

Abstract
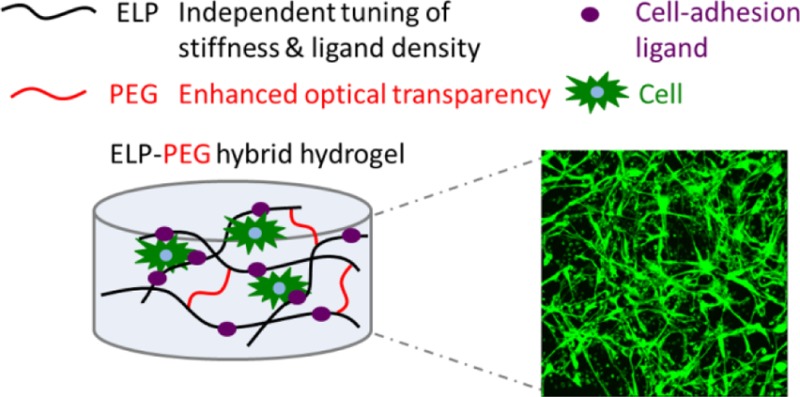
Hydrogels have been developed as extracellular matrix (ECM) mimics both for therapeutic applications and basic biological studies. In particular, elastin-like polypeptide (ELP) hydrogels, which can be tuned to mimic several biochemical and physical characteristics of native ECM, have been constructed to encapsulate various types of cells to create in vitro mimics of in vivo tissues. However, ELP hydrogels become opaque at body temperature because of ELP’s lower critical solution temperature behavior. This opacity obstructs light-based observation of the morphology and behavior of encapsulated cells. In order to improve the transparency of ELP hydrogels for better imaging, we have designed a hybrid ELP-polyethylene glycol (PEG) hydrogel system that rapidly cross-links with tris(hydroxymethyl) phosphine (THP) in aqueous solution via Mannich-type condensation. As expected, addition of the hydrophilic PEG component significantly improves the light transmittance. Coherent anti-Stokes Raman scattering (CARS) microscopy reveals that the hybrid ELP-PEG hydrogels have smaller hydrophobic ELP aggregates at 37 °C. Importantly, this hydrogel platform enables independent tuning of adhesion ligand density and matrix stiffness, which is desirable for studies of cell–matrix interactions. Human fibroblasts encapsulated in these hydrogels show high viability (>98%) after 7 days of culture. High-resolution confocal microscopy of encapsulated fibroblasts reveals that the cells adopt a more spread morphology in response to higher RGD ligand concentrations and softer gel mechanics.
Introduction
In vivo, cells grow within a complex network of extracellular matrix (ECM), which provides mechanical support while directing multiple types of cell behavior. Building three-dimensional scaffolds that recapitulate aspects of native cellular microenvironments for in vitro cell culture is of great significance to study cell and tissue physiology and to grow replacement tissue for regenerative medicine.1−3 Hydrogels, that is, cross-linked networks that possess high water content, are characterized by tissuelike elasticity and facile diffusion of biomolecules, making them attractive candidates for mimicking soft tissue microenvironments.4,5 Furthermore, many hydrogels, such as polyethylene glycol (PEG) hydrogels, can be formed under mild, cytocompatible conditions and are easily modified to possess cell adhesion ligands, specific mechanical properties, and cell-mediated degradability.6−11 Elastin-like polypeptides (ELPs), a class of artificial polypeptides inspired by the amino acid sequence of tropoelastin, are composed of the pentapeptide repeat Val-Pro-Gly-Xaa-Gly, where the guest residue Xaa can be any amino acid except Pro.12,13 Genetically engineered ELPs, with precisely controlled sequences and molecular weights,13 have been used to create a family of protein-based hydrogels for tissue engineering.14−19 Previously we reported the design of ELP hydrogels with selective bioactive sequences interspersed within the elastin-like repeats to enable tailored rates of enzymatic degradation14 and cell adhesion interactions15 to better mimic the extracellular microenvironment. However, ELP undergoes a phase transition to form hydrophobic aggregates at physiological temperature, causing increased light scattering and hence poor optical transparency.16,17 This low light transmittance obstructs the observation and in-depth investigation of the morphology and behavior of encapsulated cells. Therefore, the motivation of this study is to develop a hybrid ELP-PEG hydrogel system with good light transmittance and independently tunable cell-adhesive ligand density and matrix stiffness.
Both cell-adhesive ligand density and matrix stiffness can greatly impact cell behavior.18−23 The tripeptide RGD, which is an integrin-specific, cell-binding sequence found within fibronectin and several other ECM molecules,24 has been widely used in many types of hydrogels.25−28 Cell proliferation, adhesion, spreading, migration, and differentiation can be influenced by the overall density of matrix-bound, cell-adhesive RGD peptides,18,19 by nanoscale ligand clustering,20,21 and matrix stiffness.22,23 To date, the majority of these studies have been performed on two-dimensional (2D) cultures.29 However, cell behavior can be dramatically different between 2D and three-dimensional (3D) cultures.2,3,30−32 Within 3D cell cultures, physical inhibition of cell spreading by the surrounding polymer matrix can occur.33,34 In addition, perturbations in gene expression may arise as the cell is surrounded by a 3D microenvironment as compared to experiencing a 2D substrate.1,31,35,36
Several materials are being developed to tune cell-adhesive ligand density and matrix stiffness independently in 3D cell cultures.11,37−40 Among them, ELP hydrogels enable straightforward and independent tuning of material properties,26,41 but the maximum imaging distance observed so far is limited to approximately 100 μm, that is, ∼10 mammalian cell layers, using confocal microscopy.26 This limited imaging depth is due to the light scattering caused by ELP thermal aggregates at physiological temperature and the increased light scattering intensity of polymer gels after cross-linking.42,43 Conversely, PEG hydrogels have very good imaging properties, although elegant orthogonal chemistry is required to enable independent tuning of the matrix stiffness and cell-adhesive ligand density.44,45 Therefore, to combine the advantages of ELP hydrogels’ independent tuning of material properties and PEG hydrogels’ optical transparency, we developed a hybrid ELP-PEG hydrogel system. We hypothesized that grafting hydrophilic PEG onto the hydrophobic ELP backbone would decrease the formation of hydrophobic aggregates within the hydrogel, thereby resulting in less light scattering. Consistent with this hypothesis, compared with pure ELP hydrogels, light transmittance was greatly improved, and smaller hydrophobic aggregates were observed in the hybrid ELP-PEG hydrogels. Meanwhile, the density of the RGD integrin-binding ligand can be easily tuned by altering the primary amino acid sequence of the ELP component without influencing the scaffold stiffness. Similarly, the mechanical properties of ELP-PEG hydrogels can be easily tailored by altering the cross-linking density. Human fibroblasts were successfully encapsulated in the hybrid ELP-PEG hydrogels to study the effects of integrin ligand density and matrix stiffness on their spreading morphology in a 3D environment.
Materials and Methods
Elastin-like Polypeptide (ELP) Expression and Purification
The design and synthesis of a modular recombinant ELP was previously reported, containing bioactive domains and lysine residues to act as amine-reactive cross-linking sites.15,29 The amino acid sequences of RGD-ELP and RDG-ELP used in these experiments are shown in the Supporting Information, Figure S1. ELP was expressed and purified using standard recombinant protein technology. Briefly, protein sequences were cloned into pET15b plasmids, expressed in Escherichia coli, strain BL21(DE3), and induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at an OD600 of 0.8 for ∼6 h. The harvested cell pellets were suspended, lysed by three freeze–thaw cycles, and purified by iterative inverse temperature-cycling as previously reported.15,46 Protein molecular weight and purity were confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Purified ELP was dialyzed three times (10 000 molecular weight cutoff, 36 h, 4 °C, deionized water) to desalt. The ELP was then lyophilized and stored at 4 °C until use.
Formation of Cross-Linked Hydrogels
PEG bis(amine) (average Mn 3400, Sigma-Aldrich, St. Louis, MO), lyophilized RGD-ELP, and lyophilized RDG-ELP were solubilized in chilled phosphate buffered saline (PBS) (1×, pH 7.4) at a stock concentration of 12.5 wt % separately and dissolved by vortex. Air bubbles were removed by centrifugation, and the final solutions were kept on ice until use. An initial cross-linker stock solution of 16.1 mg/mL tris(hydroxymethyl)phosphine (THP, Sigma-Aldrich) was prepared in PBS and kept on ice. The stoichiometric cross-linking ratio (X) of cross-linker reactive hydroxyl groups (3 per THP molecule) to total primary amine groups (14 per ELP chain and 2 per PEG chain) was varied by adjusting the concentration of THP stock solution during the experiments. The hydrogel precursor solutions were mixed with the cross-linker at a 4:1 volume ratio to yield cross-linked hydrogels that consisted of 5 wt % ELP and 0, 1, 2.5, or 5 wt % of PEG. Cross-linking ratios of 1.10, 1.65, and 2.20 were used in different experiments. To control the density of RGD ligands, RGD-ELP and RDG-ELP were mixed at different ratios while maintaining a constant 5 wt % ELP.
Characterization with Fourier Transform Infrared Spectroscopy (FTIR)
For FTIR characterization, 5 wt % ELP hydrogels and 5 wt % ELP-2.5 wt % PEG hydrogels were prepared as described above. The hydrogels were submersed in PBS and incubated at 37 °C for 48 h. During the incubation period, PBS was changed every 8 h in order to fully remove any uncross-linked molecules. Measurements of PEG bis(amine), lyophilized ELP hydrogels, and ELP-PEG hydrogels were performed using an FTIR spectrometer (Vertex 70, Bruker Optics). Air was used as a background control, and a single measurement consisted of 32 scans with a resolution of 4 cm–1.
Characterization with Coherent Anti-Stokes Raman Scattering (CARS) Microscopy
In order to visualize the degree of ELP aggregate formation in the ELP versus ELP-PEG hydrogels, maps of the carbon–hydrogen vibration at 2930 cm–1,47 characteristic for proteins, were collected by CARS microscopy. The CARS microscope is described in detail elsewhere.48,49 Briefly, a Nd:Vanadate laser (Picotrain, HighQ Lasers GmbH, Hohenems, Austria) generated two ps pulsed laser beams (532 and 1064 nm, 7 ps, 76 MHz), the 532 nm beam of which pumped an optical parametric oscillator (Levante Emerald OPO, Angewandte Physik & Elektronik GmbH, Berlin, Germany, 690–900 nm). The OPO was tuned to 811 nm in order to drive the carbon–hydrogen vibration at 2930 cm–1 by overlapping it in time and space with the fundamental 1064 nm beam of the pump laser in the sample. The two excitation beams were focused onto the sample plane by an oil immersion objective (Nikon Plan Fluor, 40× NA 1.30) mounted in an inverted optical microscope (Eclipse TE2000-E with a C2 Confocal Microscope scanning head, Nikon). The near-infrared excitation beams assured deep penetration depth and the label-free approach of CARS microscopy ascertained imaging of the true ELP aggregate distribution, unbiased by photodegradation and 3D diffusion properties of labeling molecules. A spatial resolution of ∼300 nm was achieved, as the emission of the CARS signal is limited to the high-intensity region of the focal volume. A single-photon counting detector from Becker & Hickl GmbH was used to detect the CARS signal by simultaneously pixelwise scanning the two excitation beams over the sample. Dichroic mirrors and high optical-density filters were used to separate the CARS signal from the excitation beams before the detector.
Light Transmittance Measurements
To compare the transmittance of pure ELP and ELP-PEG hydrogels, 30 μL of gel solution was pipetted into 96-well plates (resulting in gels with thickness of ∼1000 μm), followed by a 15 min incubation at room temperature to initiate cross-linking, a 10 min incubation at 37 °C and submersion in PBS to mimic cell-encapsulation, and a 24 h equilibration at 4, 25, or 37 °C. Three samples were prepared in each group. The absorbance at 500 nm was determined using a SpectraMax M2 microplate reader. As the poor light transmittance in these samples is due to light scattering (and not absorbance), the absorbance values reported by the microplate reader were converted to transmittance through the Beer–Lambert Law.
Hydrogel Mechanical Characterization
Mechanical testing was performed on a stress-controlled ARG2 rheometer (TA Instruments) using a 20 mm diameter cone-on-plate geometry for gelation time characterization and an 8 mm plate-on-plate geometry for elastic and shear moduli measurements. For gelation time characterization, samples were allowed to gel in situ on the rheometer. Time sweeps were performed at an oscillatory stress of 4.74 Pa at 25 °C. The gelation time was defined as the time at which the sample strain curve reached an inflection point. For other mechanical characterizations, hydrogel cylinders were formed in silicone molds with 8 mm diameter and 1 mm depth on top of glass microscope slides. After incubation for 15 min at room temperature and 10 min at 37 °C, the hydrogels were submersed in a pool of PBS at 37 °C for 24 h. The silicone mold was carefully removed before each test. The top plate was lowered to a gap distance of 1500 μm, and the outer edge of the hydrogel was covered with PBS to prevent dehydration during experimentation.
Elastic modulus testing was performed on the hydrogels at a 2 μm/s strain rate in unconfined compression. Normal stress was calculated by dividing normal force over the hydrogel cross-sectional area, and engineering strain was calculated as the change in the gap distance divided by the original gap distance. The elastic modulus in compression mode was determined by the initial slope of the generated stress–strain curve from 0 to 7% strain. Dynamic strain sweeps and frequency sweeps were performed on samples compressed to a gap of 600 μm. A strain sweep from 0.1 to 1000% was performed at an angular frequency of 1 Hz to test the linear viscoelastic region (LVR). Angular frequency sweeps were conducted from 0.1 to 1 Hz with constant 1% strain amplitude. Storage (G′) and loss (G″) moduli at 1 Hz were selected from the frequency sweep. The mass swelling ratio was calculated from the hydrogel wet mass divided by the dry mass after lyophilization (both wet mass and dry mass were adjusted by subtracting the mass of salt). All measurements were in triplicate.
Human Fibroblast Culture
Human normal fibroblasts (ATCC CRL-2522) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 IU/mL penicillin-streptomycin at 37 °C and 5% atmospheric CO2. Cells were expanded and passaged by trypsinization for subsequent use in assays. For optical comparison experiments, the cells were encapsulated at a final concentration of 500 cells/μL to allow observation of distinct individual cells. For live/dead and cell spreading studies, cells were encapsulated at a final concentration of 3.5 × 103 cells/μL. At days 0, 4, and 7, cell viability was assessed with a fluorescent live/dead cytotoxicity kit (Molecular Probes, 2.0 mM calcein AM and 4.0 mM ethidium homodimer). Cells were stained for 45 min at 37 °C and 5% CO2. After staining, the gels were immediately imaged as a 3D stack using confocal microscopy (Leica SPE). Z-Stacks of 500 μm depth into the hydrogels were captured with 2.39-μm intervals between slices. Three-dimensional projections and maximum projection images of front view, top view, and side view were assessed using the Leica LAS AF software. For spreading analysis, cells were fixed overnight in 4% paraformaldehyde and blocked with 0.1% v/v Triton X-100 in PBS for 2 min at room temperature. After rinsing, samples were stained with Hoechst (1:5000) for cell nuclei and with rhodamine conjugated phalloidin (1:200 dilution, Invitrogen) for F-actin.
Statistical Analysis
All data (gelation time, modulus, swelling ratio, transmittance, cell viability, and cell spreading) are represented as mean ± standard deviation. Statistical difference between samples was analyzed by one way ANOVA and Tukey post test, performed using Statistical Analysis Software (SAS). For all statistical tests, a threshold value of α = 0.05 was chosen, and a p-value at or below 0.05 indicated significance.
Results and Discussion
Improved Optical Transmittance
To improve the optical properties of ELP hydrogels, we incorporated hydrophilic PEG to make hybrid ELP-PEG hydrogels (Figure 1A). FTIR spectra of ELP hydrogels (5 wt %), unreacted PEG bis(amine), and ELP-PEG hydrogels (5 and 2.5 wt %, respectively) were collected to confirm successful incorporation of PEG bis(amine) into ELP hydrogels (Figure 1B). Compared with pure ELP hydrogels, the significantly stronger absorption band at 1091.8 cm–1 in the spectrum of the hybrid gel was attributed to the stretching vibrations of the ether C–O–C in PEG, indicating the existence of covalently linked PEG in the hydrogel. In addition, the slightly stronger absorption bands at 1238.1, 2866.9, and 3286.6 cm–1 corresponded to stretching vibration of C–N, alkyl C–H, and amine N–H, respectively. These also suggested that PEG-bis(amine) was successfully reacted with the THP cross-linker during gelation. By visual comparison, the hybrid ELP-PEG hydrogels achieved substantially improved optical transparency compared with the pure ELP hydrogels (Figure 1C). While the pure ELP hydrogel became opaque at 37 °C, the hybrid ELP-PEG gel retained its partially transparent nature across the range of 4–37 °C.
Figure 1.

ELP-PEG hybrid hydrogels have improved optical transparency. (A) Schematic of ELP-PEG hydrogel structure. Cross-linker THP reacts with the amine groups in ELP and PEG bis(amine) to create hybrid ELP-PEG gels. (B) FTIR spectra of pure ELP hydrogel (5 wt %), ELP-PEG hydrogel (5 wt %-2.5 wt % respectively), and PEG bis(amine). Enhanced absorbance at the characteristic ether peak (1091.8 cm–1) indicates successful incorporation of PEG bis(amine) into ELP-PEG hydrogels. (C) Visual comparison of the optical transparencies of ELP hydrogel (5 wt %) and ELP-PEG hydrogel (5 and 2.5 wt %, respectively) at 4, 25, and 37 °C. For all gels, the cross-linking ratio, X, is kept constant at 2.20.
ELPs are thermally responsive and undergo transitions from a more soluble state to a less soluble state by hydrophobic interactions when the temperature is raised above its lower critical solution temperature (LCST).50 Uncross-linked ELP has a transition temperature (Tt) of 33.9 °C, while the Tt for the cross-linked ELP gel is below 25 °C (Supporting Information, Figure S2). This is due to the decrease in the mean polarity caused by the reaction of amine groups upon cross-linking to form a gel. In addition, studies have shown that the light scattering intensity from polymer gels is always larger than that from the solution of the same polymer at the same concentration, which is due to spatial gel inhomogeneity.42,43 By adding PEG segments, the Tt of hybrid ELP-PEG gels was significantly increased. Due to both the restricted temperature limit of the instrument (25–45 °C) and the broadened thermal transition resulting from hindered chain mobility in the hydrogels,51 the actual Tt could not be measured for the hybrid hydrogels. Nonetheless, the trend was clear that Tt increased as the PEG content was increased from 0 to 5% (Figure S2). This was consistent with previous studies showing that the addition of hydrophobic residues resulted in a lower Tt for ELP, whereas addition of polar residues increased Tt due to the tendency for hydrophilic residues to resist aggregation.52,53 Additionally, we observed increasing light transmittance at 37 °C in hydrogels with increasing PEG concentrations when keeping the cross-linker concentration constant (Figure S2). Because PEG is known to play a role in resisting cell adhesion,54 we selected a formulation with the least amount of PEG (2.5 wt %) required to still achieve a high light transmittance (0.80) at 37 °C for subsequent study.
Compared to pure ELP hydrogel, light transmittance of the hybrid ELP-PEG hydrogel was improved across a range of wavelengths from 400 to 800 nm (Supporting Information, Figure S3). Quantification of light transmittance at a wavelength of 500 nm confirmed that hybrid ELP-PEG hydrogels were significantly more transparent than pure ELP gels at 4, 25, and 37 °C (Figure 2A). These data also revealed that the light transmittance of both ELP and ELP-PEG hydrogels were decreased as temperature increased, which was attributed to the thermal aggregation of ELP. To confirm this thermal aggregation hypothesis, we observed the gel structures using CARS microscopy. In pure ELP hydrogels, small aggregates on the order of 1 μm in size were observed (Figure 2B, left panels). As this length scale is larger than the wavelength of visible light, these aggregates serve as significant light scattering centers. In contrast, a more homogeneous structure and smaller hydrophobic aggregates were observed in the hybrid ELP-PEG gels compared with pure ELP gels (Figure 2B, right panels). ELP hydrophobic aggregates presumably form as a result of chain collapse through hydrophobic interactions after the bound water molecules surrounding the nonpolar solutes are expelled.55 The incorporated hydrophilic PEG helped to retain bound water and hence resulted in smaller hydrophobic aggregates.
Figure 2.

Comparison of the optical properties of ELP hydrogels (5 wt %) and ELP-PEG hydrogels (5 and 2.5 wt %, respectively). (A) Light transmittance (λ = 500 nm) at 4, 25, and 37 °C (* p < 0.05). (B) Gel structure observed by CARS microscopy. (C, D) Confocal 3D reconstructions of encapsulated, viable human fibroblasts, stained by calcein AM, in ELP and ELP-PEG hydrogels.
To check if this improvement in light transmittance was sufficient to improve light microscopy observation of cell behavior, we encapsulated human normal fibroblasts in both pure ELP and hybrid ELP-PEG hydrogels. The 3D cell-gel constructs were immersed in full cell culture medium, DMEM with 10% fetal bovine serum, for 6 h prior to live/dead staining. The 3D reconstruction of images obtained through confocal microscopy showed that cells could be observed in the hybrid ELP-PEG gels to a depth of ∼500 μm along the Z-axis (Figure 2D). In contrast, only two to three cell layers, with a Z depth of ∼50 μm were observed in the control ELP gels (Figure 2C). Thus, a higher number of cells were observed in the maximum projection of the XY-plane in the hybrid gels, even though the initial numbers of encapsulated cells were kept the same. To prove that there were indeed cells within the higher Z-coordinates in the pure ELP hydrogel, the sample was flipped over and observed from the opposite direction (Supporting Information, Figure S4). One cell layer was observed, indicating that cells were dispersed throughout the pure ELP gel.
Independent Tuning of Ligand Density and Matrix Stiffness
In order to further optimize the hydrogel formulation for cell encapsulation, THP concentrations were altered to tune the gelation time. Generally speaking, shorter gelation times are desirable to achieve homogeneous cell encapsulation and to prevent cell sedimentation.56 By increasing the cross-linking ratio (X), that is, ratio of hydroxyls in THP to overall primary amine groups in ELP and PEG, a shorter gelation time was achieved (Figure 3A). When adjusting the cross-linking ratio from 1.10 to 1.65 to 2.20, gelation time decreased significantly from 38 to 13 to 10 min, respectively (Figure 3B). This result is consistent with previous work by Chung et al. showing that gelation time decreased with the increasing concentration of the similar cross-linker THPC.41 Cross-linking ratios of 1.65 and 2.20 were selected for further mechanical characterization and cell encapsulation experiments.
Figure 3.

Rheological analysis of ELP-PEG hydrogels (5 and 2.5 wt %, respectively) with different cross-linking ratios (X). (A) Time sweep and (B) gelation time quantification of ELP-PEG hydrogels (X = 1.10, 1.65, and 2.20). (C) Shear storage (G′) and loss (G″) moduli and (D) elastic moduli of ELP-PEG hydrogels (X = 1.65, 2.20) fabricated from RGD-ELP or RDG-ELP.
Oscillatory strain sweeps were performed on the cross-linked hydrogels to determine the linear viscoelastic region (LVR). For the ELP-PEG hybrid gels, LVR terminated at a critical strain of ∼8% (Supporting Information, Figure S5A). Within the LVR, the storage and loss moduli were largely independent of the oscillatory strain amplitude, whereas above the critical strain, the gel structure was damaged. Thus, 1% strain, which is well within the LVR, was chosen for subsequent rheological tests. The angular frequency sweep showed that the storage moduli (G′) remained constant over the frequency range tested and were much higher than the loss moduli (G″), indicating the formation of an elastic polymer network (Supporting Information, Figure S5B). In addition, compared with pure ELP hydrogels, hybrid ELP-PEG hydrogels had lower shear storage moduli and lower elastic moduli (Supporting Information, Figure S6B–D). This indicates that addition of PEG can serve as a new way to tune the mechanical properties of ELP-based hydrogels.
To show that hydrogel ligand density can be tuned without changing matrix stiffness, the shear storage moduli and elastic moduli were compared between ELP-PEG hybrid hydrogels composed either of ELP containing the cell-adhesive RGD peptide or of ELP containing a nonadhesive RDG peptide (Supporting Information, Figure S1). At the same cross-linking ratio, no significant differences were found between the shear moduli or elastic moduli of RGD-ELP/PEG hydrogels and the scrambled RDG-ELP/PEG hydrogels (Figure 3C, D). Similarly, the hydrogel mass swelling ratio was similar between RGD-ELP and RDG-ELP containing hybrid hydrogels (Supporting Information, Figure S7).
At a cross-linking ratio of 2.20, both the shear storage moduli and the elastic moduli were significantly increased compared to a cross-linking ratio of 1.65. Theoretically, the largest moduli are expected when all of the potential cross-links within a polymeric network have reacted; therefore, if a cross-linking reaction is 100% efficient, then a cross-linking ratio of exactly 1 should yield the stiffest hydrogels. However, many amine-reactive cross-linkers have reaction efficiencies less than 100%, resulting in stiffer hydrogels at cross-linker ratios larger than 1. For instance, N-hydroxysuccinimide (NHS) esters, a widely used cross-linker for lysine residues in proteins, are usually used in 2–50 fold molar excess.57,58 The increase in ELP-PEG moduli at a cross-linking ratio of 2.20 compared to 1.65 suggests that, similar to other amine-reactive cross-linkers, the THP reaction is not 100% efficient.
Taken together, these data indicate that the adhesion ligand density can be tuned independently from the matrix stiffness by altering the concentration of RGD-ELP and RDG-ELP in the hydrogel while keeping the same overall ELP mass percentage. This kind of independent variation of mechanical and biochemical signals is very important for understanding fundamental mechanisms of cell–matrix interaction.59−61
Human Fibroblast Encapsulation and Cellular Response to Matrix Stiffness and Adhesion Ligands
To analyze the cytocompatibility of the hybrid ELP-PEG hydrogels, we encapsulated human normal fibroblasts within the hydrogels and analyzed cell viability using live/dead staining. Fibroblasts encapsulated in the hybrid hydrogels had over 98% viability at days 4 and 7 after encapsulation (Figure 4B, D), indicating that the hybrid material and the selected cross-linking chemistry is suitable for 3D cell encapsulation.
Figure 4.

Viability and morphology of human fibroblasts encapsulated in hybrid ELP-PEG hydrogels (5 and 2.5 wt %, respectively) with tunable RGD ligand density and matrix stiffness. (A) Day 4 and (C) day 7 live/dead (green/red) confocal projection images of cells encapsulated in RGD-ELP/PEG, X = 1.65 (upper left); RDG-ELP/PEG, X = 1.65 (upper right); RGD-ELP/PEG, X = 2.20 (lower left); and RDG-ELP/PEG, X = 2.20 (lower right). (B) Day 4 and (D) day 7 cell viability quantification. (E) Representative day 7 confocal images of cell morphology. Cell nuclei were stained with DAPI (blue), and F-actin stained with phalloidin (red). (F) Quantification of the percentage of spread cells.
The encapsulated fibroblasts were observed to respond to changes in ligand density and matrix stiffness. At day 4, cells rarely exhibited spread morphology and remained rounded in the RDG-ELP/PEG hydrogels at cross-linking ratios (X) of both 1.65 and 2.20 (Figure 4A). In contrast, cells adopted a more spread morphology in the RGD-ELP/PEG hydrogels. In addition, more cells were found to spread in the more compliant gels, that is, those formulated with a smaller cross-linking ratio. These results are consistent with other studies showing that cells can interact with the adhesive RGD ligand when presented in a number of different contexts.8,32,62 The RGD peptide is well-known to initiate cell binding through integrin cell-surface receptors and thereby promote cell adhesion, spreading, and actin-filament organization.63
To further analyze spreading and actin cytoskeletal structure, day 7 cultures were imaged using nuclear (DAPI) and F-actin (phalloidin) stains (Figure 4E). Similar to the live/dead staining results, negligible cell spreading was observed in the absence of the RGD ligand (Figure 4F). Consistent with other reports that matrix stiffness can influence cell morphology,22,44,64 we also found that cell spreading was dependent on the matrix stiffness. About 60% of the cells spread within the more compliant hydrogel (X = 1.65, E ∼1300 Pa) at day 7, while within the stiffer hydrogel (X = 2.20, E ∼2500 Pa) only about 3% of the cells showed a spread morphology (Figure 4E, F). These results may be a consequence of the smaller hydrogel mesh size and/or the increased stiffness of hydrogels fabricated with higher cross-linker concentrations. Both smaller hydrogel mesh sizes and stiffer hydrogels have been reported to restrict 3D cell spreading.65−67 In our previously published work on pure ELP gels, decreasing the ELP content to 3 wt % and the cross-linking stoichiometry (X) to 0.5 resulted in gels with storage moduli of ∼0.5 kPa and improved cell spreading.26,41,46 In comparison, the ELP-PEG gels utilized here had an overall polymer wt % of 7.5%. Future studies will explore the creation of ELP-PEG hydrogels with lower polymer wt % and further decreased stoichiometric cross-linking, which may further enhance cell spreading.
Conclusions
We have successfully developed a new hybrid ELP-PEG hydrogel system, combining the tunability of ELP hydrogels with the optical advantages of PEG hydrogels. Human fibroblasts encapsulated in this hybrid gel system showed very high viability and uniform distribution throughout the gel. In addition, this hydrogel system enabled flexible and tailored tuning of the material stiffness and the cell-adhesive RGD ligand density. The hybrid ELP-PEG hydrogel could be further optimized by incorporating new ELP sequences with different biochemical ligands or degradation sites. Similarly, various multiarm PEG variants could be included to further tune mechanical properties. The versatility and cytocompatibility of this new family of hybrid hydrogels suggests that they have great potential for future use as in vitro tissue mimics for fundamental studies of cell–matrix interactions.
Acknowledgments
The authors acknowledge funding provided by NSF DMR-0846363, NIH R21-AR062359, NIH DP2-OD-006477, and NIH R01-DK085720 (S.C.H.), Kodak fellowship (H.W.), EU FP7 program Grant 607842 (A.P. and A.E.) and VINNOVA VINNMER grant (A.E.). We acknowledge Prof. Fan Yang at Stanford University for the generous gift of human fibroblasts.
Supporting Information Available
Amino acid sequences of RGD-ELP and RDG-ELP (Figure S1). Transmittance (λ = 500 nm) versus temperature for different formulations (Figure S2). Transmittance (λ = 400–800 nm) of pure ELP and hybrid ELP-PEG hydrogels (Figure S3). Observation of encapsulated fibroblasts from bottom and top surfaces of ELP hydrogel (Figure S4). Strain sweep and angular frequency sweep rheology (Figure S5). Rheological comparison between pure ELP and ELP-PEG hydrid hydrogels (Figure S6). Mass swelling ratio of ELP-PEG hydrogels (Figure S7). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tibbitt M. W.; Anseth K. S. Biotechnol. Bioeng. 2009, 103, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampaloni F.; Reynaud E. G.; Stelzer E. H. K. Nat. Rev. Mol. Cell Biol. 2007, 8, 839. [DOI] [PubMed] [Google Scholar]
- Lee J.; Cuddihy M. J.; Kotov N. A. Tissue Eng., Part B 2008, 14, 61. [DOI] [PubMed] [Google Scholar]
- Kharkar P. M.; Kiick K. L.; Kloxin A. M. Chem. Soc. Rev. 2013, 42, 7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvendiren M.; Burdick J. A. Curr. Opin. Biotechnol. 2013, 24, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutolf M. P.; Hubbell J. A. Nat. Biotechnol. 2005, 23, 47. [DOI] [PubMed] [Google Scholar]
- Nicodemus G. D.; Bryant S. J. Tissue Eng., Part B 2008, 14, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick J. A.; Anseth K. S. Biomaterials 2002, 23, 4315. [DOI] [PubMed] [Google Scholar]
- Zhu J. M. Biomaterials 2010, 31, 4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney M. J.; Anseth K. S. Biomaterials 2006, 27, 2265. [DOI] [PubMed] [Google Scholar]
- Beck J. N.; Singh A.; Rothenberg A. R.; Elisseeff J. H.; Ewald A. J. Biomaterials 2013, 34, 9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betre H.; Setton L. A.; Meyer D. E.; Chilkoti A. Biomacromolecules 2002, 3, 910. [DOI] [PubMed] [Google Scholar]
- McDaniel J. R.; Bhattacharyya J.; Vargo K. B.; Hassouneh W.; Hammer D. A.; Chilkoti A. Angew. Chem., Int. Ed. 2013, 52, 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley K. S.; Heilshorn S. C. Adv. Mater. 2009, 21, 4148. [Google Scholar]
- Straley K. S.; Heilshorn S. C. Soft Matter 2009, 5, 114. [Google Scholar]
- Meyer D. E.; Trabbic-Carlson K.; Chilkoti A. Biotechnol. Prog. 2001, 17, 720. [DOI] [PubMed] [Google Scholar]
- Dreher M. R.; Raucher D.; Balu N.; Colvin O. M.; Ludeman S. M.; Chilkoti A. J. Controlled Release 2003, 91, 31. [DOI] [PubMed] [Google Scholar]
- Chua P. H.; Neoh K. G.; Kang E. T.; Wang W. Biomaterials 2008, 29, 1412. [DOI] [PubMed] [Google Scholar]
- Rajagopalan P.; Marganski W. A.; Brown X. Q.; Wong J. Y. Biophys. J. 2004, 87, 2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comisar W. A.; Kazmers N. H.; Mooney D. J.; Linderman J. J. Biomaterials 2007, 28, 4409. [DOI] [PubMed] [Google Scholar]
- Koo L. Y.; Irvine D. J.; Mayes A. M.; Lauffenburger D. A.; Griffith L. G. J. Cell Sci. 2002, 115, 1423. [DOI] [PubMed] [Google Scholar]
- Engler A. J.; Sen S.; Sweeney H. L.; Discher D. E. Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- Trappmann B.; Gautrot J. E.; Connelly J. T.; Strange D. G. T.; Li Y.; Oyen M. L.; Stuart M. A. C.; Boehm H.; Li B. J.; Vogel V.; Spatz J. P.; Watt F. M.; Huck W. T. S. Nat. Mater. 2012, 11, 642. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Annu. Rev. Cell Dev. Biol. 1996, 12, 697. [DOI] [PubMed] [Google Scholar]
- Lutolf M. P.; Lauer-Fields J. L.; Schmoekel H. G.; Metters A. T.; Weber F. E.; Fields G. B.; Hubbell J. A. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe K. J.; Antaris A. L.; Heilshorn S. C. Acta Biomater. 2013, 9, 5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley J. A.; Madlambayan G.; Mooney D. J. Biomaterials 1999, 20, 45. [DOI] [PubMed] [Google Scholar]
- Shu X. Z.; Ghosh K.; Liu Y. C.; Palumbo F. S.; Luo Y.; Clark R. A.; Prestwich G. D. J. Biomed. Mater. Res., Part A 2004, 68A, 365. [DOI] [PubMed] [Google Scholar]
- Liu J. C.; Tirrell D. A. Biomacromolecules 2008, 9, 2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J.; Brugge J. S. Nat. Rev. Cancer 2005, 5, 675. [DOI] [PubMed] [Google Scholar]
- Baker B. M.; Chen C. S. J. Cell Sci. 2012, 125, 3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Tong Z.; Jia X.; Kiick K. L. Soft Matter 2013, 9, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khetan S.; Katz J. S.; Burdick J. A. Soft Matter 2009, 5, 1601. [Google Scholar]
- Burdick J. A. Nature 2009, 460, 469. [DOI] [PubMed] [Google Scholar]
- Lee J.; Abdeen A. A.; Zhang D.; Kilian K. A. Biomaterials 2013, 34, 8140. [DOI] [PubMed] [Google Scholar]
- Fu J. P.; Wang Y. K.; Yang M. T.; Desai R. A.; Yu X. A.; Liu Z. J.; Chen C. S. Nat. Methods 2010, 7, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. J.; Chung E. H.; Rodriguez R. T.; Firpo M. T.; Healy K. E. J. Biomed Mater. Res., Part A 2006, 79A, 1. [DOI] [PubMed] [Google Scholar]
- Peyton S. R.; Raub C. B.; Keschrumrus V. P.; Putnam A. J. Biomaterials 2006, 27, 4881. [DOI] [PubMed] [Google Scholar]
- Huebsch N.; Arany P. R.; Mao A. S.; Shvartsman D.; Ali O. A.; Bencherif S. A.; Rivera-Feliciano J.; Mooney D. J. Nat. Mater. 2010, 9, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich T. A.; Jain A.; Tanner K.; MacKay J. L.; Kumar S. Biomaterials 2010, 31, 1875. [DOI] [PubMed] [Google Scholar]
- Chung C.; Anderson E.; Pera R. R.; Pruitt B. L.; Heilshorn S. C. Soft Matter 2012, 8, 10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okay O.; Oppermann W. Macromolecules 2007, 40, 3378. [Google Scholar]
- Shibayama M. Macromol. Chem. Phys. 1998, 199, 1. [Google Scholar]
- Kyburz K. A.; Anseth K. S. Acta Biomater. 2013, 9, 6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. M.; Tang C.; Kottke-Marchant K.; Marchant R. E. Bioconjugate Chem. 2009, 20, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.; Lampe K. J.; Heilshorn S. C. Biomacromolecules 2012, 13, 3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez P. L.; Sweet J. A.; Fink H.; Chennazhi K. P.; Nair S. V.; Enejder A.; Heilshorn S. C. Adv. Healthcare Mater. 2013, 2, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enejder A.; Brackmann C.; Svedberg F. IEEE J. Sel. Top. Quantum Electron. 2010, 16, 506. [Google Scholar]
- Evans C. L.; Xie X. S. Annu. Rev. Anal Chem. 2008, 1, 883. [DOI] [PubMed] [Google Scholar]
- Frandsen J. L.; Ghandehari H. Chem. Soc. Rev. 2012, 41, 2696. [DOI] [PubMed] [Google Scholar]
- Martin L.; Alonso M.; Girotti A.; Arias F. J.; Rodriguez-Cabello J. C. Biomacromolecules 2009, 10, 3015. [DOI] [PubMed] [Google Scholar]
- Urry D. W.; Luan C. H.; Parker T. M.; Gowda D. C.; Prasad K. U.; Reid M. C.; Safavy A. J. Am. Chem. Soc. 1991, 113, 4346. [Google Scholar]
- DiMarco R. L.; Heilshorn S. C. Adv. Mater. 2012, 24, 3923. [DOI] [PubMed] [Google Scholar]
- Patel S.; Thakar R. G.; Wong J.; McLeod S. D.; Li S. Biomaterials 2006, 27, 2890. [DOI] [PubMed] [Google Scholar]
- Urry D. W. J. Phys. Chem. B 1997, 101, 11007. [Google Scholar]
- Kuo C. K.; Ma P. X. Biomaterials 2001, 22, 511. [DOI] [PubMed] [Google Scholar]
- Mattson G.; Conklin E.; Desai S.; Nielander G.; Savage M. D.; Morgensen S. Mol. Biol. Rep. 1993, 17, 167. [DOI] [PubMed] [Google Scholar]
- Kalkhof S.; Sinz A. Anal. Bioanal. Chem. 2008, 392, 305. [DOI] [PubMed] [Google Scholar]
- Little L.; Healy K. E.; Schaffer D. Chem. Rev. 2008, 108, 1787. [DOI] [PubMed] [Google Scholar]
- Sengupta D.; Heilshorn S. C. Tissue Eng., Part B 2010, 16, 285. [DOI] [PubMed] [Google Scholar]
- Ulrich T. A.; de Juan Pardo E. M.; Kumar S. Cancer Res. 2009, 69, 4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner J. N.; Cherry K. M.; Su R. S. C.; Liu J. C. Biomacromolecules 2012, 13, 3678. [DOI] [PubMed] [Google Scholar]
- Chen C. S.; Tan J.; Tien J. Annu. Rev. Biomed. Eng. 2004, 6, 275. [DOI] [PubMed] [Google Scholar]
- Discher D. E.; Janmey P.; Wang Y. L. Science 2005, 310, 1139. [DOI] [PubMed] [Google Scholar]
- Liu Y. X.; Chan-Park M. B. Biomaterials 2009, 30, 196. [DOI] [PubMed] [Google Scholar]
- Benoit D. S. W.; Schwartz M. P.; Durney A. R.; Anseth K. S. Nat. Mater. 2008, 7, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A.; Hanseler P.; Ranga A.; Lutolf M. P.; Voros J.; Ehrbar M.; Weber F. E. Integr. Biol. 2011, 3, 1102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.