case of apoC-II, sub-micellar concentrations of short-chain phospholipids (29, 31) and oxidized cholesterol (38) activate apoC-II fibril formation whereas low concentrations of micellar lipids and lipid bilayers, while initially inhibiting fibril formation, ultimately induce apoC-II fibrils with a distinctive straight rod-like morphology (30). The complexity of the interactions of apoC-II with lipids is further illustrated by the results of a screen of amphipathic molecules which indentified a range of submicellar modulators that exerted independent effects on fibril nucleation and elongation (32). Our present results provide new insight into the apoC-II-lipid interaction, revealing multiple lipid-binding sites. The results are consistent with mass spectrometry studies of apoC-II with lipids in the gas phase which also indicate multiple lipid-bound apoC-II species (39). The existence of multiple lipid-binding sites suggests the diversity of the effects of lipids on apoC-II fibril formation may depend on the occupancy of specific sites and the role the individual sites play in the fibril assembly process.
Our previous studies have shown that the activation of apoC-II fibril formation by DHPC occurs by the rapid induction of a discrete tetramer followed by a slow isomerization of the tetramer that precedes accelerated fibril growth (29). Activation by NBD-lyso-12-PC appears to follow this pattern with the rapid binding of NBD-lyso-12-PC accompanied by the formation of discrete tetramers. An important observation is that although NBD-lyso-12-PC is bound to apoC-II oligomers the final mature fibrils are devoid of bound lipid. Figure 7 summarizes possible pathways to explain these observations. One pathway, available to lipid-free apoC-II, involves the slow isomerization of misfolded monomers to generate species competent to rapidly assemble into mature fibrils. Evidence for this pathway is provided by studies of cross-linked apoC-II dimers, which accelerate apoC-II fibril formation (40). An alternate, NBD-lyso-12-PC-induced pathway is postulated to involve the formation of a tetrameric intermediate which nucleates fibril formation via the slow formation of a lipid-free apoC-II tetramer. The slow isomerisation of the apoC-II tetramer is based on previous studies of a slow step in the DHPC-induced acceleration of apoC-II fibril formation (29). While the molecular details of the initial NBD-lyso-12-PC tetramer complex remain unclear, a possible mechanism for the formation of this species would be a sequential process involving the transient formation of lipid-stabilised dimers followed by self-assembly to form tetrahedral “dimer-of-dimers” structures. According to this model we propose that the slow isomerisation of this tetrahedral structure is accompanied by lipid loss to generate a linear tetramer that serves as a nucleus for fibril elongation.
Figure 7. Model for lipid-induction of apoC-II fibril assembly.

The model proposes a lipid-induced tetramerisation followed by a slow isomerisation and loss of lipid to generate a nucleus for subsequent fibril elongation. An alternate slower pathway available to lipid-free apoC-II proposes an initial isomerisation of apoC-II to form a monomeric nucleus that self-assembles into fibrils. The model for apoC-II fibrils shows a linear assembly of monomers in a “letter G-like” conformation as recently described (28).
A number of naturally occurring compounds are known to affect protein folding include osmolytes such as trehalose which act in the molar concentration range (0.5–1 M), primarily by non-specific, non-ideality effects (41). A recent example is the affects of the osmolytes TMAO, proline and glycine-betaine on the huntington fibril forming pathway (42). Our previous work identified high-affinity amphipathic activators and inhibitors of human apolipoprotein (apo) C-II fibril formation, active in the micromolar concentration range, including a number of physiological metabolites (32, 38). Other disease-related proteins where amyloid fibril formation is affected by tight-binding amphiphiles include α-synuclein (43), apoA-I (44), Aβ (4), β2-microglobulin (9), islet amyloid polypeptide (13), prions (45), transthyretin (46) and medin (47). The importance of the present study is the demonstration that sub-micellar NBD-lyso-12-PC activates apoC-II amyloid fibril formation without incorporation into the final product. This suggests that small molecular weight metabolites could play hitherto unrecognised catalytic roles in protein folding and assembly with the potential to modulate aberrant protein misfolding and disease.
Acknowledgments
Funding information
This research was supported under the Australian Research Council’s Discovery Projects funding scheme (project number DP0984565. M.D.W.G is the recipient of an Australian Research Council Post Doctoral Fellowship (project number DP110103528). This work was supported in part by the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health.
Abbreviations
- apo
apolipoprotein
- CMC
critical micelle concentration
- FDS
fluorescence detection system
- FRET
fluorescence resonance energy transfer
- NBD-Lyso-12-PC
1-(dodecyl-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]lauroyl)-2-hydroxy-sn-glycero-3-phosphocholine
- ThT
thioflavin T
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 3.Gellermann GP, Appel TR, Tannert A, Radestock A, Hortschansky P, Schroeckh V, Leisner C, Lutkepohl T, Shtrasburg S, Rocken C, Pras M, Linke RP, Diekmann S, Fandrich M. Raft lipids as common components of human extracellular amyloid fibrils. Proc Natl Acad Sci U S A. 2005;102:6297–6302. doi: 10.1073/pnas.0407035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bokvist M, Lindstrom F, Watts A, Grobner G. Two types of Alzheimer’s beta-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J Mol Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 5.Chirita CN, Necula M, Kuret J. Anionic micelles and vesicles induce tau fibrillization in vitro. J Biol Chem. 2003;278:25644–25650. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- 6.Gorbenko GP, Kinnunen PK. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem Phys Lipids. 2006;141:72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Necula M, Chirita CN, Kuret J. Rapid anionic micelle-mediated alpha-synuclein fibrillization in vitro. J Biol Chem. 2003;278:46674–46680. doi: 10.1074/jbc.M308231200. [DOI] [PubMed] [Google Scholar]
- 8.Ookoshi T, Hasegawa K, Ohhashi Y, Kimura H, Takahashi N, Yoshida H, Miyazaki R, Goto Y, Naiki H. Lysophospholipids induce the nucleation and extension of beta2-microglobulin-related amyloid fibrils at a neutral pH. Nephrol Dial Transplant. 2008;23:3247–3255. doi: 10.1093/ndt/gfn231. [DOI] [PubMed] [Google Scholar]
- 9.Pal-Gabor H, Gombos L, Micsonai A, Kovacs E, Petrik E, Kovacs J, Graf L, Fidy J, Naiki H, Goto Y, Liliom K, Kardos J. Mechanism of lysophosphatidic acid-induced amyloid fibril formation of beta(2)-microglobulin in vitro under physiological conditions. Biochemistry. 2009;48:5689–5699. doi: 10.1021/bi900356r. [DOI] [PubMed] [Google Scholar]
- 10.Rangachari V, Reed DK, Moore BD, Rosenberry TL. Secondary structure and interfacial aggregation of amyloid-beta(1–40) on sodium dodecyl sulfate micelles. Biochemistry. 2006;45:8639–8648. doi: 10.1021/bi060323t. [DOI] [PubMed] [Google Scholar]
- 11.Shao H, Jao S, Ma K, Zagorski MG. Solution structures of micelle-bound amyloid beta-(1–40) and beta-(1–42) peptides of Alzheimer’s disease. J Mol Biol. 1999;285:755–773. doi: 10.1006/jmbi.1998.2348. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto S, Hasegawa K, Yamaguchi I, Tsutsumi S, Kardos J, Goto Y, Gejyo F, Naiki H. Low concentrations of sodium dodecyl sulfate induce the extension of beta 2-microglobulin-related amyloid fibrils at a neutral pH. Biochemistry. 2004;43:11075–11082. doi: 10.1021/bi049262u. [DOI] [PubMed] [Google Scholar]
- 13.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 14.Sureshbabu N, Kirubagaran R, Jayakumar R. Surfactant-induced conformational transition of amyloid beta-peptide. Eur Biophys J. 2009;38:355–367. doi: 10.1007/s00249-008-0379-8. [DOI] [PubMed] [Google Scholar]
- 15.Westermark P, Mucchiano G, Marthin T, Johnson KH, Sletten K. Apolipoprotein A1-derived amyloid in human aortic atherosclerotic plaques. Am J Pathol. 1995;147:1186–1192. [PMC free article] [PubMed] [Google Scholar]
- 16.Mucchiano GI, Haggqvist B, Sletten K, Westermark P. Apolipoprotein A-1-derived amyloid in atherosclerotic plaques of the human aorta. J Pathol. 2001;193:270–275. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH753>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 17.Mucchiano GI, Jonasson L, Haggqvist B, Einarsson E, Westermark P. Apolipoprotein A-I-derived amyloid in atherosclerosis. Its association with plasma levels of apolipoprotein A-I and cholesterol. Am J Clin Pathol. 2001;115:298–303. doi: 10.1309/PJE6-X9E5-LX6K-NELY. [DOI] [PubMed] [Google Scholar]
- 18.Medeiros LA, Khan T, El Khoury JB, Pham CL, Hatters DM, Howlett GJ, Lopez R, O’Brien KD, Moore KJ. Fibrillar amyloid protein present in atheroma activates CD36 signal transduction. J Biol Chem. 2004;279:10643–10648. doi: 10.1074/jbc.M311735200. [DOI] [PubMed] [Google Scholar]
- 19.Bergstrom J, Murphy C, Eulitz M, Weiss DT, Westermark GT, Solomon A, Westermark P. Codeposition of apolipoprotein A-IV and transthyretin in senile systemic (ATTR) amyloidosis. Biochem Biophys Res Commun. 2001;285:903–908. doi: 10.1006/bbrc.2001.5260. [DOI] [PubMed] [Google Scholar]
- 20.Coriu D, Dispenzieri A, Stevens FJ, Murphy CL, Wang S, Weiss DT, Solomon A. Hepatic amyloidosis resulting from deposition of the apolipoprotein A-I variant Leu75Pro. Amyloid. 2003;10:215–223. doi: 10.3109/13506120309041738. [DOI] [PubMed] [Google Scholar]
- 21.Yazaki M, Liepnieks JJ, Barats MS, Cohen AH, Benson MD. Hereditary systemic amyloidosis associated with a new apolipoprotein AII stop codon mutation Stop78Arg. Kidney Int. 2003;64:11–16. doi: 10.1046/j.1523-1755.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 22.Obici L, Bellotti V, Mangione P, Stoppini M, Arbustini E, Verga L, Zorzoli I, Anesi E, Zanotti G, Campana C, Vigano M, Merlini G. The new apolipoprotein A-I variant leu(174) --> Ser causes hereditary cardiac amyloidosis, and the amyloid fibrils are constituted by the 93-residue N-terminal polypeptide. Am J Pathol. 1999;155:695–702. doi: 10.1016/S0002-9440(10)65167-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obici L, Franceschini G, Calabresi L, Giorgetti S, Stoppini M, Merlini G, Bellotti V. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid. 2006;13:191–205. doi: 10.1080/13506120600960288. [DOI] [PubMed] [Google Scholar]
- 24.Bergstrom J, Murphy CL, Weiss DT, Solomon A, Sletten K, Hellman U, Westermark P. Two different types of amyloid deposits--apolipoprotein A-IV and transthyretin--in a patient with systemic amyloidosis. Lab Invest. 2004;84:981–988. doi: 10.1038/labinvest.3700124. [DOI] [PubMed] [Google Scholar]
- 25.MacRaild CA, Hatters DM, Howlett GJ, Gooley PR. NMR structure of human apolipoprotein C-II in the presence of sodium dodecyl sulfate. Biochemistry. 2001;40:5414–5421. doi: 10.1021/bi002821m. [DOI] [PubMed] [Google Scholar]
- 26.MacRaild CA, Howlett GJ, Gooley PR. The structure and interactions of human apolipoprotein C-II in dodecyl phosphocholine. Biochemistry. 2004;43:8084–8093. doi: 10.1021/bi049817l. [DOI] [PubMed] [Google Scholar]
- 27.Hatters DM, MacPhee CE, Lawrence LJ, Sawyer WH, Howlett GJ. Human apolipoprotein C-II forms twisted amyloid ribbons and closed loops. Biochemistry. 2000;39:8276–8283. doi: 10.1021/bi000002w. [DOI] [PubMed] [Google Scholar]
- 28.Teoh CL, Pham CL, Todorova N, Hung A, Lincoln CN, Lees E, Lam YH, Binger KJ, Thomson NH, Radford SE, Smith TA, Muller SA, Engel A, Griffin MD, Yarovsky I, Gooley PR, Howlett GJ. A structural model for apolipoprotein C-II amyloid fibrils: experimental characterization and molecular dynamics simulations. J Mol Biol. 405:1246–1266. doi: 10.1016/j.jmb.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Ryan TM, Howlett GJ, Bailey MF. Fluorescence detection of a lipid-induced tetrameric intermediate in amyloid fibril formation by apolipoprotein C-II. The Journal of biological chemistry. 2008;283:35118–35128. doi: 10.1074/jbc.M804004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffin MD, Mok ML, Wilson LM, Pham CL, Waddington LJ, Perugini MA, Howlett GJ. Phospholipid interaction induces molecular-level polymorphism in apolipoprotein C-II amyloid fibrils via alternative assembly pathways. J Mol Biol. 2008;375:240–256. doi: 10.1016/j.jmb.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 31.Hatters DM, Lawrence LJ, Howlett GJ. Sub-micellar phospholipid accelerates amyloid formation by apolipoprotein C-II. FEBS Lett. 2001;494:220–224. doi: 10.1016/s0014-5793(01)02355-9. [DOI] [PubMed] [Google Scholar]
- 32.Ryan TM, Griffin MD, Teoh CL, Ooi J, Howlett GJ. High-affinity amphipathic modulators of amyloid fibril nucleation and elongation. J Mol Biol. 2011;406:416–429. doi: 10.1016/j.jmb.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 33.Wilson LM, Pham CL, Jenkins AJ, Wade JD, Hill AF, Perugini MA, Howlett GJ. High density lipoproteins bind Abeta and apolipoprotein C-II amyloid fibrils. J Lipid Res. 2006;47:755–760. doi: 10.1194/jlr.C500022-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 36.Schuck P. Sedimentation patterns of rapidly reversible protein interactions. Biophys J. 98:2005–2013. doi: 10.1016/j.bpj.2009.12.4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryan TM, Griffin MD, Teoh CL, Ooi J, Howlett GJ. High-affinity amphipathic modulators of amyloid fibril nucleation and elongation. J Mol Biol. 406:416–429. doi: 10.1016/j.jmb.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 38.Stewart CR, Wilson LM, Zhang Q, Pham CL, Waddington LJ, Staples MK, Stapleton D, Kelly JW, Howlett GJ. Oxidized cholesterol metabolites found in human atherosclerotic lesions promote apolipoprotein C-II amyloid fibril formation. Biochemistry. 2007;46:5552–5561. doi: 10.1021/bi602554z. [DOI] [PubMed] [Google Scholar]
- 39.Hanson CL, Ilag LL, Malo J, Hatters DM, Howlett GJ, Robinson CV. Phospholipid complexation and association with apolipoprotein C-II: insights from mass spectrometry. Biophys J. 2003;85:3802–3812. doi: 10.1016/S0006-3495(03)74795-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pham CL, Hatters DM, Lawrence LJ, Howlett GJ. Cross-linking and amyloid formation by N- and C-terminal cysteine derivatives of human apolipoprotein C-II. Biochemistry. 2002;41:14313–14322. doi: 10.1021/bi026070v. [DOI] [PubMed] [Google Scholar]
- 41.Singer MA, Lindquist S. Multiple effects of trehalose on protein folding in vitro and in vivo. Mol Cell. 1998;1:639–648. doi: 10.1016/s1097-2765(00)80064-7. [DOI] [PubMed] [Google Scholar]
- 42.Borwankar T, Rothlein C, Zhang G, Techen A, Dosche C, Ignatova Z. Natural osmolytes remodel the aggregation pathway of mutant huntingtin exon 1. Biochemistry. 50:2048–2060. doi: 10.1021/bi1018368. [DOI] [PubMed] [Google Scholar]
- 43.Stockl M, Fischer P, Wanker E, Herrmann A. Alpha-synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J Mol Biol. 2008;375:1394–1404. doi: 10.1016/j.jmb.2007.11.051. [DOI] [PubMed] [Google Scholar]
- 44.Andreola A, Bellotti V, Giorgetti S, Mangione P, Obici L, Stoppini M, Torres J, Monzani E, Merlini G, Sunde M. Conformational switching and fibrillogenesis in the amyloidogenic fragment of apolipoprotein a-I. J Biol Chem. 2003;278:2444–2451. doi: 10.1074/jbc.M204801200. [DOI] [PubMed] [Google Scholar]
- 45.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao H, Tuominen EK, Kinnunen PK. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 2004;43:10302–10307. doi: 10.1021/bi049002c. [DOI] [PubMed] [Google Scholar]
- 47.Olofsson A, Borowik T, Grobner G, Sauer-Eriksson AE. Negatively charged phospholipid membranes induce amyloid formation of medin via an alpha-helical intermediate. J Mol Biol. 2007;374:186–194. doi: 10.1016/j.jmb.2007.08.064. [DOI] [PubMed] [Google Scholar]