

Abstract
Amphiphilic macromolecules (AMs) composed of sugar backbones modified with branched aliphatic chains and a poly(ethylene glycol) (PEG) tail can inhibit macrophage uptake of oxidized low-density lipoproteins (oxLDL), a major event underlying atherosclerosis development. Previous studies indicate that AM hydrophobic domains influence this bioactivity through interacting with macrophage scavenger receptors, which can contain basic and/or hydrophobic residues within their binding pockets. In this study, we compare two classes of AMs to investigate their ability to promote athero-protective potency via hydrogen-bonding or hydrophobic interactions with scavenger receptors. A series of ether-AMs, containing methoxy-terminated aliphatic arms capable of hydrogen-bonding, was synthesized. Compared to analogous AMs containing no ether moieties (alkyl-AMs), ether-AMs showed improved cytotoxicity profiles. Increasing AM hydrophobicity via incorporation of longer and/or alkyl-terminated hydrophobic chains yielded macromolecules with enhanced oxLDL uptake inhibition. These findings indicate that hydrophobic interactions and the length of AM aliphatic arms more significantly influence AM bioactivity than hydrogen-bonding.
1. Introduction
Atherosclerosis, a major cause of mortality worldwide, is an inflammatory disease characterized by arterial plaque development.1−5 During the early stages of atherosclerosis, low-density lipoprotein (LDL) accumulates in the subendothelial space where various cells catalyze its oxidative modification.4,6−8 This oxidized LDL (oxLDL) initiates an inflammatory response, in which monocytes are recruited to sites of endothelial dysfunction, migrate into the subendothelial space, and subsequently differentiate into macrophages.1,3,4,6,7,9 Macrophages then internalize oxLDL primarily through scavenger receptors A (SRA) and B (CD36), resulting in unregulated modified lipid accumulation and foam cell formation.2,3,5−7,9 Foam cells promote the inflammatory process and lead to atherosclerotic plaque formation, narrowing the artery, and cardiovascular events, including hypertension, stroke, and myocardial infarction.2,3,6,9 In this work, we seek to mitigate atherogenesis via new designs of macromolecules that interfere with oxLDL uptake, and thus de-escalate the atherosclerotic development.
Statins are the most well-known and widely prescribed therapeutic for treating coronary artery disease.10,11 They slow the atherosclerotic cascade through inhibiting hepatic cholesterol biosynthesis and subsequently increase the expression of hepatic LDL receptors to lower serum LDL levels. However, statins can have undesirable side effects, including muscle toxicity, cognitive problems, and metabolic issues (e.g., liver toxicity or thyroid conditions), and as a result of their systemic administration and mechanism of action statins do not directly treat atherogenic sites in the arteries.10−12 When statins are not tolerated by patients or when patients are genetically predisposed to increased LDL levels as in familial hypercholesterolemia, lipid apheresis therapies can be used to extracorporeally remove plasma lipoproteins (i.e., LDL) from the blood.13−17 Apheresis methods often utilize adsorbents, which contain ligands that interact with and retain LDL, including dextran sulfate, polyacrylate, heparin, and phosphates, and carriers such as PVA microspheres, cellulose beads, nonwoven fabrics, and other polymer systems.13,14,16−19 While these therapies lower LDL levels and improve atherosclerosis outcomes, problems remain: long-term, expensive treatments ($40 000–100 000 USD annually) are required to maintain efficacy, treatment access is limited, and many current adsorbents have low LDL selectivity and poor mechanical properties.14−17,19,20 Consequently, researchers are currently targeting various steps in the atherosclerotic cascade described above, including monocyte recruitment, macrophage-mediated cholesterol metabolism, and plaque regression to impede the inflammatory progression and improve treatment efficacy.6
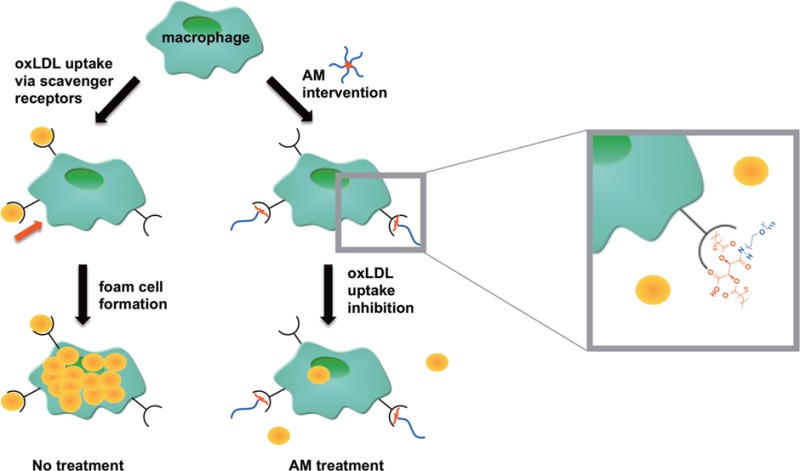
As an alternative strategy to treat atherosclerosis, researchers are investigating means to abrogate the atherosclerotic cascade by preventing oxLDL trafficking and uptake within the blood vessel walls.21,22 In vivo studies have indicated that apolipoprotein E-null mice deficient in certain scavenger receptors (e.g., SRA or CD36) result in significantly smaller atherosclerotic lesions and a decreased uptake of modified LDL (e.g., oxLDL).21−23 Given that oxLDL uptake can lead to foam cell formation and atherosclerotic plaque development, inhibiting oxLDL uptake could impede atherogenesis. Previously, our lab demonstrated that amphiphilic macromolecules (AMs) inhibit scavenger receptor-mediated oxLDL uptake, particularly through competitive inhibition of SRA and CD36.24 These sugar-based, PEGylated AMs are comprised of a sugar backbone that is acylated with aliphatic chains and conjugated to a poly(ethylene) glycol (PEG) tail.25 Given their amphiphilicity, AMs self-assemble into nanoscale micelles in aqueous environments25 with a PEG shell that may shield uptake by the reticuloendothelial system, potentially prolonging in vivo blood circulation times.26 Upon discovering AMs’ antiatherosclerotic activity, various studies were conducted to elucidate their bioactive mechanism. Dynamic light scattering (DLS) studies indicated that AMs containing an anionic charge (e.g., carboxylate moiety) within their hydrophobic domain complex with unmodified LDL, but do not complex with oxLDL, likely due to charge repulsion resulting from oxLDL’s increased net negative charge.27 As these AMs did not interact with oxLDL yet prevented its accumulation in macrophages, further immunolocalization and antibody blocking assays were conducted and demonstrated that AMs interact with macrophage scavenger receptors and subsequently prevent oxLDL uptake through these receptors.24,28−30 A library of AMs was generated by systematically modifying AM structural elements, and quantitative structure–activity relationship (QSAR) models were developed to determine the most prominent athero-protective AM features.31−34 The hydrophobic domain plays a key role; the presentation of the aliphatic arms influences AM athero-protective bioactivity.33−35
Though structure–activity relationships provided significant insights regarding AM efficacy, a more rational approach for developing bioactive AMs with increased potency would be inspired by the physicochemical attributes of the scavenger receptor binding pockets. While certain scavenger receptors contain basic residues in their oxLDL binding domains,36,37 others contain hydrophobic residues near their oxLDL binding sites.38 Increasing the AM hydrophobicity through extending alkyl chain lengths or decreasing PEG tail lengths could increase AM interactions with hydrophobic receptor pockets, whereas the addition of heteroatoms into the hydrophobic domain could enable hydrogen-bonding interactions with basic residues, ultimately reducing oxLDL uptake by mimicking scavenger receptor interactions with hydrophobic oxidized lipids.39−41 To decipher which interactions more effectively influence athero-protective bioactivity through repressing oxLDL uptake, a series of novel ether-containing AMs (ether-AMs) capable of hydrogen-bonding was synthesized based on a linear tartaric acid (TA) backbone and compared to analogous AMs containing no ether moieties (alkyl-AMs) that would exhibit stronger hydrophobic interactions (Figure 1). The relative hydrophobicity of all AMs was varied by altering both the aliphatic chain and PEG tail lengths to determine whether more lipophilic AMs would mimic the interactions of hydrophobic, oxidized lipids with scavenger receptors and thereby exhibit increased athero-protective bioactivity.
Figure 1.

Chemical structures of ether- and alkyl-AMs.
2. Materials and Methods
2.1. Materials
All reagents and solvents were purchased from Sigma-Aldrich (Milwaukee, WI) and used as received unless otherwise noted. Hydrochloric acid (HCl, 1 N), dibenzyl tartrate (DBT), and polytetrafluoroethylene (PTFE) syringe filters were purchased from Fisher Scientific (Fair Lawn, NJ). Silica gel was purchased from VWR (Radnor, PA). Monomethoxy-poly(ethylene glycol)-amine (mPEG-amine) was purchased from Laysan Bio (Arab, AL) and azeotropically distilled with toluene prior to use. Reagents for cell culture, toxicity studies, and oxLDL uptake studies include human buffy coats purchased from The Blood Center of New Jersey (East Orange, NJ), Ficoll-Paque premium 1.077 g/mL purchased from GE Healthcare (Fairfield, CT), RPMI-1640 purchased from ATCC (Manassas, VA), macrophage colony stimulating factor purchased from PeproTech (Rocky Hill, NJ), penicillin/streptomycin purchased from Lonza (Basel, Switzerland), alamarBlue assay, fetal bovine serum, and Hoechst 33342 purchased from Life Technologies (Carlsbad, CA), unlabeled oxLDL purchased from Biomedical Technologies Inc. (Ward Hill, MA), and 3,3′-dioctadecyloxacarbocyanine (DiO) labeled oxLDL purchased from Kalen Biomedical (Montgomery Village, MD).
2.2. Characterization
Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were obtained using a Varian 400 or 500 MHz spectrophotometer. Samples were dissolved in deuterated chloroform (CDCl3), and a few drops of dimethyl sulfoxide (DMSO-d6) added, if necessary; trimethylsilane was used as an internal reference. Fourier transform infrared (FT-IR) spectra were recorded on a Thermo Scientific Nicolet iS10 spectrophotometer using OMNIC software with an average of 32 scans. FT-IR samples were either pressed into potassium bromide (KBr) discs (1 wt % sample) or solvent-cast onto sodium chloride plates.
AM precursor molecular weights were determined using a ThermoQuest Finnigan LCQ-DUO system equipped with a syringe pump, an optional divert/inject valve, an atmospheric pressure ionization (API) source, a mass spectrometer (MS) detector, and the Xcalibur data system. Samples were prepared at a concentration of 10 μg/mL in methanol (MeOH) or dichloromethane (DCM) using 1% acetic acid or 1% ammonia for positive or negative ion modes, respectively. AM weight-averaged molecular weight (Mw) and polydispersity index (PDI) data were determined by gel permeation chromatography (GPC) using a Waters LC system (Milford, MA), equipped with a 2414 refractive index detector, 1515 isocratic HPLC pump, 717plus autosampler, and a Jordi divinylbenzene mixed-bed GPC column (7.8 × 300 mm, Alltech Associates, Deerfield, IL). Samples were prepared at 10 mg/mL in DCM and filtered with 0.45 μm PTFE syringe filters prior to autoinjection. DCM was used as the eluent at a flow rate of 1 mL/min. An IBM ThinkCentre computer with WaterBreeze version 3.20 software was used for data collection and processing, with Mw calibrated against broad PEG standards (Waters, Milford, MA).
2.3. Synthesis
2.3.1. Synthesis of n-Methoxyalkanoic Acid Chains (2)
The preparation of 8-methoxyoctanoic acid (2a) is presented as an example. According to modified literature procedures,42 anhydrous MeOH (8 mL) was cooled to 0 °C, potassium hydroxide (KOH, 13.80 mmol) added, and the solution stirred for 30 min. A solution of 8-bromooctanoic acid (1a, 4.60 mmol) in anhydrous MeOH (7 mL) was then added via syringe. The reaction mixture was heated to reflux temperatures and stirred overnight. After cooling to room temperature, MeOH was removed in vacuo and the resulting crude mixture reconstituted in 1 N HCl (25 mL) and diethyl ether (5 mL). The crude product was extracted using diethyl ether (4 × 30 mL), the combined organic layers were washed with 50:50 brine/H2O (30 mL) and dried over magnesium sulfate (MgSO4), and then the solvent removed in vacuo. 2a was then purified on silica gel via column chromatography using a hexanes/acetic acid/ethyl acetate gradient (99.8:0.2:0 to 98:1:1).
8-Methoxyoctanoic Acid (2a)
Yield: 0.70 g, 87% (pale yellow oil). 1H NMR (500 MHz, CDCl3): δ 3.32 (t, 2H, OCH2), 3.27 (s, 3H, OCH3), 2.26 (t, 2H, CH2CO), 1.53 (m, 4H, CH2), 1.27 (b, 6H, CH2). 13C NMR (500 MHz, CDCl3): δ 179.57, 72.95, 58.51, 34.16, 29.53, 29.21, 29.14, 26.03, 24.80. IR (cm–1, thin film from diethyl ether): 3600–3100 (OH, COOH), 1709 (C=O, COOH). ESI-MS m/z: 173.3 (M – 1).
10-Methoxydecanoic Acid (2b)
Yield: 1.93 g, 84% (light orange solid). 1H NMR (400 MHz, CDCl3): δ 3.35 (t, 2H, OCH2), 3.32 (s, 3H, OCH3), 2.32 (t, 2H, CH2CO), 1.57 (m, 4H, CH2), 1.28 (b, 10H, CH2). 13C NMR (500 MHz, CDCl3): δ 179.53, 73.11, 58.53, 34.19, 29.64, 29.55, 29.52, 29.34, 29.21, 26.24, 24.94. IR (cm–1, thin film from DCM): 3600–3100 (OH, COOH), 1709 (C=O, COOH). ESI-MS m/z: 201.3 (M – 1).
12-Methoxydodecanoic Acid (2c)
Yield: 0.70 g, 80% (white solid). 1H NMR (400 MHz, CDCl3): δ 3.38 (t, 2H, OCH2), 3.34 (s, 3H, OCH3), 2.34 (t, 2H, CH2CO), 1.60 (m, 4H, CH2), 1.27 (b, 14H, CH2). 13C NMR (500 MHz, CDCl3): δ 180.08, 73.12, 58.61, 34.32, 29.77, 29.73, 29.68, 29.66, 29.59, 29.42, 29.25, 26.30, 24.90. IR (cm–1, KBr): 3600–3100 (OH, COOH), 1731 (C=O, COOH). ESI-MS m/z: 229.3 (M – 1).
2.3.2. Synthesis of 2,3-Bis(n-methoxyalkanoyl) Dibenzyl Tartrate (DBT) (3)
The synthesis of 2,3-bis(8-methoxyoctanoyl) DBT (3a) is presented as an example. DBT (0.67 mmol), 2a (1.40 mmol), and catalytic dimethylaminopyridine (DMAP, 0.13 mmol) were dissolved in anhydrous DCM (10 mL) under argon. Upon complete dissolution, 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI, 2.80 mmol) was added as a coupling reagent and the reaction was stirred overnight under argon. The reaction mixture was diluted with DCM (25 mL) and washed with aqueous solutions of 10% potassium bisulfite (3 × 40 mL) and saturated sodium bicarbonate (3 × 40 mL) to remove the EDCI urea byproduct and unreacted 2a, respectively. The organic layer was then washed with brine (40 mL) and dried over MgSO4, and the product (3) isolated in vacuo. As the product appeared as a viscous liquid for the shorter aliphatic chain lengths, yield was not calculated. Instead, a two-step yield was calculated following the next synthetic step. The detailed characterization of 3a–c can be found in the Supporting Information.
2.3.3. Synthesis of 2,3-Bis(n-methoxyalkanoyl) TA (4)
The synthesis of 2,3-bis(8-methoxyoctanoyl) TA (4a) is presented as an example. 3a (0.67 mmol, theoretical) was deprotected following modified literature procedures,34,43 using H2(g) and a 10% w/w palladium on carbon (Pd/C) catalyst in a 1:1 DCM/MeOH solvent system (HPLC grade, 6 mL total). The reaction mixture was passed through a Celite filter using 1:1 DCM/MeOH (HPLC grade, 300 mL total) to remove the catalyst, and the filtrate concentrated in vacuo. Pure product was precipitated from (4a) or triturated in (4b and 4c) hexanes and isolated via vacuum filtration.
2,3-Bis(8-methoxyoctanoyl) TA (4a)
Two-step yield: 0.24 g, 77% (off-white solid). 1H NMR (400 MHz, CDCl3): δ 5.72 (s, 2H, CH), 3.57 (m, 2H, OCH2), 3.49 (m, 2H, OCH2), 3.40 (s, 6H, OCH3), 2.59 (quin, 2H, CH2CO), 2.39 (quin, 2H, CH2CO), 1.66 (m, 8H, CH2), 1.33 (m, 12H, CH2). 13C NMR (500 MHz, CDCl3): δ 172.86, 168.10, 73.07, 70.90, 58.34, 34.06, 28.39, 28.20, 28.18, 25.55, 24.85. IR (cm–1, KBr): 3650–3300 (OH, COOH), 1762 (C=O, ester), 1736 (C=O, COOH). ESI-MS m/z: 461.1 (M – 1).
2,3-Bis(10-methoxydecanoyl) TA (4b)
Two-step yield: 0.53 g, 85% (white powder). 1H NMR (500 MHz, CDCl3): δ 5.76 (s, 2H, CH), 3.46 (t, 4H, OCH2), 3.37 (s, 6H, OCH3), 2.45 (t, 4H, CH2CO), 1.62 (m, 8H, CH2), 1.31 (b, 20H, CH2). 13C NMR (500 MHz, CDCl3): δ 172.90, 168.51, 73.26, 70.74, 58.35, 33.79, 29.12, 28.87, 28.74, 28.59, 28.39, 25.76, 24.72. IR (cm–1, KBr): 3650–3300 (OH, COOH), 1760 (C=O, ester), 1736 (C=O, COOH). ESI-MS m/z: 517.3 (M – 1).
2,3-Bis(12-methoxydodecanoyl) TA (4c)
Two-step yield: 0.17 g, 99% (white powder). 1H NMR (500 MHz, CDCl3 with DMSO): δ 5.69 (s, 2H, CH), 3.36 (t, 4H, OCH2), 3.33 (s, 6H, OCH3), 2.42 (m, 4H, CH2CO), 1.59 (m, 8H, CH2), 1.26 (b, 28H, CH2). 13C NMR (500 MHz, CDCl3): δ 171.89, 167.42, 72.23, 70.22, 57.85, 33.15, 29.00, 28.93, 28.89, 28.84, 28.79, 28.62, 28.36, 25.50, 24.11. IR (cm–1, KBr): 3650–3300 (OH, COOH), 1761 (C=O, ester), 1737 (C=O, COOH). ESI-MS: m/z: 573.9 (M – 1).
2.3.4. Synthesis of Ether-AMs (5)
The synthesis of ether-AMs is presented as an example (5a2). Following a modified literature procedure,254a (0.45 mmol) and catalytic 4-(dimethylamino)pyridinium 4-toluenesulfonate (DPTS, 0.15 mmol) were dissolved in a mixture of anhydrous DCM (10 mL) and anhydrous dimethylformamide (DMF, 3 mL). This solution was added to 2 kDa mPEG-amine (0.15 mmol). Upon complete dissolution of PEG, dicyclohexylcarbodiimide (DCC, 1 M in DCM, 0.48 mmol) was added dropwise via syringe and the reaction stirred for 48 h at room temperature under argon. The reaction mixture was cooled to −20 °C, and the white solid precipitate (dicyclohexylurea) removed via vacuum filtration. The filtrate was then diluted with DCM (25 mL) and washed with 0.1 N HCl (1 × 40 mL) and brine (2 × 40 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. Ether-AM (5a2) was then precipitated from diethyl ether (50 mL) and isolated via centrifugation (Hettich EBA 12, Beverly, MA; 3500 rpm, 5 min), and the diethyl ether decanted. The product was washed with diethyl ether (50 mL × 4) and isolated with centrifugation and decanting, as above. The PEG Mw used to synthesize the ether-AMs will be denoted numerically in kilodaltons as a subscript (e.g., 5a2).
5a2
Yield: 0.28 g, 78% (beige waxy solid). 1H NMR (400 MHz, CDCl3): δ 6.79 (b, 1H, CONH), 5.62 (dd, 2H, CH), 3.63 (m, ∼200H, CH2CH2O), 3.35 (m, ∼13H, 2OCH3, OCH2), 2.41 (m, 4H, CH2CO), 1.59 (m, 8H, CH2), 1.32 (b, 12H, CH2). Mw, 2.5 kDa; PDI, 1.1.
5a5
Yield: 1.34 g, 85% (off-white powder). 1H NMR (400 MHz, CDCl3): δ 6.66 (b, 1H, CONH), 5.62 (dd, 2H, CH), 3.64 (m, ∼500H, CH2CH2O), 3.35 (m, ∼13H, 2OCH3, OCH2), 2.42 (m, 4H, CH2CO), 1.60 (m, 8H, CH2), 1.33 (b, 12H, CH2). Mw, 6.2 kDa; PDI, 1.1.
5b2
Yield: 0.22 g, 63% (beige waxy solid). 1H NMR (500 MHz, CDCl3): δ 6.62 (b, 1H, CONH), 5.58 (dd, 2H, CH), 3.65 (m, ∼200H, CH2CH2O), 3.34 (m, ∼13H, 2OCH3, OCH2), 2.44 (m, 4H, CH2CO), 1.60 (m, 8H, CH2), 1.30 (b, 20H, CH2). Mw, 2.3 kDa; PDI, 1.1.
5b5
Yield: 0.34 g, 98% (off-white solid). 1H NMR (400 MHz, CDCl3): δ 6.70 (b, 1H, CONH), 5.58 (dd, 2H, CH), 3.64 (m, ∼500H, CH2CH2O), 3.34 (m, ∼13H, 2OCH3, OCH2), 2.41 (m, 4H, CH2CO), 1.59 (m, 8H, CH2), 1.29 (b, 20H, CH2). Mw, 6.3 kDa; PDI, 1.1.
5c2
Yield: 0.27 g, 55% (beige waxy solid). 1H NMR (500 MHz, CDCl3): δ 6.90 (b, 1H, CONH), 5.56 (dd, 2H, CH), 3.65 (m, ∼200H, CH2CH2O), 3.38 (m, ∼13H, 2OCH3, OCH2), 2.40 (m, 4H, CH2CO), 1.58 (m, 8H, CH2), 1.27 (b, 28H, CH2). Mw, 2.6 kDa; PDI, 1.1.
5c5
. Yield: 0.13 g, 78% (off-white solid). 1H NMR (500 MHz, CDCl3): δ 6.67 (b, 1H, NH), 5.54 (dd, 2H, CH), 3.65 (m, ∼500H, CH2CH2O), 3.38 (m, ∼13H, 2OCH3, OCH2), 2.41 (m, 4H, CH2CO), 1.60 (m, 8H, CH2), 1.27 (b, 28H, CH2). Mw, 6.4 kDa; PDI, 1.1.
2.3.5. Synthesis of Aliphatic TA Derivatives (6)
The synthesis of aliphatic TA derivatives is presented as an example (6a). Aliphatic TA derivatives were synthesized following a modified literature procedure.44 In brief, L-TA (7.00 mmol) and zinc chloride (2.20 mmol) were suspended neat in decanoyl chloride (52.50 mmol) and heated to 95 °C. After stirring 24 h, the reaction mixture was cooled to room temperature and quenched with H2O (30 mL) and diethyl ether (100 mL), then vigorously stirred for 30 min. This solution was washed with H2O (5 × 100 mL), and the organic layer concentrated in vacuo to yield a viscous brown liquid. Pure product (6) was precipitated from 1 L of stirring hexanes and isolated via vacuum filtration. The length of the product’s aliphatic chains will be indicated by the lettering a–c, with a given TA derivative (e.g., 6a) having aliphatic chains of analogous length to the previously discussed n-methoxyalkanoyl derivatives (e.g., 4a). The detailed characterization of 6a–c can be found in the Supporting Information.
2.3.6. Synthesis of Alkyl-AMs (7)
The synthesis of alkyl-AMs is presented as an example (7a2). Alkyl-AMs were prepared in the same manner as were the previously discussed ether-AMs, using 6a (0.50 mmol), DPTS (0.17 mmol), mPEG-amine (0.17 mmol), and DCC (0.53 mmol). Additional anhydrous DMF was used if necessary to fully solubilize 6 prior to adding it to mPEG-amine. The PEG Mw used to synthesize the alkyl-AM will also be denoted numerically in kilodaltons as a subscript (e.g., 7a2).
7a2
Yield: 0.25 g, 60% (off-white waxy solid). 1H NMR (400 MHz, CDCl3): δ 6.97 (b, 1H, CONH), 5.55 (dd, 2H, CH), 3.65 (m, ∼200H, OCH2), 3.39 (s, 3H, OCH3), 2.40 (m, 4H, CH2CO), 1.62 (m, 4H, CH2), 1.26 (b, 24H, CH2), 0.88 (t, 6H, CH3). Mw, 1.9 kDa; PDI, 1.1.
7a5
Yield: 0.40 g, quantitative (off-white solid). 1H NMR (400 MHz, CDCl3): δ 6.90 (b, 1H, CONH), 5.53 (dd, 2H, CH), 3.66 (m, ∼500H, OCH2), 3.38 (s, 3H, OCH3), 2.39 (m, 4H, CH2CO), 1.63 (m, 4H, CH2), 1.26 (b, 24H, CH2), 0.88 (t, 6H, CH3). Mw, 5.3 kDa; PDI, 1.1.
7b2
Yield: 0.30 g, 60% (off-white waxy solid). 1H NMR (400 MHz, CDCl3): δ 6.84 (b, 1H, CONH), 5.54 (dd, 2H, CH), 3.66 (m, ∼200H, OCH2), 3.38 (s, 3H, OCH3), 2.40 (m, 4H, CH2CO), 1.62 (m, 4H, CH2), 1.25 (b, 32H, CH2), 0.88 (t, 6H, CH3). Mw, 2.1 kDa; PDI, 1.1.
7b5
Yield: 0.36 g, quantitative (off-white solid). 1H NMR (400 MHz, CDCl3): δ 6.85 (b, 1H, CONH), 5.56 (dd, 2H, CH), 3.65 (m, ∼500H, OCH2), 3.38 (s, 3H, OCH3), 2.41 (m, 4H, CH2CO), 1.61 (m, 4H, CH2), 1.25 (b, 32H, CH2), 0.89 (t, 6H, CH3). Mw, 5.4 kDa; PDI, 1.1.
7c2
Yield: 0.28 g, 55% (off-white waxy solid). 1H NMR (500 MHz, CDCl3): δ 6.91 (b, 1N, CONH), 5.55 (dd, 2H, CH), 3.66 (m, ∼200H, OCH2), 3.38 (s, 3H, OCH3), 2.41 (m, 4H, CH2CO), 1.62 (m, 4H, CH2), 1.26 (b, 40H, CH2), 0.88 (t, 6H, CH3). Mw:, 2.1 kDa; PDI, 1.1.
7c5
Yield: 0.50 g, quantitative (off-white solid). 1H NMR (400 MHz, CDCl3): δ 6.92 (b, 1H, CONH), 5.53 (dd, 2H, CH), 3.64 (m, ∼500H, OCH2), 3.38 (s, 3H, OCH3), 2.40 (m, 4H, CH2CO), 1.62 (m, 4H, CH2), 1.25 (b, 40H, CH2), 0.88 (t, 6H, CH3). Mw, 5.8 kDa; PDI, 1.1.
2.4. Critical Micelle Concentration (CMC) Measurements
AMs were dissolved in HPLC grade H2O and diluted to a series of concentrations ranging from 1 × 10–3 to 1 × 10–10 M. Separately, a stock solution of pyrene was prepared in HPLC grade acetone (5 × 10–6 M) and 0.5 mL of this solution was added to a series of vials. Acetone was removed in vacuo, and AM solutions (5 mL) were added. AM-pyrene solutions were incubated for 48 h at 37 °C with gentle agitation (60 rpm) to allow pyrene to partition into the AM micelles. Fluorescence studies were then conducted on a RF-5301PC spectrofluorometer (Shimadzu Scientific Instruments, Columbia, MD), using pyrene as the fluorescent probe. Emission was measured from 300–360 nm with a 390 nm excitation wavelength. Upon micelle formation, pyrene partitions into the micelle hydrophobic core and the maximum wavelength emission shifts from 332 to 334.5 nm. The ratio of absorption of pyrene in micelles (334.5 nm) to pyrene alone (332 nm) was thus plotted against the logarithm of AM concentration, and the inflection point of this curve was taken as the CMC.25
2.5. Dynamic Light Scattering (DLS) Measurements
DLS analysis was performed on Zetasizer Nano ZS90 instrument (Malvern Instruments, Southboro, MA) in triplicate with a 90° scattering angle. AM samples were dissolved in HPLC grade H2O (10 mg/mL) and equilibrated for 24 h at 37 °C with gentle agitation (60 rpm). Solutions were passed through 0.45 μm PTFE syringe filters prior to size measurements, and Z-average sizes were collected and analyzed.
2.6. Cell Culture
Peripheral blood mononuclear cells (PBMCs) were isolated from human buffy coats (Blood Center of New Jersey; East Orange, NJ) by centrifugation through Ficoll-Paque density gradient (GE Healthcare). PBMCs were plated into T-175 flasks, and monocytes selected via adherence after 24 h. Monocytes were cultured for 7 days in RPMI 1640 (ATCC) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 50 ng/mL macrophage colony-stimulating factor (M-CSF) for differentiation into human monocyte-derived macrophages (HMDMs).30,35
2.7. Cell Viability Studies
To screen cellular toxicity of the AMs, the alamarBlue assay was carried out according to manufacturer’s protocol. In brief, HMDMs were plated in a 96-well plate at 150 000 cells/mL in basal media (RPMI 1640 supplemented with 10% FBS and 1% penicillin/streptomycin) and allowed to rest for 24 h. Cells were then treated with the desired concentration of AM (10–5 - 10–3 M) diluted in basal media for 24 h. Following incubation, the treatment (media containing specific AM concentrations) was removed and alamarBlue (diluted 1 to 10 in basal media) was added to each well and cells incubated for 24 h. The supernatant was then transferred to a new plate and absorbance read on a spectrophotometer (Infinite 200 Pro, Tecan, Männedorf, Switzerland) at 570 and 600 nm.
2.8. OxLDL Uptake by Macrophages
HMDMs were cocultured with 3,3′-dioctadecyloxacarbocyanine- (DiO-)labeled oxLDL (1 μg/mL) and unlabeled oxLDL (4 μg/mL) with or without different AM concentrations, ranging from 10–5 to 10–3 M, in basal media (RPMI 1640 supplemented with 10% FBS and 1% penicillin/streptomycin) for 24 h. Treatments were then removed and replaced with cold phosphate buffered saline (PBS) and placed on ice packs. HMDMs were removed from wells by vigorous pipetting and transferred to 5 mL tubes, centrifuged at 1000 rpm for 10 min, and fixed in 1% paraformaldehyde (150 μL). The oxLDL fluorescence associated with HMDMs was quantified on a FACScalibur (Beckton Dickenson, Franklin Lakes, NJ) flow cytometer, collecting 10 000 events/sample, and analyzed with Flow Jo software (Treestar, Ashland, OR). This study included a minimum of three experimental replicates. Data is presented as percent of oxLDL uptake as determined by the following equation:
2.9. Statistical Analysis
OxLDL uptake studies were conducted in experimental triplicate. The results were then evaluated using Student’s t test, with significance criteria assuming a 95% confidence level (P < 0.05). Standard error of the mean is reported in the form of error bars on the graphs of the final data.
3. Results and Discussion
3.1. AM Synthesis and Characterization
Ether-AMs were synthesized to assess whether incorporating heteroatoms, specifically ethers, into the terminal-end of alkyl chains of the hydrophobic domain would enhance AM bioactivity by promoting hydrogen-bonding interactions with scavenger receptor binding pockets, potentially reducing oxLDL uptake even further. To this end, a series of methoxy-terminated long-chain carboxylic acids was first synthesized to serve as the AMs’ hydrophobic arms. Using the Williamson ether synthesis, potassium methoxide, generated from MeOH and KOH, was reacted with bromo-terminated alkanoic acids (1) to yield methoxy-terminated alkanoic acids (2) via an SN2 reaction.45 To acylate the TA backbone with 2, typical AM synthetic methods were attempted in which methoxy-terminated alkanoyl chlorides were prepared from thionyl chloride and reacted with L-TA in the presence of a Lewis acid catalyst.25 These conditions, however, required an excess of 2 and resulted in incomplete acylation. As an alternative, 2 was coupled to a protected TA backbone (DBT, Scheme 1) through EDCI coupling, resulting in complete acylation while using near stoichiometric amounts of 2. The acylated DBT derivative (3) was subsequently deprotected via hydrogenolysis, using a 10% w/w Pd/C catalyst, to give the acylated TA product (4). To finally generate the ether-AMs (5), 4 was coupled to mPEG-NH2 using DCC coupling with a DPTS catalyst. A stoichiometric excess of 4 and DCC ensured that PEG coupled to only one of 4’s two carboxylic acids. Ether-AM precursors’ chemical structures were confirmed via NMR and FT-IR spectroscopies and MS, while ether-AM synthesis was verified by 1H NMR spectroscopy and GPC.
Scheme 1. Synthetic Scheme for Ether-AMs.

The PEG Mw used will be denoted numerically in kilodaltons as a subscript with x = 45 yielding 2 kDa PEG and x = 113 yielding 5 kDa PEG (e.g., 5a2 and 5a5 signify an ether-AM with a 2 kDa and 5 kDa PEG tail, respectively).
1H NMR spectroscopy was critical in affirming successful synthesis of ether-AMs and their precursors. Figure 2 presents the NMR spectra obtained during the synthesis of 5a2, as an example. Successful 8-methoxyoctanoic acid (2a) synthesis was confirmed by the appearance of a triplet and singlet at 3.32 and 3.27 ppm (d in Figure 2A), corresponding to the methylene and methyl protons adjacent to the methoxy oxygen atom. The relative integration of DBT’s methine singlet (g in Figure 2B) to signals associated with the 8-methoxyoctanoyl arms demonstrated complete acylation to form 3a, with two aliphatic arms present per DBT backbone. Disappearance of the aromatic and benzyl proton signals (e and f in Figure 2B) illustrated the complete deprotection of 3a to produce 4a (Figure 2C). Finally, successful PEGylation to yield 5a2 was confirmed by the appearance of a large ∼200 proton PEG multiplet (i in Figure 2D). The 1:2 ratio of the amide proton signal (h in Figure 2D) to the methine proton signal of the TA backbone further indicated that PEG was only conjugated to one side of the TA derivative.
Figure 2.

1H NMR spectra for 5a2 ether-AM synthesis as an example: 8-methoxyoctanoic acid 2a (A), 2,3-bis(8-methoxyoctanoyl) DBT 3a (B), 2,3-bis(8-methoxyoctanoyl) TA 4a (C), and ether-AM 5a2 (D).
In addition to synthesizing ether-AMs, a series of analogous alkyl-AMs (Scheme 2) was prepared to compare the influence of hydrogen-bonding and hydrophobic interactions on AM physicochemical and biological properties. These alkyl-AMs (7, Figure 1) only differed from ether-AMs in that the methoxy oxygen atom was replaced with a methylene group, yielding AMs with saturated aliphatic arms of analogous lengths to the ether-AMs. To synthesize these analogues, previously reported methods were used in which L-TA was reacted with an acyl chloride to generate a modified TA hydrophobe (6) that was subsequently coupled to mPEG-amine using DCC. AM and AM precursor chemical structures were confirmed via the aforementioned methods.
Scheme 2. Synthetic Scheme for Alkyl-AMs.

The PEG Mw used will be denoted numerically in kilodaltons as a subscript with x = 45 yielding 2 kDa PEG and x = 113 yielding 5 kDa PEG (e.g., 7a2 and 7a5 signify an alkyl-AM with a 2 kDa and 5 kDa PEG tail, respectively).
Once the synthesis of all ether- and alkyl-AMs was confirmed, their physicochemical properties were evaluated. When AM concentrations in aqueous environments exceed a CMC, they self-assemble into micelles; this transition was measured using an established fluorimetry assay.25 In evaluating ether-AMs alone, it was observed that, while keeping the PEG Mw constant, 5c AMs exhibited slightly lower CMC values than those of 5a and 5b, which were comparable (Table 1). As 5c AMs contained the most methylenes within the hydrophobic domain, these AMs were likely less soluble in water as compared to 5a and 5b, resulting in lower CMC values. Furthermore, as micellization is entropically driven by the displacement of water molecules from the hydrophobic domain, these results likely stem from 5c AMs having more water molecules associated with their hydrophobic domains prior to micellization, resulting in a larger entropic increase and thus a greater free energy decrease upon micellization.46 Although 5c AMs’ CMC values were lower than those of other ether-AMs, all ether-AMs exhibited CMCs near 10–4 M. In comparing ether-AMs to their respective alkyl-AM analogues (e.g., 5a2 vs 7a2), all alkyl-AMs exhibited CMC values lower than the analogous ether-AMs, ranging approximately from 10–6 to 10–4 M (Table 1). Similar to the trends among the ether-AMs (5), the analogues’ (7) lower CMC values likely result from the increased hydrophobicity of 7 as compared to 5. Given that lower CMCs can provide greater stability against dilution, the alkyl-AMs would be more likely to remain in micellar assemblies when diluted under physiological conditions.47
Table 1. Physicochemical Properties of Ether-AMs (5) and Alkyl-AMs (7), with 2000 Mw Compounds Shown on the Left and 5000 Mw Compounds Shown on the Right.
| 2000 Mw |
5000 Mw |
||||
|---|---|---|---|---|---|
| AM | CMC (M) | size (nm) | AM | CMC (M) | size (nm) |
| 5a2 | 1.97 × 10–4 | 102.3 ± 0.3 | 5a5 | 2.19 × 10–4 | 111.4 ± 0.8 |
| 7a2 | 9.79 × 10–5 | 8.0 ± 0.1 | 7a5 | 5.79 × 10–5 | 15.3 ± 0.1 |
| 5b2 | 1.99 × 10–4 | 93.5 ± 1.4 | 5b5 | 2.02 × 10–4 | 119.8 ± 2.0 |
| 7b2 | 3.43 × 10–5 | 7.3 ± 0.1 | 7b5 | 3.20 × 10–5 | 11.2 ± 0.1 |
| 5c2 | 1.24 × 10–4 | 12.5 ± 0.0 | 5c5 | 9.01 × 10–5 | 28.7 ± 0.5 |
| 7c2 | 6.94 × 10–6 | 8.1 ± 0.1 | 7c5 | 7.49 × 10–6 | 10.6 ± 0.2 |
To determine whether AM micelles exhibited nanoscale sizes suitable for biomedical applications, the micelles were next measured using DLS. Prior to DLS measurements, AMs were incubated in water for 24 h at 37 °C to mimic physiological conditions. In comparing the ether-AMs, 5a and 5b exhibited sizes near 100 nm while 5c exhibited smaller sizes, regardless of PEG Mw (Table 1). The smaller sizes of 5c AMs may result from enhanced hydrophobic interactions upon micellization due to their larger hydrophobic domains,48 which could overcome potential repulsion caused by the methoxy moieties. Furthermore, other investigators have reported that different micelle morphologies (e.g., spindle-like, rod-like, or bowl-like) result when increasing the length of block copolymers’ hydrophobic domains.49−52 Although the DLS method employed assumes Brownian motion of sphere-shape particles, it is plausible that alternate micelle morphologies are present, giving rise to the smaller particle sizes. Despite the range in sizes (12–119 nm), all ether-AMs remained within a size range (10–200 nm) considered optimal for enhanced stability in vivo.47 In contrast to ether-AMs, all alkyl-AMs exhibited smaller sizes, ranging from 8 to 15 nm. As these analogues are more hydrophobic than the ether-AMs, these results correlate well with the ether-AM size trends, suggesting larger hydrophobic domains yield stronger hydrophobic interactions and smaller micelle sizes. Finally, all 2000 Mw AMs exhibited smaller sizes than their 5000 Mw counterparts (e.g., 5a2 vs 5a5). This phenomenon likely resulted from 2000 Mw AMs’ smaller PEG size, as seen in previous literature.53,54 Despite variations in sizes, ether-AMs exhibited suitable sizes for drug delivery applications, while some alkyl-AMs displayed sizes slightly smaller than the desirable size range.
3.2. AM Biological Properties
Prior to assessing ether- and alkyl-AMs’ antiatherogenic potential, cytotoxicity was screened in HMDMs at concentrations ranging from 10–5 to 10–3 M (Supporting Information Figures S1 and 2). Treatments that resulted in 70% or more viable cells were considered nontoxic. While the most hydrophobic ether-AM, 5c2, was cytotoxic only at the highest concentration administered (10–3 M), all other ether-AMs were nontoxic at all concentrations tested. Of the alkyl-AMs, 7b2 and 7c5 were cytotoxic at 10–3 M and 7c2 exhibited cytotoxicity at both 10–3 and 10–4 M. In agreement with previously published results on nanoscale systems,55−57 these results suggest that as AM hydrophobicity is increased, the macromolecules become more cytotoxic. Conversely, AMs with larger PEG tails showed improved cell viability over AMs with smaller molecular weight PEG tails (i.e., 5000 vs 2000 Da, respectively). Furthermore, ether-AMs, containing two additional, ethereal oxygen atoms within their hydrophobic domain, are better tolerated by HMDMs than alkyl-AMs when administered at higher concentrations.
To assess the impact of the hydrophobic chain architecture on AM antiatherogenic bioactivity, HMDMs were coincubated with AMs at concentrations ranging from 10–5 to 10–3 M and fluorescently labeled oxLDL. This broad range of concentrations (e.g., above and below CMC values) was investigated to determine the influence of concentration and the presence of micelles (or unimers) on AM bioactivity. Furthermore, all in vitro studies were conducted in the presence of serum proteins to mimic physiological conditions; however, serum proteins are capable of disrupting micelle integrity30,58−60 and may impact AM bioactivity. Previous studies have indicated that in comparison to serum-free conditions AMs’ efficacy decreases in the presence of serum proteins, which may result from serum protein interactions with AMs.32 The presence of serum proteins, therefore, allows for a more realistic understanding of ether- and alkyl-AM bioactivity.
As shown in Figure 3A, the hydrophobic chain composition and AM concentration play an integral role in 5000 Mw PEG tail AM antiatherogenic bioactivity. As the ether-AMs’ aliphatic chain length increases, their ability to inhibit oxLDL uptake increases, such that 5c5 > 5b5 > 5a5, with 5a5 exhibiting no bioactivity. This phenomenon is concentration-dependent, with higher ether-AM concentrations resulting in more oxLDL uptake inhibition, except for 5a5. For example, while HMDMs treated with 10–4 M 5c5 exhibit 73% oxLDL uptake, those cells incubated with 10–3 M 5c5 significantly repress oxLDL uptake to less than 2% (Figure 3A). Furthermore, 10–3 M 5c5 reduced the amount of oxLDL internalized by cells by such a significant magnitude that only 5.3% of the cells had any oxLDL in them at all (Figure 4A). Given that 5b5 administered at 10–3 M significantly inhibited oxLDL uptake (Figure 3A) despite its larger-sized micelles, it appears that micelle size alone does not dictate AMs’ antiatherogenic potential. Further, as 5a5 exhibited no bioactivity at 10–3 M despite its micellar assembly, it is likely that the chemical composition of the AM more strongly influences bioactivity than the corresponding micellar configuration. Although AM micellar structure does not demonstrate a pronounced effect on AM bioactivity, the increased size and PEG shielding provided by the micellar assembly would likely increase the AMs biological stability when administered in a clinical setting.26,47
Figure 3.

Effect of administering varying concentrations of 5000 Mw (A) and 2000 Mw (B) ether-AMs (dark gray) and alkyl-AM analogues (light gray) on percent of oxLDL uptake in HMDMs. AMs of specific alkyl lengths are grouped between the dashed lines, and the AM treatments not investigated due to cytotoxicity issues are indicated as text on the graph. Significant deviations from the oxLDL positive control (black) are denoted by asterisks (∗) on the graph.
Figure 4.

Percent of HMDMs positive for oxLDL after incubation with varying concentrations of 5000 Mw (A) and 2000 Mw (B) ether-AMs (dark gray) and alkyl-AM analogues (light gray). AMs of specific alkyl lengths are grouped between the dashed lines, and the AM treatments not investigated due to cytotoxicity are indicated as text on the graph. Significant deviations from the oxLDL positive control (black) are denoted by asterisks (∗) on the graph.
Alkyl-AMs with 5000 Mw PEG tails (Figure 3A, light gray) show similar trends to the aforementioned ether-AMs (Figure 3A, dark gray), exhibiting reduced oxLDL uptake as AM concentration and/or alkyl chain length are increased; however, they were much more efficacious in preventing oxLDL uptake than the ether-AMs. When administered at 10–4 M, for example, 7a5 (29%), 7b5 (4.7%), and 7c5 (1.5%) showed significantly lower oxLDL uptake than their corresponding ether-AMs 5a5 (112%), 5b5 (108%), and 5c5 (73%). In fact, the 10–3 M concentrations of the 5000 Mw alkyl-AMs were so potent that less than 7% of HMDMs were positive for any oxLDL at all (Figure 4A). As alkyl-AMs repressed more oxLDL uptake than analogous ether-AMs and AM potency increased with increasing aliphatic arm length, these results indicate that hydrophobicity and the length of AMs’ aliphatic arms play a more significant role than hydrogen-bonding in modulating athero-protective bioactivity. Given that macrophage scavenger receptors (e.g., CD36) contain hydrophobic residues near their oxLDL binding pockets,38 it is plausible that AMs primarily interact with scavenger receptors through hydrophobic interactions, resulting in reduced oxLDL uptake. Furthermore, previous literature suggests that the scavenger receptor ligands of different lengths exhibit varying activity, likely resulting from how the ligands arrange within the receptor pocket.61 As previous research demonstrated that increasing AM hydrophobicity does not always improve bioactivity,34 it is plausible that the longer aliphatic chains arrange more favorably within scavenger receptor binding pockets through the aforementioned hydrophobic interactions.
In comparing the 2000 Mw ether-AMs to the corresponding alkyl-AMs (Figure 3B), similar trends were apparent with AMs showing reduced oxLDL uptake as their concentration, hydrophobicity, and aliphatic chain length increases. Only 7a2 significantly reduced the number of oxLDL positive HMDMs (1.4%, Figure 4B) when administered at 10–3 M; fewer AM concentrations were investigated for these studies, however, due to toxicity. This data suggests that the amphiphilic balance provided by the higher molecular weight PEG chains is critical for minimizing cellular toxicity and highly hydrophobic domains are detrimental to cellular viability. Although AMs containing 2000 Mw PEG are relatively more hydrophobic than their 5000 Mw counterparts, PEG Mw did not have a pronounced effect on AM bioactivity. These results agree with previously published results, suggesting that while PEG size can modulate cytotoxicity, the AM hydrophobic domain dominates antiatherogenic bioactivity.32 While relatively high micromolar concentrations are required to achieve significant oxLDL uptake inhibition (i.e., 10–3–10–4 M) and may pose biocompatibility concerns, previous in vivo studies demonstrated that a previously synthesized AM containing a larger hydrophobic domain with a 5000 Mw PEG tail exhibits no significant toxicity in mice when administered via intraperitoneal injection at approximately 4500 μM (2000 mg/kg).62 As the tested in vivo concentration is higher than in vitro concentrations used in this work, the most potent ether- and alkyl-AMs could serve as viable atherosclerosis treatments.
Collectively, these studies demonstrate that increasing hydrophobicity through longer aliphatic chains and minimal heteroatoms improves bioactivity. Although certain AM treatments, such as 10–4 M 7a5, exhibited a significant reduction in oxLDL uptake (29%, Figure 3A), the majority of HMDMs (98%, Figure 4A) were still positive for oxLDL. While this treatment reduced oxLDL uptake, oxLDL was still able to accumulate in macrophages that could, over time, elicit the atherosclerotic cascade. In comparing the dose response of the most efficacious AMs tested (Figure 5), 7c5 is the most promising candidate for antiatherosclerotic therapies: it has the lowest CMC value, effectively inhibits both oxLDL uptake and accumulation in macrophages at a lower concentration (10–4 M) than the other AMs tested, and was not cytotoxic at this concentration. Studies assessing this candidate’s in vivo bioactivity and biocompatibility are the focus of future work, aiming to identify an appropriate administration route that maximizes antiatherosclerotic efficacy with minimal adverse effects.
Figure 5.

Dose response of most efficacious AM treatments.
4. Conclusions
Ether- and alkyl-AMs were synthesized to assess the relative contributions of hydrogen-bonding and hydrophobic interactions in oxLDL uptake inhibition in primary human macrophages. Ether-AMs displayed higher CMCs and larger hydrodynamic diameters than corresponding alkyl-AM analogues, likely due to their decreased hydrophobicity and the presence of hydrophilic moieties in the hydrophobic domain, respectively. Hydrophobicity and aliphatic chain length also played a critical role in the antiatherogenic potential of the AMs to inhibit oxLDL accumulation, with more hydrophobic AMs (i.e., alkyl-AMs and/or AMs with longer aliphatic arms) showing a greater reduction in oxLDL uptake. However, the amphiphilic balance provided by the higher Mw PEG tails and ether moieties were beneficial for minimizing cellular toxicity. Therefore, AMs with larger PEG components (i.e., 5000 Mw PEG tails) and larger hydrophobic domains (i.e., longer alkyl chains and/or no ether moieties) were the lead candidates due to their combined biocompatibility and high oxLDL inhibition potential. These findings reinforce the significance of hydrophobicity and aliphatic chain length in modulating bioactivity, critical for the design of next-generation antiatherogenic AMs.
Acknowledgments
This study was supported by the National Institutes of Health (NIH R21093753 to P.V.M. and NIH R01 HL107913 to P.V.M. and K.E.U.) and the U.S. Department of Education fellowship for Graduate Assistance in Areas of National Need (GAANN, A.F.).
Supporting Information Available
Characterization measurements for 2,3-bis(n-methoxyalkanoyl) dibenzyl tartrate intermediates and alkyl-AM precursors are included, as well as results from cell viability screenings. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Orford J. L.; Selwyn A. P.; Ganz P.; Popma J. J.; Rogers C. Am. J. Cardiol. 2000, 86, 6H. [DOI] [PubMed] [Google Scholar]
- Ross R. N. Engl. J. Med. 1999, 340, 115. [DOI] [PubMed] [Google Scholar]
- Li A. C.; Glass C. K. Nat. Med. 2002, 8, 1235. [DOI] [PubMed] [Google Scholar]
- Pirillo A.; Norata G. D.; Catapano A. L. Mediators Inflammation 2013, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.-H.; Fu Y.-C.; Zhang D.-W.; Yin K.; Tang C.-K. Clin. Chim. Acta 2013, 424, 245. [DOI] [PubMed] [Google Scholar]
- Saha P.; Modarai B.; Humphries J.; Mattock K.; Waltham M.; Burnand K. G.; Smith A. Curr. Opin. Pharmacol. 2009, 9, 109. [DOI] [PubMed] [Google Scholar]
- Choudhury R. P.; Lee J. M.; Greaves D. R. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 309. [DOI] [PubMed] [Google Scholar]
- Steinberg D. J. Biol. Chem. 1997, 272, 20963. [DOI] [PubMed] [Google Scholar]
- Tiwari R. L.; Singh V.; Barthwal M. K. Med. Res. Rev. 2008, 28, 483. [DOI] [PubMed] [Google Scholar]
- Ahmad S.; Madsen C. S.; Stein P. D.; Janovitz E.; Huang C.; Ngu K.; Bisaha S. R.; Kennedy L. J.; Chen B. C.; Zhao R. L.; Sitkoff D.; Monshizadegan H.; Yin X.; Ryan C. S.; Zhang R. G.; Giancarli M.; Bird E.; Chang M.; Chen X.; Setters R.; Search D.; Zhuang S. B.; Nguyen-Tran V.; Cuff C. A.; Harrity T.; Darienzo C. J.; Li T.; Reeves R. A.; Blanar M. A.; Barrish J. C.; Zahler R.; Robl J. A. J. Med. Chem. 2008, 51, 2722. [DOI] [PubMed] [Google Scholar]
- Maron D. J.; Fazio S.; Linton M. F. Circulation 2000, 101, 207. [DOI] [PubMed] [Google Scholar]
- Golomb B. A.; Evans M. A. Am. J. Cardiovasc. Drugs 2008, 8, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Xie H.; Sun L.; Ou L.; Wang L.; Yu Y.; Kong D. Biomedical Materials 2009, 4, 065007. [DOI] [PubMed] [Google Scholar]
- Ma K.-w.; Dai X.-z.; Feng S.-y.; Jing A.-h.; Yang J.-y. Transfus. Apheresis Sci. 2011, 44, 3. [DOI] [PubMed] [Google Scholar]
- Robinson J. G. J. Manag. Care Pharm. 2013, 19, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page M. M.; Bell D. A.; Hooper A. J.; Watts G. F.; Burnett J. R. Best Pract. Res., Clin. Endocrinol. Metab. 2014, 28, 387. [DOI] [PubMed] [Google Scholar]
- Li J.; Hou Y.; Chen X.; Ding X.; Liu Y.; Shen X.; Cai K. J. Mater. Sci.: Mater. Med. 2014, 25, 1055. [DOI] [PubMed] [Google Scholar]
- Heuck C.-C.Ger. Med. Sci. 2011, 9, Doc02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.; Wang H.; Yang C.; Zhong R.; Lei Y.; Sun K.; Liu J. Appl. Surf. Sci. 2011, 257, 7521. [Google Scholar]
- Taseva K.; Fischer S.; Passauer J.; Weiss N.; Bornstein S. R.; Julius U. Atheroscler. Suppl. 2013, 14, 45. [DOI] [PubMed] [Google Scholar]
- Goyal T.; Mitra S.; Khaidakov M.; Wang X. W.; Singla S.; Ding Z. F.; Liu S. J.; Mehta J. L. Curr. Atherosclerol. Rep. 2012, 14, 150. [DOI] [PubMed] [Google Scholar]
- Febbraio M.; Podrez E. A.; Smith J. D.; Hajjar D. P.; Hazen S. L.; Hoff H. F.; Sharma K.; Silverstein R. L. J. Clin. Invest. 2000, 105, 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H.; Kurihara Y.; Takeya M.; Kamada N.; Kataoka M.; Jishage K.; Ueda O.; Sakaguchi H.; Higashi T.; Suzuki T.; Takashima Y.; Kawabe Y.; Cynshi O.; Wada Y.; Honda M.; Kurihara H.; Aburatani H.; Doi T.; Matsumoto A.; Azuma S.; Noda T.; Toyoda Y.; Itakura H.; Yazaki Y.; Horiuchi S.; Takahashi K.; Kruijt J. K.; vanBerkel T. J. C.; Steinbrecher U. P.; Ishibashi S.; Maeda N.; Gordon S.; Kodama T. Nature 1997, 386, 292. [DOI] [PubMed] [Google Scholar]
- Chnari E.; Nikitczuk J. S.; Wang J.; Uhrich K. E.; Moghe P. V. Biomacromolecules 2006, 7, 1796. [DOI] [PubMed] [Google Scholar]
- Tian L.; Yam L.; Zhou N.; Tat H.; Uhrich K. E. Macromolecules 2004, 37, 538. [Google Scholar]
- Yamamoto Y.; Nagasaki Y.; Kato Y.; Sugiyama Y.; Kataoka K. J. Controlled Release 2001, 77, 27. [DOI] [PubMed] [Google Scholar]
- Chnari E.; Lari H. B.; Tian L.; Uhrich K. E.; Moghe P. V. Biomaterials 2005, 26, 3749. [DOI] [PubMed] [Google Scholar]
- Iverson N. M.; Plourde N. M.; Sparks S. M.; Wang J. Z.; Patel E. N.; Shah P. S.; Lewis D. R.; Zablocki K. R.; Nackman G. B.; Uhrich K. E.; Moghe P. V. Biomaterials 2011, 32, 8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plourde N. M.; Kortagere S.; Welsh W.; Moghe P. V. Biomacromolecules 2009, 10, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York A. W.; Zablocki K. R.; Lewis D. R.; Gu L.; Uhrich K. E.; Prud’homme R. K.; Moghe P. V. Adv. Mater. 2012, 24, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Plourde N. M.; Iverson N.; Moghe P.; Uhrich K. E. Int. J. Nanomed. 2007, 2, 697. [PMC free article] [PubMed] [Google Scholar]
- Iverson N. M.; Sparks S. M.; Demirdirek B.; Uhrich K. E.; Moghe P. V. Acta Biomater. 2010, 6, 3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehir S.; Plourde N. M.; Gu L.; Poree D. E.; Welsh W. J.; Moghe P. V.; Uhrich K. E. Acta Biomater. 2012, 8, 3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poree D. E.; Zablocki K.; Faig A.; Moghe P. V.; Uhrich K. E. Biomacromolecules 2013, 14, 2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D. R.; Kholodovych V.; Tomasini M. D.; Abdelhamid D.; Petersen L. K.; Welsh W. J.; Uhrich K. E.; Moghe P. V. Biomaterials 2013, 34, 7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt N.; Gordon S. J. Clin. Invest. 2001, 108, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki I.; Ishigaki T.; Oyama T.; Matsunaga S.; Xie Q. H.; Ohnishi-Kameyama M.; Murata T.; Tsuchiya D.; Machida S.; Morikawa K.; Tate S. Structure 2005, 13, 905. [DOI] [PubMed] [Google Scholar]
- Collot-Teixeira S.; Martin J.; McDennott-Roe C.; Poston R.; McGregor J. L. Cardiovasc. Res. 2007, 75, 468. [DOI] [PubMed] [Google Scholar]
- Boullier A.; Bird D. A.; Chang M. K.; Dennis E. A.; Friedman P.; Gillotte-Taylor K.; Horkko S.; Palinski W.; Quehenberger O.; Shaw P.; Steinberg D.; Terpstra V.; Witztum J. L. In Atherosclerosis Vi; Numano F., Gimbrone M. A., Eds.; New York Academy of Sciences: New York, 2001; Vol. 947, p 214. [DOI] [PubMed] [Google Scholar]
- Chisolm G. M.; Steinberg D. Free Radical Biol. Med. 2000, 28, 1815. [DOI] [PubMed] [Google Scholar]
- Podrez E. A.; Poliakov E.; Shen Z. Z.; Zhang R. L.; Deng Y. J.; Sun M. J.; Finton P. J.; Shan L.; Gugiu B.; Fox P. L.; Hoff H. F.; Salomon R. G.; Hazen S. L. J. Biol. Chem. 2002, 277, 38503. [DOI] [PubMed] [Google Scholar]
- Goodman M. M.; Callahan A. P.; Knapp F. F. J. Med. Chem. 1985, 28, 807. [DOI] [PubMed] [Google Scholar]
- Ihre H.; De Jesus O. L. P.; Frechet J. M. J. J. Am. Chem. Soc. 2001, 123, 5908. [DOI] [PubMed] [Google Scholar]
- Tian L. Ph.D. Thesis, Rutgers University, 2004. [Google Scholar]
- Li J. J., Ed. In Name Reactions; Springer: New York, 2014; p 628. [Google Scholar]
- Rosen M. J.; Kunjappu J. T.. Surfactants and interfacial phenomena; John Wiley & Sons: Hoboken, New Jersey, 2012. [Google Scholar]
- Kim S.; Shi Y.; Kim J. Y.; Park K.; Cheng J.-X. Expert Opin. Drug Delivery 2010, 7, 49. [DOI] [PubMed] [Google Scholar]
- Yang J. S.; Zhou Q. Q.; He W. Carbohydr. Polym. 2013, 92, 223. [DOI] [PubMed] [Google Scholar]
- Hamley I. W.Block copolymers in solution: fundamentals and applications; Wiley: New York, 2005. [Google Scholar]
- Cai C.; Zhu W.; Chen T.; Lin J.; Tian X. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5967. [Google Scholar]
- Owen S. C.; Chan D. P. Y.; Shoichet M. S. Nano Today 2012, 7, 53. [Google Scholar]
- Xiao M. Y.; Xia G. J.; Wang R.; Xie D. Q. Soft Matter 2012, 8, 7865. [Google Scholar]
- Gao H. J.; Liu J. J.; Yang C. H.; Cheng T. J.; Chu L. P.; Xu H. Y.; Meng A. M.; Fan S. J.; Shi L. Q.; Liu J. F. Int. J. Nanomed. 2013, 8, 4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. G.; Lukyanov A. N.; Singhal A.; Torchilin V. P. Nano Lett. 2002, 2, 979. [Google Scholar]
- Nogueira D. R.; Moran M. C.; Mitjans M.; Martinez V.; Perez L.; Vinardell M. P. Eur. J. Pharm. Biopharm. 2013, 83, 33. [DOI] [PubMed] [Google Scholar]
- Halets I.; Shcharbin D.; Klajnert B.; Bryszewska M. Int. J. Pharm. 2013, 454, 1. [DOI] [PubMed] [Google Scholar]
- Richards D.; Ivanisevic A. Chem. Soc. Rev. 2012, 41, 2052. [DOI] [PubMed] [Google Scholar]
- Gaucher G.; Dufresne M. H.; Sant V. P.; Kang N.; Maysinger D.; Leroux J. C. J. Controlled Release 2005, 109, 169. [DOI] [PubMed] [Google Scholar]
- Savic R.; Azzam T.; Eisenberg A.; Maysinger D. Langmuir 2006, 22, 3570. [DOI] [PubMed] [Google Scholar]
- Chen H.; Kim S.; He W.; Wang H.; Low P. S.; Park K.; Cheng J. X. Langmuir 2008, 24, 5213. [DOI] [PubMed] [Google Scholar]
- Guaderrama-Diaz M.; Solis C. F.; Velasco-Loyden G.; Laclette J.; Mas-Oliva J. Mol. Cell. Biochem. 2005, 271, 123. [DOI] [PubMed] [Google Scholar]
- Lewis D. R. Ph.D. Thesis, Rutgers University, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.