

Abstract
Dynactin is a highly conserved, multiprotein complex that works in conjunction with microtubule-based motors to power a variety of intracellular motile events. Dynamitin (p50) is a core element of dynactin structure. In the present study, we use targeted mutagenesis to evaluate how dynamitin’s different structural domains contribute to its ability to self-associate, interact with dynactin, and assemble into a complex with its close binding partner, p24. We show that these interactions involve three distinct structural elements: (1) a previously unidentified dimerization motif in the N-terminal 100 amino acids, (2) an α-helical motif spanning AA 106–162, and (3) the C-terminal half of the molecule (AA 213–406), which is predicted to fold into an anti-parallel α-helix bundle. The N-terminal half of dynamitin by itself is sufficient to disrupt dynactin, although very high concentrations are required. The ability of mutations in dynamitin’s interaction domains to disrupt dynactin in vitro was found to correlate with their inhibitory effects when expressed in cells. We determined that the dynactin subunit, p24, governs dynamitin oligomerization by binding dynamitin along its length. This suppresses aberrant multimerization and drives formation of a protein complex that is identical to the native dynactin shoulder.
Keywords: dynein, dynactin, motor, motility, microtubule
INTRODUCTION
First identified as a major component of dynactin, dynamitin (p50) is an important reagent for perturbing dynein/dynactin function in living cells. Four dynamitin subunits sit at the junction between dynactin’s two major structural domains, the cargo-binding Arp1 filament and the p150Glued arm that supports interactions with motors and microtubules. When free dynamitin monomers are mixed with dynactin, they bind and trigger release of p150Glued from the Arp1 filament, which renders the remaining dynactin “remnant” non-functional (1). This remarkable property has led dynamitin to become a widely used inhibitor of dynein/dynactin-dependent phenomena.
From a structural perspective, dynamitin is an intriguing subject for study. Its primary sequence predicts several structural motifs that may participate in protein-protein interactions. Highly purified, recombinant dynamitin shows a strong propensity for self-association, a property we have proposed is the basis for its ability to disrupt dynactin (2). Dynamitin’s other close binding partner in dynactin is the p24 subunit. These two proteins remain tightly associated in a heterotrimeric complex (2:1 dynamitin:p24; dynactin “shoulder”) when dynactin is disassembled biochemically (2, 3). The dynamitin/p24 shoulder complex interacts with p150Glued as well as components of the Arp1 filament (3, 4) to hold dynactin together. Both pure, recombinant dynamitin and dynamitin/p24 shoulder complexes isolated from dynactin are elongated structures with high axial ratios, yet the structure they form in dynactin is compact (5), suggesting great flexibility.
Given dynamitin’s importance to dynactin integrity, a detailed analysis of mechanism by which dynamitin interacts with itself and p24 is clearly warranted. In the present study, we explore dynamitin domain organization and function using limited proteolysis and targeted mutagenesis. The behavior of a battery of mutant dynamitin species was evaluated using three different assays for protein-protein interactions. Homo-oligomerization was studied using sedimentation equilibrium analytical ultracentrifugation. The ability of dynamitin mutants to bind dynactin and disrupt its structure was examined using sucrose gradient velocity sedimentation. The mutants were also evaluated using a novel assay for p24 binding and solubilization. In the course of this work, we discovered a previously unidentified dimerization motif in the dynamitin N-terminus and established that this motif, plus a predicted coiled-coil in dynamitin’s N-terminal half, are required for proper self-association and p24 binding. For dynamitin species to be able to disrupt dynactin, the following requirements must be satisfied: they must maintain a pool of monomers, they must be able to at least dimerize, and they must contain the first predicted coiled-coil motif, even if this motif is mutated. Finally, we showed that the interaction between dynamitin and p24 involves contacts along the length of the dynamitin molecule, as neither the N- nor the C-terminal half alone can bind and solubilize p24. Together, our findings provide important new information about the nature and extent of the dynamitin-dynamitin and dynamitin-p24 interfaces that are the basis of dynactin structure.
RESULTS
In silico analysis of dynamitin structure
Our previous work revealed that both native and recombinant dynamitin molecules exist primarily as oligomeric assemblies that are trimers or larger (2, 3). To learn more about the structural features that might govern dynamitin associations, we examined its primary sequence to identify potential protein-protein interaction motifs. We began by using the program Multicoil (6) to determine which of dynamitin’s three strongly predicted coiled-coil motifs (1); C1, C2 and C3 in Figure 1) has the potential to form assemblies made up of three or more α-helices. This analysis indicated that the third motif (C3; AA 281–308) has a very high likelihood of forming triple coils or larger assemblies. Multicoil propensity is shared, to a lesser extent, by the predicted coiled-coil motifs C1 and C2.
Figure 1.

Schematic of the predicted structural features of human dynamitin and the mutants used in the analytical ultracentrifugation studies. The cartoons represent full length dynamitin and engineered fragments and indicate the positions of point mutants. The strongly predicted coiled-coil motifs C1 (AA 105–135), C2 (AA 219–251) and C3 (AA 281–308) are gray; C3, which also has a high propensity for multicoil formation, is indicated in black. The stippled box (AA 186–213) indicates a region that is predicted to be natively disordered. The three a-helices (H1, AA 221–243; H2, AA 261–299; and H3, AA 308–406) that are predicted to assume an anti-parallel, spectrin-like fold are indicated at the top. The binding sites for MacMARCKS, calmodulin and ZW10 map are indicated as M (AA 1–58), C (AA 59–83), and Z (AA 121–143), respectively (13, 20).
To learn more about how these motifs might contribute to dynamitin’s tertiary structure, we subjected its sequence to the mGenTHREADER feature of PSIPRED (7), an algorithm that threads amino acid sequences to find secondary structure patterns that exhibit similarities to known protein folds. This analysis strongly predicted that the C-terminal half of dynamitin (AA 222–406) folds into an antiparallel α-helix bundle similar to a spectrin repeat. This prediction is entirely compatible with both the coiled-coil and multi-coil predictions, as neither distinguishes between parallel and anti-parallel arrangements. Dynamitin sequences from a wide range of organisms (human, chicken, zebrafish, C.elegans, D. melanogaster, S. cerevisiae, A. nidulans, T. thermophila) all yielded similar mGenTHREADER results despite their lack of conservation (ClustalW analysis; (8)). Helices 1 and 2 of the predicted α-helix bundle (H1 and H2; Figure 1), which overlap with the predicted coiled-coils C2 and C3, are short, as in conventional spectrin. In an anti-parallel a-helix bundle, the middle helix (H2/C3) is intimately associated with both of the other α-helices. This organization is completely consistent with the high propensity of C3 for multicoil formation. The last α-helix, H3 (AA 308–406), lacks any obvious bends or breaks and is predicted to be considerably longer than H1 and H2.
Analysis of the potential native disorder of dynamitin using the program DISOPRED (9) revealed a disordered region near the middle of the molecule (Figure 1), which might represent a hinge between structural domains. The program FoldIndex (10), which predicts how well different regions of proteins fold, predicted the same disordered region and also suggested that the N-terminal ≈110 amino acids might be relatively unstructured. The rest of the protein, particularly the C-terminal half, was predicted to be well-folded. The idea that the dynamitin C-terminus folds into a discrete structural element that is physically distinct from the N-terminus is consistent with our finding (see next section; Figure 2) that the C-terminal half of dynamitin is a protease-stable domain.
Figure 2.
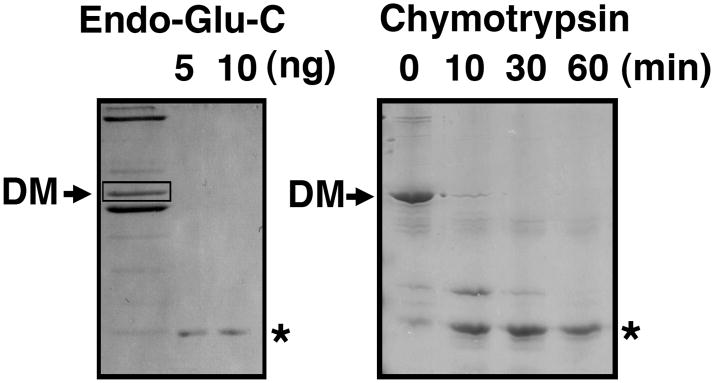
Identification of a protease-stable domain of dynamitin. Dynamitin was subjected to in-gel proteolysis (12) using endoproteinase-Glu-C (Endo-Glu-C, also known as “V8” protease; left) or proteolysis in solution using chymotrypsin (right). Left panel: Chick brain dynactin (left lane) was subjected to SDS-PAGE to isolate individual subunits, then gel bands corresponding to dynamitin (DM) were excised and re-electrophoresed in the presence of Endo-Glu-C (5 or 10 ng as indicated). Right panel: Purified recombinant dynamitin (1 nmol) was mixed with 10 pmol chymotrypsin and incubated for the times indicated, then the samples were subjected to SDS-PAGE. Both gels are stained with Coomassie blue. The asterisks mark the positions of the predominant digestion products (≈ 30 kDa).
Limited proteolysis of dynamitin
When we originally cloned chicken dynamitin (11), we verified the identity of the cDNA clone by obtaining the N-terminal amino acid sequence of a proteolytic fragment of native dynamitin generated by in-gel digestion (12) with endoproteinase Glu-C (“V8” protease). This fragment was about half the mass of intact dynamitin and corresponded to AA 213 – 406 (Figure 2). The protein fragments that result from in-gel proteolysis typically correspond to exceptionally stable pieces of the parent protein, suggesting that the C-terminal half of dynamitin might be a protease-stable domain. To verify this, we subjected recombinant human dynamitin to limited digestion in solution with chymotrypsin, a protease that typically yields large fragments. Once again, we obtained a stable fragment about half the size of the starting protein (Figure 2). MALDI-MS revealed that this fragment, AA 213–406, was nearly identical to the stable Endo-Glu-C fragment. Because two different proteases used on dynamitins from two different species yielded essentially identical fragments, we conclude that the C-terminal half of dynamitin is a protease-stable domain. Accordingly, we engineered and expressed recombinant proteins corresponding to the N- and C-terminal halves of human dynamitin (Figure 1) for further use.
Dynamitin mutagenesis
Equilibrium analytical ultracentrifugation analysis indicates that pure recombinant human dynamitin readily oligomerizes (2). The data fit well to a monomer – 3-mer/4-mer distribution, with 3- /4-mers being considerably more abundant than monomers. A small amount of higher order oligomers (6/8-mer) are also observed. To determine how dynamitin’s predicted interaction motifs and structural domains contribute to oligomerization, we engineered a series of point mutants in which hydrophobic residues (L or V) that are strongly predicted to participate in coil-coil formation were replaced with prolines (Figure 1). Some of these mutations were also engineered into the N- and C-terminal fragments.
Oligomerization behavior of dynamitin fragments and point mutants
The ability of the different mutant dynamitin species to oligomerize was evaluated using sedimentation equilibrium analytical ultracentrifugation (Table I). We began by analyzing the behavior of the isolated N- and C-terminal halves. Samples of the N-terminal half (AA 1–212) contained monomers and 3- or 4-mers (the data fit equally well to both assembly states) and a minor population of 6- or 8-mers (Table I). Nearly half the protein remained monomeric, in contrast to full length dynamitin which maintains less than 20% as monomer. The C-terminal fragment exhibited dramatically different behavior. It self-associated extensively; large oligomers (6/8-mers and beyond) were the major constituent and monomers were not detected. Samples of the C-terminal half containing point mutations designed to disrupt C2 (L236P) or C3 (V295P) exhibited the same behavior (Table I). This suggests that the introduction of a bend in either H1 or H2 of the anti-parallel α-helix bundle does not significantly perturb the structure of the C-terminal domain. We conclude that the C-terminal domain acts as a structural unit that has a strong tendency to self-associate. However, the C-terminal half by itself shows indiscriminate association into very large, non-native oligomers. This suggests that the N-terminal half plays an important role in governing oligomerization.
Table I.
Activities of dynamitin mutants
| Dynamitin species | Mutation | N-mers | Dynactin disruption (in vitro) | p24 binding | Dynamitin binding (Y2H) | Inhibition of dynein/dynactin function in vivo |
|---|---|---|---|---|---|---|
| Full length | None | 1 – 3/41 | ++++ | + | +++ | + |
| Full length (6XHis) | None | 1 – 3/41 | ++++ | + | ND | ND |
| Full length (6XHis) | L118P (C1) | ND | +++ | ND | ND | + |
| Full length (6XHis) | V122P (C1) | 1 – 3/41 | +++ | + | ND | + |
| Full length (6XHis) | L236P (C2) | 1 – 3/41 | ++ | + | ND | + |
| Full length (6XHis) | V295P (C3) | 1 – 3/41 | ++ | + | ND | + |
| Full length (6XHis) | C1/C2/C3 | 1 – 3/41 | + | + | ND | ND |
| AA 1–78 | None | 1 – 2 | – | – | ND | +3 |
| AA 1– 116 | None | 1 – 2 | – | – | +++ | ND |
| AA 1–212 | None | 1 – 3/41 | + | – | ND | – |
| AA 1–212 | L118P (C1) | 1 – 2 | ++ | ND | ND | ND |
| AA 1–212 | V122P (C1) | 1 – 2 | ++ | ND | ND | ND |
| AA 1–212 | P140A (HTH) | 1 – 2 – 3/4 | ++ | ND | ND | ND |
| AA 137–406 | None | ND | ND | ND | ND | – |
| AA 160–406 | None | ND | ND | ND | +++ | ND |
| AA 213–406 | None | ≤ 6/82 | – | – | ND | ND |
| AA 213–406 | L236P (C2) | ≤ 6/82 | ND | ND | ND | ND |
| AA 213–406 | V295P (C3) | ≤ 6/82 | ND | ND | ND | ND |
All dynamitin species used for in vitro experiments (analytical ultracentrifugation, disruption of purified dynactin, p24 binding) carried C/A point mutations at AA 245 and 261 (as appropriate) to eliminate formation of covalently cross-linked species. “Cys-lite” dynamitin is as active as wild type dynamitin for dynactin disruption (2) and p24 solubilization (data not shown). Oligomerization state (“N-mers”) was assessed by sedimentation equilibrium analytical ultracentrifugation. All species were evaluated in at least two independent experiments in which at least two different concentrations were used. The data obtained with full length dynamitin species fit equally well to a monomer/trimer (with a minor population of hexamer) or a monomer/tetramer (with a minor population of octamers) assembly mode. This is indicated as 1 – 3/4. Dynactin disruption was evaluated by examining p150Glued release via velocity sedimentation (as in Figure 3). p150Glued release was scored as follows: species that caused complete release at a 25X molar excess were scored (++++), species that caused most but not all p150Glued to be released were scored (+++), species that showed about 50% release were scored (++), species that showed minimal (but detectable) release at either 25X or 100X molar excess were scored (+). Species that showed no release at any concentration were scored (–). p24 binding was evaluated using the shoulder reconstitution assay described in the text (Results and Methods). Dynamitin species that allowed p24 to remain soluble were scored (+); species that did not prevent p24 from precipitating were scored (–). If the parent wild type fragment had no disruption or p24 binding activity the corresponding mutants were not evaluated (ND). The ability of some dynamitin fragments (described in (20) to interact with dynamitin was also evaluated by yeast two-hybrid (Y2H) analysis; (+++) indicates species that showed a strong interaction. The ability of mutants to interfere with dynein/dynactin activity in vivo was evaluated by overexpression in HeLa cells followed by immunofluorescence analysis of Golgi complex and mitotic spindle integrity. Mutants that showed effects at low or high expression levels were scored (+) and mutants that did not show consistent effects even at high expression levels were scored (–). ND: Not determined. Footnotes:
Full length dynamitin was typically 15–20% monomer, 60–70% trimer/tetramer, with the remainder being 6-mer/8-mer and beyond, whereas the N-terminal fragment (AA 1–212) was typically 45% monomer, 40% trimer/tetramer, with the remainder being 6-mer/8-mer and beyond.
Little, if any monomer was detected in these samples.
From (11).
We also characterized the behavior of full length dynamitin carrying point mutations in C1, C2 or C3 (Table I). All the mutants exhibited behavior that fit to the standard monomer-3-/4-mer assembly mode, as did the triple mutant (C1/C2/C3). That dynamitin carrying mutations in one or all of its coiled-coil motifs oligomerizes normally indicates that its self-assembly mechanism is robust. The benign effects of these mutations suggest that none of them alter dynamitin structure or surface features dramatically. These data also support the hypothesis that the N-terminal half of dynamitin contributes to proper self-association.
The N-terminal half of dynamitin contains two distinct self-association motifs
The N-terminal half of dynamitin contains a single predicted coiled-coil motif, C1 which is not strongly predicted to form triple or higher order coils, yet this fragment readily forms 3-/ 4-mer and some 6/8-mers (Table I). This suggests either that C1 is sufficient for oligomerization or that the dynamitin N-terminus contains another structural feature that contributes to dynamitin-dynamitin binding. To explore this question, we evaluated the behavior of several mutant species derived from the N-terminal half.
C1 (AA 105–135; Figure 1) is the first part of a longer predicted a-helix (AA 105–162) that contains a proline residue at AA 140. To determine how this bend in a potentially longer helix impacted oligomerization we mutated AA P140 to alanine (P140A). The mutant sequence predicts a coiled-coil that is longer than in the wild type protein. The P140A mutant formed dimers and 3-/4-mers , but showed no evidence of higher order oligomers (Table I). The fact that this mutant completely suppresses higher order oligomerization suggests that the bent α-helix in the wild type protein contributes to the non-physiological oligomers (i.e., 6/8-mers) that can form in vitro.
To further evaluate the role played by C1 in dynamitin self-association, we examined the behavior of two other point mutants, L118P and V122P. Both residues contribute to the hydrophobic heptad repeat motif that characterizes coiled-coil proteins. L118 is very highly conserved among between species (identical in most metazoans) whereas V122 is slightly less conserved (identical in mammals, chicken and worm, but not zebrafish, D. melanogaster, T. thermophila or yeast). Sedimentation equilibrium analysis indicated that both proteins exhibited behavior that fit well to a monomer – dimer assembly mode (Table I). No larger oligomers were detected. We conclude that coiled-coil associations of the C1 motif drives higher order oligomerization of the N-terminal fragment. These data further suggest that the N-terminal half of dynamitin contains a self-association motif besides C1 that supports dimerization.
To test this possibility we prepared two shorter fragments, AA 1–78 (11) and AA 1–116. AA 1–78 contains sequence motifs that have been proposed to interact with other proteins including cytoplasmic dynein, calmodulin and MacMARCKS (11, 13). Its sequence predicts several short α-helices and β-strands but contains no strongly predicted folds or other motifs; FoldIndex (10) predicts that this entire part of the protein may be relatively unstructured. AA 1– 116 contains eleven amino acids of C1 which is not sufficient for coiled-coil formation. Sedimentation equilibrium analysis of both fragments indicated the presence of monomers and dimers only (Table I). These findings indicate that the extreme N-terminus of dynamitin contains a previously unidentified dimerization motif.
The ability of dynamitin point and truncation mutants to associate with full length dynamitin was also analyzed by yeast two-hybrid analysis (Table I). An N-terminal fragment (AA 1–116) and a C-terminal fragment (AA 160–406) both showed binding to dynamitin in this assay, consistent with our findings that both fragments contain self-association motifs.
Identification of structural features required for dynactin disruption
A well-known activity of dynamitin is its ability to disrupt dynactin. This process also depends on dynamitin interactions, as it involves binding of free dynamitin monomers to the dynactin shoulder/sidearm structure. Binding of exogenous dynamitin monomers leads to a remodeling of subunit/subunit contacts within the dynactin shoulder and ultimately, subunit release (2). To determine how dynamitin’s self-association motifs and structural domains contribute to dynactin disruption, we tested the activity of point mutant and truncated forms of dynamitin in an vitro disruption assay. Purified bovine brain dynactin was mixed with a 25X molar excess of various dynamitin mutants, a ratio at which wild type dynamitin causes complete release of p150Glued from dynactin (2). Following incubation for 30 minutes on ice, the mixtures were sedimented into 5 – 20% sucrose gradients. Individual fractions were collected and dynactin disruption was evaluated by determining the sedimentation behavior of the dynactin subunits p150Glued and Arp1 on immunoblots (Figure 3).
Figure 3.

Dynactin disruption mediated by wild-type, full length dynamitin and representative dynamitin mutants. A: Fifteen μg of purified bovine brain dynactin was left untreated (top panel) or mixed with a 25X molar excess of recombinant, full length, human dynamitin (DM), either wild type (WT) or carrying one or more point mutations, as indicated (C1/C2/C3 carried the V122P mutation). Following incubation, the samples were subjected to velocity sedimentation into 5 – 20% sucrose gradients and the gradient fractions were evaluated by immunoblotting. The behavior of the dynactin subunit p150Glued is shown here. In all cases, Arp1 sedimented at 19–20S (as seen in B; data not shown for the other mutants). B: Purified dynactin was treated with a 25X molar excess of full length dynamitin (top) or 100X molar excess of the C-terminal half (bottom; AA 213–406), then subjected to velocity sedimentation as in A. p150Glued and Arp1 were detected by immunoblotting. The C-terminal half was detected using Coomassie blue staining. C: Purified dynactin was treated with a 100X molar excess of the N-terminal half (AA 1–212). The behaviors of p150Glued, endogenous dynamitin (DM) and the AA 1–212 fragment were followed by immunoblotting. Arp1 sedimented at 19–20S in this experiment (as in B; data not shown).
First, we evaluated whether full length dynamitin carrying single L/V →P mutations in C1, C2 or C3 could disrupt dynactin (Figure 3A, Table I). These mutants all showed relatively normal self-association behavior in sedimentation equilibrium experiments, so we reasoned that they would retain disruption activity. All caused release of p150Glued, although the C2 and C3 mutants were less active than wild type. The triple mutant (C1/C2/C3; V122P/L236P/V295P) also had reduced activity in this assay.
Next, we tested the ability of dynamitin fragments to disrupt dynactin. The isolated C-terminal half did not cause release of p150Glued (Figure 3B, Table I), consistent with the fact that this species does not maintain the pool of free monomers that is necessary for disruption (2). The N-terminal half, by contrast, did disrupt dynactin, but only when very large amounts were used (100X vs. 25X molar excess). The effect on dynactin structure was the same as what is seen with full length dynamitin; both p150Glued and endogenous dynamitin were released (Figure 3C). Samples of the N-terminal half bearing point mutations in C1 (either L118P or V122P), or the P140A mutant, also caused p150Glued release (Table I). The shorter N-terminal fragments (AA 1–78 and AA 1–116), by contrast, did not disrupt dynactin even when used at 100X molar excess (Table I). This suggests that the region spanning AA 117–212 (i.e., C1 and beyond) is required for shoulder-sidearm remodeling and subunit release.
During the process of dynactin disruption in vitro, two exogenous dynamitin monomers become stably associated with the remaining Arp1 minifilament (the dynactin “remnant”) (2). It is not known what elements of dynamitin structure are required for this, but it can be assumed that one or more of the oligomerization motifs are involved. Because the N-terminal fragment contains two oligomerization motifs we reasoned that it might remain bound to the dynactin remnant after disruption. When we tested this hypothesis directly using the disruption assay we found that the N-terminal half (AA 1–212 in Figure 3C) did not cosediment with Arp1, indicating that it did not remain associated with the dynactin remnant. None of the N-terminal fragments tested (the two C1 mutants, or the two shorter fragments, AA 1–78 or 1–116) cosedimented with Arp1 in these experiments (data not shown). These data indicate that the N-terminal half is able to interact transiently with dynactin and trigger subunit release. However, stable binding of dynamitin to the dynactin remnant requires interactions mediated by the C-terminal half.
In vivo activity of dynamitin mutants
Overexpression of dynamitin in cultured cells causes very well-characterized defects in mitotic spindle structure and mitotic delays, as well as dispersion of the Golgi complex and endocytic organelles toward the cell periphery (1, 11, 14). These defects arise because dynactin lacking p150Glued is no longer able to interact with dynein. We extended our analysis of the dynamitin mutants by evaluating their effects on dynactin integrity and function in vivo. We focused on a representative subset of truncation and point mutants in this work. Cells were transfected with expression constructs encoding the different mutants, then fixed and stained for Golgi markers (α-mannosidase and/or giantin) and tubulin to evaluate Golgi morphology and mitotic spindle integrity, respectively. Dynactin integrity was evaluated by sedimenting detergent lysates into sucrose gradients and following the behavior of p150Glued by immunoblotting. In a previous study, we demonstrated that the N-terminal fragment AA 1–78 did not cause dynactin disruption when overexpressed in vivo (11), in agreement with our present in vitro findings. Overexpression of a C–terminal fragment (AA 137–406) had no discernable effect on the structure of the Golgi complex or mitotic spindle integrity and did not yield dynactin disruption (Table I). These observations are in keeping with our finding that the dynamitin C–terminal half (AA 213–406) cannot disrupt dynactin in vitro. Overexpression of point mutants in C1 (L118P or V122P) caused Golgi dispersion (Figure 4), mitotic spindle defects and dynactin disruption (Table I), in agreement with our finding that C1 mutants readily disrupt dynactin in vitro. Point mutants in C2 (L236P) and C3 (V295P) also caused Golgi disruption and spindle abnormalities (Table I). Although these two mutants had slightly reduced activity when tested in vitro (Figure 3), apparently they are expressed at high enough levels in vivo to interfere with dynein/dynactin function. For all the active mutants, the effects of dynamitin mutants on Golgi complex morphology (Figure 4) and mitotic spindle formation were identical to what has been reported for full length, wild type dynamitin (1, 11, 15).
Figure 4.
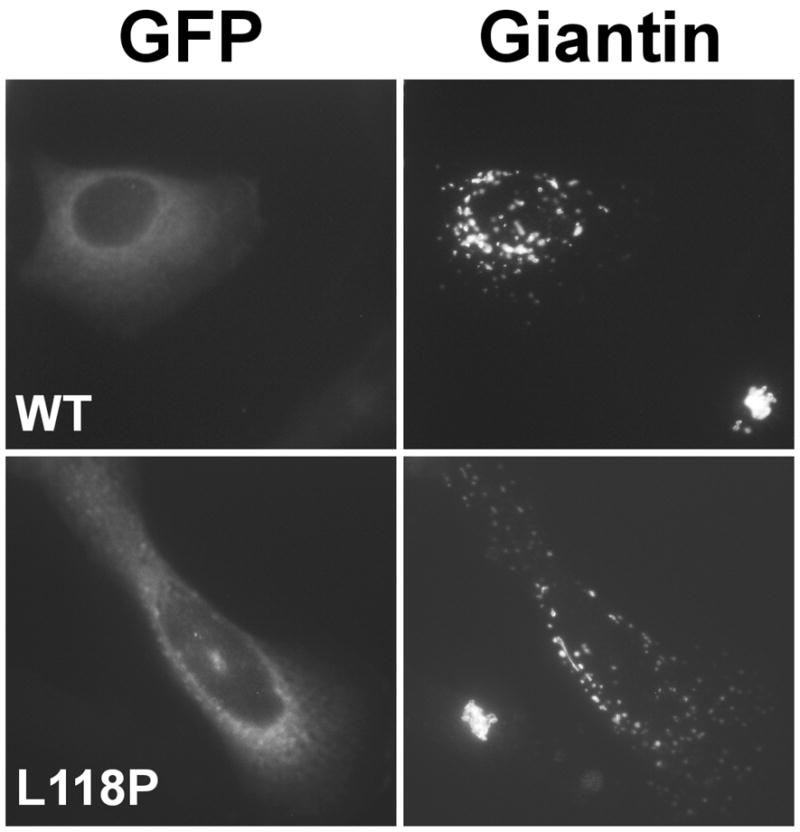
Effects of wild type and mutant dynamitin in vivo. Cells were transfected with plasmids encoding GFP-tagged wild type or the L118P mutant forms of dynamitin, then fixed and stained for the Golgi complex marker, giantin. Note the compact Golgi complex in the control cell in each panel. All dynamitin mutants that caused Golgi complex dispersion yielded similar results.
Dynamitin binds to and solubilizes p24
When dynactin molecules are disrupted completely using dynamitin (2) or the chaotropic salt KI (3), dynamitin is released together with p24 in a trimeric complex known as the dynactin shoulder. That two different disruption strategies yield the same structure suggests that the dynactin shoulder is a stable and fundamental building block of dynactin. When expressed in bacteria, recombinant dynamitin is soluble, whereas recombinant p24 is not ((16); our unpublished observations). Given the fact that dynamitin and p24 are so intimately associated in the native dynactin molecule, we reasoned that dynamitin might be able to bind and enhance the solubility of p24 in vitro. To test this hypothesis, we denatured p24 together with dynamitin using guanidine-HCl in the presence of a stabilizing salt, arginine. The mixture was then dialyzed to remove the guanidine-HCl and arginine (17, 18). Any insoluble protein that formed during dialysis was removed by centrifugation. When p24 was subjected to this treatment by itself, it precipitated completely and none remained soluble. Dynamitin, by contrast, remained soluble throughout these manipulations, and denatured/renatured dynamitin was able to disrupt dynactin normally (data not shown), indicating that it was properly re-folded.
When dynamitin (2X molar excess) was added to denatured, His-tagged p24 prior to dialysis, most of the p24 could be recovered with dynamitin in the soluble pool. Multiple lines of evidence indicated that dynamitin and p24 form a stable, biochemically well-behaved complex. First, affinity purification of His-tagged p24 also yielded dynamitin (Figure 5A). Secondly, when samples were subjected to MonoQ chromatography or velocity sedimentation, p24 and dynamitin coeluted from the MonoQ column (Figure 4B) or cosedimented at about 5S (Figure 4C). Quantitative densitometry of Coomassie blue-stained gels of fractions purified by metal-affinity chromatography followed by MonoQ (as in Figure 4B) yielded a stoichiometry of 2:1 dynamitin:p24, the same composition as the dynactin shoulder. To further verify that the reconstituted sample was the same as dynactin shoulder, we subjected it to sedimentation equilibrium analytical ultracentrifugation. This yielded data that fit well to a heterotrimeric species with the predicted mass of a 2:1 dynamitin:p24 complex (see also (2)). Reconstituted shoulder behaved ideally in this analysis; neither smaller (i.e., monomers or dimers) nor larger (i.e., large oligomers) species were seen, in marked contrast to what was seen for full length, wild type dynamitin and most of the mutants. Apparently, p24 controls oligomerization of recombinant dynamitin and causes it to form a highly homogeneous, well-behaved complex that corresponds to the native dynactin shoulder.
Figure 5.

Reconstitution of recombinant dynactin shoulder (dynamitin:p24 complex). His-tagged p24 was denatured and renatured in the presence of excess untagged dynamitin as described in the text. The resulting soluble protein was analyzed by column chromatography (A and B) or (C) velocity sedimentation into a sucrose gradient. (A) A HiTrap column was used to separate dynamitin/p24 complexes (fractions 15–29) from free dynamitin (fraction 6). (B) Dynamitin/p24 complexes isolated as in B were purified further by MonoQ chromatography. For both panels, the lane labeled L is the column load and the numbers indicate different column fractions. (C) The sedimentation profile of the dynamitin/p24 mixture prior to any chromatography is shown at the top; dynamitin alone is shown below for comparison. The bottom of the sucrose gradient is at the left. BSA (4.6S) was added to both samples prior to sedimentation to provide an internal standard; this did not alter the sedimentation behavior of dynamitin or the dynamitin/p24 complex. All gels were stained with Coomassie blue.
To learn more about the structural basis of the dynamitin/p24 interaction, we evaluated the ability of dynamitin mutants to bind and solubilize p24 in this assay (Table I). We began with point mutants in C1, C2 and C3 of full length dynamitin. All were able to solubilize p24. Next, we tested whether either the N- or C-terminal half could solubilize p24. Neither was able to solubilize p24 to a detectable degree. This suggests that the interaction of p24 with dynamitin requires the N-terminal dimerization motif and/or C1, as well as the C-terminal oligomerization domain. We conclude that this interaction is extensive and involves contacts along the length of both molecules.
DISCUSSION
The present study provides new information on dynamitin structure that has important implications regarding the organization and assembly of dynactin. Point and truncation mutants targeting different elements of dynamitin structure were analyzed using a series of in vitro assays designed to highlight distinct protein-protein interactions. The ability of dynamitin mutants to self-associate was determined using sedimentation equilibrium analytical ultracentrifugation. For some mutants, these findings were verified by yeast two-hybrid analysis. The ability of mutant forms of dynamitin to interact with and disrupt dynactin was evaluated both in vitro and in vivo. p24 binding was evaluated using a novel protein solubilization assay. Taken together, our results suggest new models of dynamitin structure and self-assembly (cartooned in Figure 6).
Figure 6.

Model of dynamitin structural domains (see box; shading is as in Figure 1), plus proposed modes of self-association of wild type and mutant species. The N-terminal dimerization domain is depicted as a shaded oval, the N-terminal α-helix as a thin zig-zag, the disordered region as a dashed line, and the C-terminal domain as a thick zig-zag. The different sized arrows indicate steps that are more or less highly favored.
Dynamitin exhibits three distinct modes of homotypic interaction involving motifs positioned along the length of the protein. The tendency of free dynamitin to self-associate is robust, and our analysis reveals that the N- and C-terminal halves play different roles in this process. Oligomerization appears to be initiated by dimerization of the N-terminal 78 AA. Additional dynamitin monomers are recruited by interactions mediated by the predicted coiled-coil, C1. These interactions govern the process of oligomerization and prevent indiscriminate associations. Once assembly has been properly templated by the N-terminal half, the oligomers gain added stability from interactions among the C-terminal domains. Unlike the N-terminal half, which behaves well and does not aggregate, the C-terminal half of dynamitin by itself undergoes aberrant self-association into very large oligomeric species (Table I, Figure 6).
Dynamitin self-association is profoundly altered by p24. p24 appears to “tame” dynamitin self-assembly and causes it to form a single, ideally behaved molecular species. Remarkably, this heterotrimeric complex is identical to a native dynactin sub-structure, the dynactin shoulder.
Sequence analysis suggests that C1 is especially important for dynamitin structure and function. Dynamitin sequences from a variety of organisms contain predicted coiled-coil motifs at similar positions, but only C1 is universal. C1 is a part of longer predicted α-helix that is interrupted by a single proline (P140); this was referred to earlier as a helix-turn-helix (HTH) motif (1). This entire α-helix appears to play a role in oligomerization, because a P140A mutant shows attenuated oligomerization and introduction of a second proline into C1 prevents oligomerization altogether (Table I, Figure 6).
Unlike C1 which is strongly predicted in all species examined, C2 and C3 are not always observed, However, mGenTHREADER analysis of all available dynamitin sequences predicts that their C-terminal halves adopt a conserved fold, the spectrin-like, anti-parallel α-helix bundle cartooned in Figure 6. Introduction of a proline into C2 or C3 has minimal impact on dynamitin behavior. This is not surprising. Many of the α-helices found in canonical spectrin family proteins contain prolines indicating that this residue does not prevent formation of anti-parallel α-helix bundles. Thus, our results provide support for the structural model cartooned in Figure 6.
Structure predictions on dynamitin have provided additional information beyond illuminating the structure of C-terminal domain. DISOPRED predicts that the N-and C-terminal domains of dynamitin are connected by an unstructured hinge, suggesting that this may be a site for extreme flexibility. Rotary shadow images of the isolated dynactin shoulder complex reveal a highly flexible, pleiomorphic structure (3) as opposed to a stiff rod. Some degree of flexibility must be required to allow the elongated shoulder complex to fold up into the compact, triangular shoulder that is seen in EM images of intact dynactin (5, 19).
The C-terminal half of dynamitin is clearly important for the stability of dynamitin/dynamitin and dynamitin/p24 oligomers. Point mutants in C1, which prevent oligomerization of fragments that lack the C-terminal domain, do not interfere with oligomerization of the full length protein or p24 binding. The strong tendency of the dynamitin C-terminal half to self-associate is not a normal property of spectrin family proteins. Few, if any classical spectrin repeats undergo homotypic interactions and those that do are limited to those repeats found at the ends of these elongated proteins. Furthermore, the dimers that result are invariably antiparallel. The parallel association of the C-terminal domain in dynamitin oligomers (the only polarity that is compatible with our data) thus appears to represent a novel type of quaternary structure.
The N-terminal portion of dynamitin has been reported to associate with the proteins MacMARCKS, calmodulin and ZW10 (13, 20), suggesting that it is exposed on the surface. We showed previously that free AA 1–78 causes dispersion of the Golgi complex without disrupting dynactin structure (11) and we show here that this fragment does not disrupt dynactin in vitro. This small, dimeric protein fragment might interact with dynein directly to interfere with dynein/dynactin binding, as we originally proposed (11).
Unlike AA 1–78, AA 1–212 does not alter Golgi complex or mitotic spindle structure. The apparent lack of effect of AA 1–212 in vivo (Table I) probably reflects its profoundly reduced ability to disrupt dynactin (Figure 3). Unlike full length dynamitin, AA 1–212 does not remain bound to the dynactin “remnant” following disruption. Apparently, the dynamitin C-terminal half is required for stable association with dynactin. p24 binding also requires both the halves of the dynamitin molecule. This raises the possibility that the binding of exogenous dynamitin to dynactin involve interactions with p24.
In the native dynactin molecule, two dynamitin/p24 shoulder complexes are bound to a p150Glued dimer to form a structure known as the shoulder/sidearm complex (3). Our work provides important new information regarding the structural basis of the dynamitin/p24 interaction, but the question of how shoulder binds p150Glued remains unanswered. mGenTHREADER structure predictions for p24 and p150Glued may shed some light on this question. Both proteins are strongly predicted to form α-helices that are further predicted to fold into anti-parallel, spectrin-like bundles. p24 is the least well-conserved of all dynactin subunits (21), yet it is consistently predicted to fold into a pair of spectrin-like repeats. Some p150Glued sequences also predict a spectrin-like fold in the central portion of the molecule (AA ≈ 650 – 1040). This predicted motif ends in a highly conserved coiled-coil (AA 926–1049, referred to as CC2 in (22)). Rigorous determination of the shoulder/sidearm structure will obviously have to await high resolution analysis, but these predictions provide the basis for models of p24 and p150Glued structure that we are currently testing. Finally, the availability of large amounts of highly homogenous, recombinant dynactin shoulder is a critical first step toward obtaining a high resolution structure of this fundamental dynactin subcomplex.
MATERIALS AND METHODS
Dynamitin mutagenesis and purification
Dynamitin point mutants were generated using QuickChange mutagenesis and fragments were engineered using PCR. Human dynamitin species (wild type, mutant, untagged or tagged with the His-/XPress tag; Invitrogen) were expressed in bacteria and purified by ammonium sulfate precipitation followed by MonoQ ion exchange chromatography and Superose12 gel filtration chromatography, as described (2, 23). Elution peaks were identified by A280. The Superose12 column was eluted with 25mM Tris-HCl, pH 8.0, 100 mM KCl, 1 mM EDTA and 1 mM 2-mercaptoethanol; the isolated dynamitin was dialyzed overnight against the same buffer used for the analytical ultracentrifugation analyses. All of the mutant proteins expressed well and could be easily purified according to this standard procedure. Disruption experiments were performed in 50 mM Tris-Cl, pH 7.5, 150 mM NaCl and 1 mM EDTA, as described (2).
Analytical ultracentrifugation
The partial specific volume (v) was calculated from the amino acid composition using the method of Cohn and Edsall (24). The solvent density (r) was calculated from buffer composition as described (25). These calculations along with corrections for temperature were calculated using the computer program SEDNTERP (26).
Equilibrium sedimentation experiments were carried out at 4°C and later at 20°C using cells fitted with 12 mm double sector charcoal-filled epon centerpieces and sapphire windows. Cell sectors were loaded with 120 ml of either solvent or protein solution. Samples at two or three different concentrations over the range of 0.4 to 2 mg/ml were run in duplicate in an eight-place An-50 Ti rotor. The lower temperature runs were allowed to reach equilibrium at rotor speeds between 9,500 and 13,000 rpm. The runs at 20°C were made at 18 – 19,000 rpm, at which speeds the concentration distributions at equilibrium were essentially depleted at the menisci. Equilibrium was assumed to have been established when no detectable changes in the concentration distributions could be detected over 2 to 3 hour intervals. Data from the 4°C runs were collected by both the absorption (at 280 nm) and interference optical systems; for the runs at 20°C only the interference system was used. The data from both sets of experiments were analyzed using the ORIGIN-based self-assembly computer program (XL-A/XL-I Data Analysis Software, v. 4.0, Beckman Instruments, Inc.). Values for v and r were calculated as described above. Dynamitin samples analyzed at 4°C were in 25 mM Tris-Cl, pH 8.0, 100 mM M KCl, 2 mM β-mercaptoethanol, 1 mM EDTA. Those analyzed at 20°C were in either the solvent just described or in 100 mM KPO4, pH 6.5, 2 mM β-mercaptoethanol, 1 mM EDTA.
Dynactin disruption
Fifteen μg of purified bovine brain dynactin was mixed with a 25X molar excess (moles exogenous per mole of dynamitin in dynactin; each mole of dynactin contains 4 moles dynamitin) of the different recombinant human dynamitin species, diluted to 0.1 or 0.5 ml in sedimentation buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, and 1 mM EDTA) and then incubated on ice for 30 min. The N-terminal fragment, which triggered only partial p150Glued release at a 25X molar excess, was also evaluated at a 100X molar excess. The C-terminal fragment was also tested at both 25X and 100X molar excess. All buffers included 1000 fold dilutions of a protease inhibitor cocktail containing 1 mg/ml pepstatin, 1 mg/ml leupeptin, 40 mg/ml PMSF (phenylmethylsulfonyl fluoride), 10 mg/ml TAME (N α –p-tosyl-L-arginine methyl ester), 10 mg/ml BAME (α-benzoyl-L-arginine methyl ester), 10 mg/ml TPCK (α-p-phenylalanine chloromethyl ketone), and 10 mg/ml TLCK (N a-p-tosyl-L-lysine chloromethyl ketone). Following incubation, the samples were diluted further to 1.5 ml in sedimentation buffer and sedimented into a 5–20% continuous sucrose gradient in a Beckman SW41 rotor at 150,000 x g for 15 hours at 4°C. Fractions were collected, then chloroform/methanol precipitated and analyzed by SDS-PAGE and/or immunoblotting (p150Glued, Arp1 and dynamitin). Dynamitin species were determined to be active for dynactin disruption if any p150Glued was seen to sediment at 8-9S.
Polyacrylamide gel electrophoresis, immunoblotting and densitometry
SDS-PAGE was performed according to Laemmli (27). Proteins were separated on 11% polyacrylamide gels using the Mini-Protean II system (Bio-Rad, Hercules, CA) and were detected by Coomassie blue staining. For immunoblots, proteins were transferred to PVDF (Millipore, Bedford, MA) as described in (28) using a Mini Trans-Blot apparatus (Bio-Rad). p150Glued was detected using a mAb to the N-terminus (BD Transduction Laboratories), Arp1 was detected using mAb 45A (19) and dynamitin (both endogenous and exogenous) was detected using a polyclonal serum (2). All antibodies were used in conjunction with the Tropix Western-LightTM protein detection kit (Tropix, Bedford, MA).
In vivo analysis
Yeast-two hybrid analysis was performed as described (20). Transfection of cDNAs encoding different dynamitin species and immunofluorescence was performed as in (1, 11, 14), using antibodies to giantin, a-mannosidase and tubulin to assess Golgi complex and mitotic spindle integrity. Dynactin disruption was evaluated by sucrose gradient velocity sedimentation as in (1, 22).
p24 purification and renaturation with p50 and p50 mutants
One liter of Luria-Bertani (LB) with ampicillin (final concentration 50μg/mL) and chloramphenicol (final concentration 35μg/mL) was inoculated with 4 mL of a glycerol sample of pRSETp24 that had been frozen at exponential growth. The culture was grown at 37° C to an OD600 of 0.4 – 0.6, induced with 1mM IPTG, and then grown at 37° C overnight. After centrifugation the pellet was lysed with Bugbuster (Novagen) following the manufacturer’s recommendation to isolate inclusion bodies. The final pellet was resuspended in 5mL/pellet of 6M guanidine, 0.5M NaCl, 20mM NaPhosphate pH 7.4, centrifuged at 15,000 x G for 15 min, filtered and loaded on a NI HiTrap column (Amersham). The protein was eluted by running a pH gradient from 20mM NaPhosphate pH 7.4 to 20mM NaPhosphate pH 2.1.
Fractions containing p24 from the Ni HiTrap column were dialyzed against 3M guanidine-HCl, 0.5M arginine, 20mM Tris pH 7.4, 1mM DTT and further against 2M guanidine-HCl, 0.5M arginine, 20mM Tris pH 7.4, 1mM DTT. This sample of p24 was combined with a 2X molar amont of dynamitin that had been dialyzed against the same buffer. The p24/dynamitin mixture was incubated on ice for 30 minutes and then dialyzed against 1M guanidine-HCl, 0.5M arginine, 20mM Tris pH 7.4, 1mM DTT. The mixture was further dialyzed against 0.5M arginine, 20mM Tris pH 7.4, 1mM DTT and finally against 100mM NaCl, 20mM Tris pH 7.4, 1mM DTT. Solubilization of p24 was determined by visual inspection of the dialysate for the presence of a white precipitate. p24 by itself, or mixed with the dynamitin N- or C-terminal half, precipitated out in the second dialysis step (1M to no guanidine-HCl).When p24 was renatured in the presence of wild type or mutant full length dynamitin no precipitate was observed. The p24/p50 complex was further purified on a second Ni HiTrap column and/or a MonoQ column.
Acknowledgments
We thank Dr. Evangelos Moudrianakis for maintenance of the Biology Department analytical ultracentrifuge facility, Dr. Ting-Yu Yeh for help with figure preparation, Ms. Julie Dohm for suggesting the p24 reconstitution assay, Ms. Hatice Basdag for technical assistance and members of the Schroer lab for comments on the manuscript. This work was supported by NIH grants RO1GM44589 (to TAS) and R37GM047434 (to Dr. Richard Vallee, Worcester Foundation).
References
- 1.Echeverri CJ, Paschal BM, Vaughan KT, Vallee RB. Molecular characterization of 50kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J Cell Biol. 1996;132:617–633. doi: 10.1083/jcb.132.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melkonian KA, Maier KC, Godfrey JE, Rodgers M, Schroer TA. Mechanism of dynamitin-mediated disruption of dynactin. J Biol Chem. 2007;282:19355–19364. doi: 10.1074/jbc.M700003200. [DOI] [PubMed] [Google Scholar]
- 3.Eckley DM, Gill SR, Melkonian KA, Bingham JB, Goodson HV, Heuser JE, Schroer TA. Analysis of dynactin subcomplexes reveals a novel actin-related protein associated with the Arp1 filament pointed end. J Cell Biology. 1999;147:307–319. doi: 10.1083/jcb.147.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark SW, Rose MD. Arp10p is a pointed–end-associated component of yeast dynactin. Mol Biol Cell. 2006;17:738–748. doi: 10.1091/mbc.E05-05-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imai H, Narita A, Schroer TA, Maeda Y. Two-dimensional averaged images of the dynactin complex revealed by single particle analysis. J Mol Biol. 2006;359:833–839. doi: 10.1016/j.jmb.2006.03.071. [DOI] [PubMed] [Google Scholar]
- 6.Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997;6:1179–1189. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 8.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 10.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 11.Valetti C, Wetzel DM, Schrader M, Hasbani MJ, Gill SR, Kreis TE, Schroer TA. Role of dynactin in endocytic traffic: Effects of dynamitin overexpression and colocalization with CLIP-170. Mol Biol Cell. 1999;10:4107–4120. doi: 10.1091/mbc.10.12.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cleveland DW. Peptide mapping in one dimension by limited proteolysis of sodium dodecyl sulfate-solubilized proteins. Meth Enzymol. 1983;96:222–229. doi: 10.1016/s0076-6879(83)96020-2. [DOI] [PubMed] [Google Scholar]
- 13.Yue L, Lu S, Garces J, Jin T, Li J. Protein kinase C-regulated dynamitin-macrophage-enriched myristoylated alanine-rice C kinase substrate interaction is involved in macrophage cell spreading. J Biol Chem. 2000;275:23948–56. doi: 10.1074/jbc.M001845200. [DOI] [PubMed] [Google Scholar]
- 14.Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintanance of membrane organelle distribution. J Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burkhardt JK. The role of microtubule-based motor proteins in maintaining the structure and function of the Golgi complex. Biochim Biophys Acta. 1998;1404:113–126. doi: 10.1016/s0167-4889(98)00052-4. [DOI] [PubMed] [Google Scholar]
- 16.Scheich C, Niesen FH, Seckler R, Bussow K. An automated in vitro protein folding screen applied to a human dynactin subunit. Protein Sci. 2004;13:370–380. doi: 10.1110/ps.03304604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Bernardez Clark E, Schwarz E, Rudolph R. Inhibition of aggregation side reactions during in vitro protein folding. Methods Enzymol. 1999;309:217–236. doi: 10.1016/s0076-6879(99)09017-5. [DOI] [PubMed] [Google Scholar]
- 18.Middelberg AP. Preparative protein refolding. Trends Biotechnol. 2002;20:437–443. doi: 10.1016/s0167-7799(02)02047-4. [DOI] [PubMed] [Google Scholar]
- 19.Schafer DA, Gill SR, Cooper JA, Heuser JE, Schroer TA. Ultrastructural analysis of the dynactin complex: An actin-related protein is a component of a filament that resembles f-actin. J Cell Biol. 1994;126:403–412. doi: 10.1083/jcb.126.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Starr DA, Williams BC, Hays TS, Goldberg ML. ZW10 helps recruit dynactin and dynein to the kinetochore. J Cell Biol. 1998;142:763–774. doi: 10.1083/jcb.142.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eckley DM, Schroer TA. Interactions between the evolutionarily conserved, actin-related protein, Arp11, protein, actin and Arp1. Mol Biol Cell. 2003;14:2645–2654. doi: 10.1091/mbc.E03-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintyne NJ, Gill SR, Eckley DM, Crego CL, Compton DA, Schroer TA. Dynactin is required for microtubule anchoring at fibroblast centrosomes. J Cell Biol. 1999;147:321–334. doi: 10.1083/jcb.147.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wittman T, Hyman A. Recombinant p50/dynamitin as a tool to examine the role of dynactin in intracellular processes. San Diego, CA: Academic Press; 1999. [DOI] [PubMed] [Google Scholar]
- 24.Cohn EJ, Edsall JT. Proteins, Amino Acids and Peptides as Ions and Dipolar Ions. Rheinhold; NY: 1943. Chapter 16: Density and apparent specific volume of proteins; pp. 370–377. [Google Scholar]
- 25.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding S, Rowe A, editors. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Cambridge, England: Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 26.Hayes D, Laue T, Philo J. Sedimentation interpretation (SENDTERP) 1.06. 2001. http://www.jphilo.mailway.com/
- 27.Laemmli UK. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci (USA) 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]