

Abstract
CD8+ T cells respond to TCR stimulation by producing proinflammatory cytokines, and destroying infected or malignant cells through the production and release of cytotoxic granules. Scaffold protein Discs large homolog 1 (Dlg1) specifies TCR-dependent functions by channeling proximal signals toward the activation of p38-dependent proinflammatory cytokine gene expression and/or p38-independent cytotoxic granule release. Two Dlg1 variants are expressed in CD8+ T cells via alternative splicing, Dlg1AB and Dlg1B, which have differing abilities coordinate TCR-dependent functions. Although both variants facilitate p38-independent cytotoxicity, only Dlg1AB coordinates p38-dependent proinflammatory cytokine expression. In this study, we identify TCR-induced Dlg1 tyrosine phosphorylation as a key regulatory step required for Dlg1AB-mediated p38-dependent functions, including proinflammatory cytokine expression. We find that Dlg1AB but not Dlg1B is tyrosine phosphorylated by proximal tyrosine kinase Lck in response to TCR stimulation. Furthermore, we identify Dlg1 tyrosine 222 (Y222) as a major site of Dlg1 phosphorylation required for TCR-triggered p38 activation and NFAT-dependent expression of proinflammatory cytokines, but not for p38-independent cytotoxicity. Taken together, our data support a model where TCR-induced phosphorylation of Dlg1 Y222 is a key point of control that endows Dlg1AB with the ability to coordinate p38 activation and proinflammatory cytokine production. We propose blocking Dlg1AB phosphorylation as a novel therapeutic target to specifically block proinflammatory cytokine production but not cytotoxicity.
Introduction
Proper activation of CD8+ CTLs is essential for maintaining adaptive immunity to intracellular pathogens. In response to TCR stimulation, CTLs produce and release proinflammatory cytokines and destroy infected or transformed cells through targeted release of cytotoxic granules. TCR engagement is coupled to downstream effector function through the recruitment and activation of proximal tyrosine kinases Lck and Zap70. These early activation events initiate a variety of signaling networks including MAPKs ERK, JNK, and p38, which each regulate transcription factors including NFAT and NF-κB. This group of transcription factors controls the expression of genes that dictate TCR-dependent events including: proliferation, differentiation and effector function (1, 2). Recent studies demonstrate that CD8+ T cell activation is not a binary event, but instead represents a spectrum of tightly controlled biological responses, suggesting that TCR-proximal activation events are selectively coupled to specific downstream signaling networks and functions (3). However, the mechanism(s) by which CTL functionality is specified downstream of the TCR remains incompletely understood.
Scaffold proteins have emerged as key points of control that couple extracellular stimuli to intracellular signaling networks and downstream functions, through the formation multicomponent signaling complexes. One such scaffold protein, Discs Large Homolog 1 (Dlg1), localizes to the junction between T cells and APCs known as the immunological synapse, where it regulates Ag-dependent cytoskeletal and signaling events (4–9). The ability of Dlg1 to regulate cellular functions is attributed to its ability to associate with important cytoskeletal regulators and signal transducers through one or more of its modular protein interaction domains (4, 5, 7). These domains include three PSD-95/Dlg/ZO-1 (PDZ) domains, an SH3 domain and a catalytically inactive guanylate kinase (GUK) domain common to all membrane-associated GUK (MAGUK) scaffolds, as well as an L27 oligomerization domain, an N-terminal proline rich region and a C-terminal HOOK domain, which are unique to Dlg1. Dlg1 domain structure can be modified by alternative splicing events that are thought to effect Dlg1 stability, localization and function (10–15).
We and others have demonstrated that Dlg1 specifies TCR signal transduction, by coordinating the activation of p38 through the alternative pathway (5, 7, 16). The alternative p38 pathway is initiated downstream of the TCR, and requires the activity of proximal tyrosine kinase Lck and Zap70 (17). TCR engagement triggers activation of Zap70, which phosphorylates p38 at Y323, triggering p38 autophosphorylation at T180 and subsequent upregulation of p38 kinase activity (17, 18). This pathway is distinct from the canonical p38 pathway, which is triggered by environmental stress and results in direct phosphorylation of p38 at T180 and Y182 by MKK3 or MKK6 (19). A vital downstream target of alternatively activated p38 is the transcription factor NFATc1 (NFAT2), which when phosphorylated at S54 upregulates numerous CD8+ T cell effector functions including proinflammatory cytokine gene expression (20, 21).
We recently demonstrated that at least two Dlg1 variants are expressed in T cells because of alternative splicing, Dlg1AB and Dlg1B, that differ in the inclusion or exclusion of the proline-rich i1A region (O. Silva, J. Crocetti, L. Humphries, H. Elaesser, D. Brooks, J. Burkhardt, and M.C. Miceli, submitted for publication). Although both variants can mediate cytoskeletal reorganization and release of cytotoxic granules, only the Dlg1AB variant can facilitate alternative p38 activation leading to induction of NFAT-dependent genes including IFN-γ and TNF-α (O. Silva et al., submitted for publication). The ability of Dlg1AB to activate p38 has been attributed to interaction with proximal tyrosine kinase Lck; however, the mechanism by which Dlg1:Lck association facilitates p38 activation was unclear.
Despite having no inherent enzymatic activity, Dlg1 is dynamically regulated by posttranslational modifications and intramolecular interactions that induce changes in protein stability and binding partners that dictate the scope of Dlg1 function (22–25). Tyrosine phosphorylation of scaffold proteins can lead to conformational changes and creation of new binding sites that play a vital role in scaffold protein function (26, 27). In CD8+ T cells, Dlg1 associates with several tyrosine kinases, including proximal tyrosine kinase Lck and Zap70 (5). However, a role for tyrosine phosphorylation in regulating Dlg1 function in T cells has not been described previously.
In this paper, we report that scaffold protein Dlg1 is phosphorylated on tyrosine residues in response to TCR stimulation. This phosphorylation event occurred specifically on the Dlg1AB splice variant and can be mediated by Dlg1-asociated proximal tyrosine kinase Lck. We identified Y222 is a major site of TCR-dependent Dlg1 phosphorylation and demonstrated that loss of Y222 phosphorylation in the Y222F mutant significantly impaired TCR-induced p38 phosphorylation and NFAT-dependent expression of proinflammatory cytokines IFN-γ and TNF-α. Conversely, Dlg1 Y222 phosphorylation was not required for functions mediated by Dlg1B including degranulation. Taken together, our data identify Dlg1 Y222 phosphorylation as a key point of control that selectively impacts Dlg1AB-mediated functions such as proinflammatory cytokine expression. We propose Dlg1 Y222 phosphorylation as a novel therapeutic target to selectively block proinflammatory cytokine production while not impacting other immune functions such as target cell killing.
Materials and Methods
Mice
The generation of Dlg1flox/flox mice has been described previously (28). These mice were crossed with C57BL/6 mice expressing the OT-1 TCR. All mice were maintained and used in accordance with the University of California Los Angeles Chancellor’s Animal Research Committee.
In vitro kinase assays
OT-1 hybridoma cells (40 × 106) were lysed using an immunoprecipitation lysis buffer (Pierce) in the presence of protease and phosphatase inhibitors (Pierce). Resulting lysates were cleared by centrifugation and incubated with wild-type (WT) or Y222F GST-Dlg1 fusion proteins bound to glutathione–Sepharose bead slurry and then washed twice with kinase buffer (Cell Signaling Technology). Resulting bead complexes were incubated at 30°C for 20 min in 50 μl kinase buffer in the presence or absence of ATP (Cell Signaling Technology). Alternatively, WT or Y222F GST-Dlg1 fusion proteins were bound to glutathione Sepharose beads and incubated with recombinant Lck or Zap70 (Active Motif) in 50μl kinase buffer in the presence or absence of ATP at 30°C for 20 min.
Dlg1 knockdown and expression
Stable Dlg1 knockdown lines were created in both OT-1 hybridomas using an mIR-155–based retroviral knockdown vector (29). Sense and antisense sequences specific for the 3′-untranslated region (3′-UTR) of Dlg1 (5′-GTCCTCCACACTGACACAGAT-3′) were cloned in to the mIR-155–based knockdown vector. Virus was produced by transfecting 293T cells with 25 μg knockdown vector and 25 μg pCL-Eco vector (Mirus). Resulting virus was used to spin infect OT-1 hybridomas for 90 min at 1250 × g on two consecutive days. Cells were allowed to rest for 1 d and then assessed for Dlg1 knockdown. Dlg1 was reintroduced into these cell lines using an murine stem cell virus–based retroviral vector expressing a Dlg1 variant or mutant using the same viral production and transduction methods used for the knockdown virus.
Flow cytometry
For measurement of Dlg1, 1 × 106 cells were fixed and permeabilized using FoxP3 staining buffer set (00-5523-00; eBioscience), according to the manufacturer’s instructions. Dlg1 was detected using αDlg1 Ab (610875; BD Biosciences) at 1:1000, followed by Alexa Fluor 647–conjugated donkey anti-mouse IgG F(ab′)2 (715-606-150; Jackson ImmunoResearch Laboratories) at 1:1000. For measurement of p38 phosphorylation, expanded primary CD8+ T cells were harvested, counted, and placed in fresh medium at a concentration of 2 × 106 cells/ml. Cells were allowed to rest for 4 h at 37°C in 6-well dishes at volume of 2 ml/well and were transferred to 12-well plates coated with 5 μg anti-CD3 (clone 145-2C11; 553057; BD Biosciences) and 5 μg anti-CD28 (clone 37.51; 553295; BD Biosciences) with a final volume of 1 ml/well (2 × 106 cells) and incubated for 5 or 15 min at 37°C to stimulate. Cells were fixed in paraformaldehyde (4% final concentration) for 30 min, followed by permeabilization using the FoxP3 staining buffer set (00-5523-00; eBioscience), according to manufacturer’s instructions. Phosphorylated p38 T180/Y182 was detected using Alexa Fluor 647–conjugated anti–p-p38 Ab (T180/Y182; Phosflow 612595; BD Biosciences) at 1:20. For CD107a staining: primary 1 × 105 CD8+ T cells were incubated with EG.7 thymoma cells at indicated concentrations for 2–4 h in the presence of GolgiPlug, GolgiStop, and CD107a-allophycocyanin Ab (560646; BD Biosciences). Cells were surface stained using CD8a-PE and fixed. Events were collected using FACSCalibur (BD Biosciences) and analyzed using FlowJo software.
Immunoprecipitation and blotting
For immunoprecipitation, T cell lysates were incubated with 20 μl 50% (v/v) protein G–Sepharose beads (17-5132-01; GE Healthcare) for 2 h at 4°C. Sepharose beads were removed, and lysates were incubated with 2 μg anti-Dlg1 Ab (610875; BD Biosciences) or IgG1κ control Ab (554121; BD Pharmingen) for 1 h at 4°C, and then, 40 μl 50% protein G–Sepharose beads in PBS were added and incubated overnight (∼12 h) at 4°C. Beads were washed and boiled with Laemmli buffer. Blotting was performed using Abs directed against Dlg1 (610875; BD Biosciences), phosphotyrosine (clone 4G10; Millipore), p38 (clone C20; SC535; Santa Cruz Biotechnology), GST (2625; Cell Signaling Technology), or phosphorylated p38 (T180/Y182 clone D3F9; 4511; Cell Signaling Technology). Blots were imaged using ECL Plus Western blotting substrate (32132; Pierce) or LiCor Odyssey Imaging System.
RNA isolation, Reverse transcription, and quantitative PCR
RNA was isolated using TRIzol reagent as described previously (5). Two micrograms of RNA was reverse transcribed using Superscript II reverse transcriptase (Invitrogen), according to the manufacturer’s instructions, using random hexamer and oligo(dT)20 as primers. The iCycler thermocycler was used for quantitative PCR analysis according to the manufacturer’s instructions (Bio-Rad). A final volume of 25 μl was used for each quantitative PCRs as described previously (5). The following gene-specific primers were used for amplification: L32(F), 5′-AAG CGAAACTGGCGGAAAC-3′, and L32(R), 5′-TAACCGATGTTGGGCATCAG-3′; NFATc1(F), 5′-GCCTCGTAT CAGTGGGCGAAG-3′, and NFATc1(R), 5′-CGAAGCTCGTATGGACCA-3′; IκBα(F), 5′-CTGCAGGCCACCAACTACAA-3′, and IκBα(R), 5′-CAGCACCCAAAGTCACCAAGT-3′; IFN-γ(F), 5′-GTCAAC AACCCACAGGTCCAG-3′, and IFN-γ(R), 5′-CCTTTTCCGCTTCCTGAGG-3′; TNF-α(F), 5′-AATGGCCTCCCTCTCATCAGT-3′, and TNF-α(R), 5′-GCTACAGGCTTGTCACTCGAATT-3′; and IL-2(F), 5′-CCTGAGCAGGATGGAGAATTACA-3′, and IL-2(R), 5′-TCCAGAACATGCCGCAGAG-3′. All quantitative PCR products were normalized to L32 ribosomal protein quantitative PCR products.
Dlg1 knockout
CD8+ T cells were sorted from the spleens of Dlg1flox/flox mice using negative selection (Miltenyi Biotec) and expanded on anti-CD3 and anti-CD28 Ab for 72 h, followed by transduction with cre-expressing retrovirus. Cellular function was tested 5 d after retroviral transduction.
Lactate dehydrogenase assay
T cell cytotoxicity was determined using the Cyto96 nonradioactive assay (Promega). A total of 10,000 EG.7 thymoma cells were incubated with OT-1 CD8+ CTLs at indicated concentrations for 4 h, followed by analysis, according to the manufacturer’s instructions.
gDNA Isolation and Amplification
Genomic DNA was isolated from total splenocytes using a DNeasy Blood and Tissue Kit (Qiagen). PCR amplification of genomic DNA was performed using primers to amplify dlg1 exon 8.
Statistical methods
SD was calculated using Microsoft Excel. Statistical significance was obtained with a two-sided Student t test assuming equal variances.
Results
Dlg1AB is tyrosine phosphorylated by a Dlg1-associated kinase in response to TCR stimulation
Dlg1 is rich in tyrosine residues and is known to associate with several tyrosine kinases; therefore, we considered the possibility that the signaling potential of Dlg1 may be regulated by phosphorylation of specific tyrosines (5). To determine whether Dlg1 was phosphorylated in T cells, we performed in vitro kinase assays using Dlg1 complexes formed using T cell lysates. Specifically, GST fusion proteins expressing full-length Dlg1AB (GST-Dlg1AB) or full-length Dlg1B (GST-Dlg1B) were incubated with T cell lysates to form Dlg1 complexes with T cell–specific ligands. Upon addition of ATP to these complexes, robust tyrosine phosphorylation (pY-Dlg1) of Dlg1AB but not Dlg1B was observed (Fig. 1A, 1B). These data indicate that Dlg1AB but not Dlg1B associates with and is phosphorylated by at least one kinase found in T cell lysates. To determine whether Dlg1 phosphorylation was TCR induced, Dlg1 complexes were immunoprecipitated from resting or TCR-stimulated T cells, and the level of Dlg1 tyrosine phosphorylation was assessed. We observed an increase in tyrosine phosphorylation of the top band of Dlg1 in TCR-stimulated samples compared with resting samples, which we have previously identified as the Dlg1AB splice variant in T cells (Fig. 1C, 1D; O. Silva et al., submitted for publication). On the basis of these data, we concluded that Dlg1AB but not Dlg1B is phosphorylated by a Dlg1-associated kinase in response to TCR stimulation.
FIGURE 1.

A Dlg1-associated kinase phosphorylates Dlg1AB in response to TCR stimulation. (A and B) In vitro kinase assay where GST-Dlg1AB (A and B) or GST-Dlg1B (B) fusion proteins were bound to glutathione–Sepharose beads and incubated with T cell lysates, centrifuged, washed, and incubated with 10 mM ATP for 20 min at 30°C. Protein complexes were boiled off beads and subjected SDS-PAGE and blotted using anti-4G10 (pY-Dlg1) and anti-GST (GST-Dlg1). (C and D) OT-1 hybridomas were stimulated using 5 μg/ml anti-CD3 and 5 μg/ml CD28, followed by cross-linking with donkey anti-Armenian hamster Ab for 15 min at 37°C, followed by lysis. Dlg1 was immunoprecipitated from resulting lysates and subjected to SDS-PAGE and then blotted using anti-4G10 (pY-Dlg1) and αDlg1. Arrows refer to the Dlg1AB and Dlg1B variants. (D) The blot was quantified using ImageJ software. Error bars represent the SD from the mean. Data represent two independent experiments.
Tyrosine 222 (Y222) is a major site of TCR-induced Dlg1 phosphorylation
Dlg1AB and Dlg1B differ in the inclusion or exclusion of the proline-rich i1A region, which permits association of Dlg1 with proximal tyrosine kinase Lck (Fig. 2A; O. Silva et al., submitted for publication). The Dlg1AB protein variant contains 927 aa, 29 of which are tyrosines (GenBank accession number AAH57118.1) (Fig. 2A). To narrow our search for specific sites of Dlg1 tyrosine phosphorylation, we used the ScanSite algorithm to determine the most likely candidate tyrosine residues (30). Tyrosine 222 (Y222) was identified as the most likely site of tyrosine phosphorylation using high-stringency search criteria. Y222 is located in the N-terminal region of Dlg1 in the flexible linker region between the i1B and PDZ1 domains. In addition, Y222 is part of a highly conserved Tyr-Glu-Glu-Iso (YEEI) motif that is a predicted target of known Dlg1-associated tyrosine kinases Lck and Zap70 (5, 31, 32) (http://ppsp.biocuckoo.org) (Fig. 2A).
FIGURE 2.

Dlg1-associated Lck phosphorylates Dlg1 at position tyrosine 222. (A) Domain structure of Dlg1AB and Dlg1B. Asterisks indicate the location of the 29 tyrosine residues along the Dlg1 sequence. The bold asterisk indicates the location of Y222. (A, bottom inset) Amino acid sequence alignment of the region of Dlg1 surrounding Y222 from various all species listed in the National Center for Biotechnology Information Database. Shaded region indicates evolutionarily conserved linker region of Dlg1. (B) In vitro kinase assays using GST-Dlg1AB WT or GST-Dlg1 Y222F (Tyr to Phe mutation) fusion proteins bound to glutathione–Sepharose beads and incubated with T cell lysates, centrifuged, washed, and incubated with ATP. Protein complexes subjected SDS-PAGE and blotted using anti-4G10 (pY-Dlg1) and anti-GST (GST-Dlg1). (C) In vitro kinase assay using GST-Dlg1AB fusion proteins bound to glutathione–Sepharose beads and incubated with T cell lysates, centrifuged, washed, and incubated with ATP in the presence or absence of 10 μM PP2 inhibitor. Protein complexes were subjected SDS-PAGE and blotted using anti-4G10 (pY-Dlg1) and anti-GST (GST-Dlg1). (D) GST-Dlg1AB WT or Y222F fusion proteins were bound to glutathione–Sepharose beads and incubated with ATP and rLck, rZap70, or buffer only (none). Proteins were subjected to SDS-PAGE and blotted for anti-4G10 (pY-GST-Dlg1 and pY-GST-LAT) or anti-GST (GST-Dlg1). (B, right; D, right) Blots were quantitated using Li-Cor Odyssey software. pY-Dlg1 was normalized to total Dlg1 for each lane, and WT phosphorylation was set to 1.0; error bars represent the SD of four independent experiments. **p < 0.05.
To determine whether this candidate tyrosine was a site of phosphorylation, we created GST-Dlg1AB fusion proteins in which Y222 was mutated to phenylalanine (Y222F) to disrupt phosphorylation at this position. GST-Dlg1AB (WT) or GST-Dlg1AB Y222F (Y222F) fusion proteins were used to form complexes with T cell ligands and subsequently in vitro phosphorylated. Upon the addition of ATP, GST-Dlg1AB Y222F consistently showed a 40% decrease in tyrosine phosphorylation signal relative to GST-Dlg1AB demonstrating that Y222 was a major site of Dlg1 phosphorylation (Fig. 2B). Although the region surrounding Y222 is conserved in both variants (Fig. 2A), we found that Dlg1B is not tyrosine phosphorylated, suggesting that the tyrosine kinase responsible for Y222 phosphorylation may not be present in Dlg1B complexes (Fig. 1).
Dlg1-associated Lck can mediate Dlg1 phosphorylation
Dlg1 Y222 is part of a YEEI motif predicted to be a target of Src and Syk family kinases (Fig. 2A). To determine a possible role for Src kinases in Dlg1 phosphorylation, we in vitro phosphorylated GST-Dlg1AB complexes in the presence or absence of Src kinase family inhibitor PP2. Dlg1 tyrosine phosphorylation was observed upon the addition of ATP, which was completely blocked by the addition of PP2, demonstrating that Src family kinase activity was required for Dlg1 phosphorylation (Fig. 2C). However, because Lck kinase activity regulates Zap70 activation, these data cannot rule out a role for Zap70 in Dlg1 phosphorylation.
To assess the role of these kinases in phosphorylating Dlg1, we in vitro phosphorylated GST-Dlg1AB or GST-Dlg1AB Y222F fusion proteins using active recombinant Lck or Zap70 to determine the ability of each kinase to directly phosphorylate Dlg1. We found that rLck but not rZap70 directly phosphorylated Dlg1 in vitro. Furthermore, phosphorylation of GST-Dlg1AB Y222F by rLck was significantly reduced compared with GST-Dlg1AB, demonstrating that Lck is likely responsible for Y222 phosphorylation (Fig. 2D). Although rZap70 did not phosphorylate Dlg1, it did phosphorylate our positive control (GST-LAT), demonstrating that the kinase was active in our system (Fig. 2D). These results rule out a role for Syk family member Zap70 in Dlg1 phosphorylation. Taken together, our data demonstrate that Src family kinases play an important role in Dlg1 phosphorylation and identify Lck as one possible kinase capable of phosphorylating Dlg1. Although we cannot rule out a role for Src family kinase Fyn, previous studies demonstrating that Lck but not Fyn binds Dlg1 suggest that Lck is likely the preferred Src family member responsible for Dlg1 tyrosine phosphorylation (4).
Dlg1 Y222 phosphorylation is required for TCR-induced alternative p38 activation in CD8+ T cells
Dlg1AB coordinates the activation of p38 in response to TCR stimulation via the alternative activation pathway (5). To assess the requirement of Dlg1 Y222 in coordinating alternative p38 activation, we formed Dlg1 complexes by incubating GST-Dlg1AB or GST-Dlg1AB Y222F with T cell lysates and assessed the ability of each Dlg1 scaffold to facilitate phosphorylation of associated p38. We observed a significant decrease in the level of p38 phosphorylation (pY-p38) of Dlg1-associated p38 in Dlg1AB Y222F complexes compared with GST-Dlg1AB complexes, whereas the total amount of Dlg1-associated p38 did not change (Fig. 3A). These data provide evidence that Dlg1 Y222 phosphorylation is required for proper coordination of Dlg1-associated p38 phosphorylation. To determine whether Dlg1 Y222 was required for TCR-triggered p38 phosphorylation, we created stable cell lines expressing predominately Dlg1AB (WT) or Dlg1AB Y222F (Y222F). In these cells, total endogenous Dlg1 was knocked down using a microRNA-based retroviral construct specific for the 3′-UTR of Dlg1, yielding stable cell lines that expressed <10% of endogenous of Dlg1 protein. Dlg1 was re-expressed in knockdown cells using a murine stem cell virus–based retrovirus expressing Dlg1AB, Dlg1AB Y222F, or a control vector. The resulting CD8+ T cells expressed comparable levels of Dlg1AB or Dlg1AB Y222F (Fig. 3B). Control or Dlg1AB- or Dlg1AB Y222F-expressing cells were stimulated through their TCR, and the level of Dlg1-associatd p38 phosphorylation was measured. A low level of TCR-inducible p38 phosphorylation was present in the control cells, which was significantly enhanced in cells expressing Dlg1AB but not in cells expressing Dlg1AB Y222F (Fig. 3C). The low level of p38 phosphorylation in Y222F cells is likely due to a small amount of residual Dlg1AB. Additional studies in primary CD8+ CTLs demonstrated that overexpression of Dlg1AB caused a significant enhancement of TCR-induced total cellular p38 phosphorylation compared with control cells whereas Dlg1AB Y222F did not (Fig. 3D, 3E). Taken together, our results highlight an essential role for Dlg1 Y222 phosphorylation in the regulation of TCR-induced alternative p38 activation in CD8+ T cells.
FIGURE 3.

Dlg1 Y222 phosphorylation is required for TCR-induced alternative p38 activation. (A) In vitro kinase assay were GST-Dlg1AB WT, or GST-Dlg1 Y222F fusion proteins were bound to glutathione–Sepharose beads and incubated with T cell lysates, centrifuged, washed, and incubated with 10 mM ATP for 20 min at 30°C. Protein complexes were boiled off beads and subjected SDS-PAGE, and blotted using anti-4G10 (pY-p38) and anti-p38. Blots were quantitated using Li-Cor Odyssey software. pY-p38 was normalized to total p38 for each lane, and WT phosphorylation was set to 1.0; error bars represent the SD of three independent experiments. **p < 0.05. (B and C) Control, Dlg1-deficient OT-1 hybridomas (3′-UTR) re-expressing vector only (3′-UTR+Vector), Dlg1AB (3′-UTR+Dlg1AB), or Dlg1AB Y222F (3′-UTR+Dlg1Y222F) were lysed and subjected to SDS-PAGE and blotted using anti-Dlg1 or anti-p38 (B) or stimulated using 5 μg/ml anti-CD3 and 5 μg/ml CD28 (C), followed by cross-linking with donkey anti-Armenian hamster Ab for 15 min at 37°C, followed by lysis. Dlg1 was immunoprecipitated from resulting lysates and subjected to SDS-PAGE then blotted using anti–p-p38 180/182 and anti-p38. (D and E) CD8+ T cells were isolated from the spleens of OT-1 mice and expanded on plate-bound anti-CD3 and anti-CD28 Ab for 72 h, followed by overexpression of Dlg1AB or Dlg1Y222F. Resulting cells were permeabilized and stained for intracellular Dlg1 (D) or stimulated with plate-bound anti-CD3 and CD28 (E) for 15 min, followed by fixation, permeabilization, and staining for intracellular p-p38 180/182. Data presented are representative of three independent experiments.
Loss of Dlg1 Y222 phosphorylation results in decreased NFAT-, but not NF-κB-dependent expression
Previous studies from our group and others have demonstrated that Dlg1-mediated alternative p38 activation induces NFAT- but not NF-κB–dependent gene expression (5, 7, 16). Recently, we have demonstrated that Dlg1AB but not Dlg1B can facilitate p38-dependent NFAT activation (O. Silva et al., submitted for publication). To determine whether Dlg1 Y222 plays a role in TCR-induced NFAT or NF-κB activation, CD8+ T cells re-expression Dlg1AB or Dlg1AB Y222F were stimulated through their TCR, and the level of includible Nfatc1 (NFAT-dependent gene) and Iκbα (NF-κB–dependent gene) gene expression was measured via quantitative PCR. In agreement with published results, CD8+ T cells lacking Dlg1 demonstrated impaired induction of Nfatc1 upon TCR stimulation (Fig. 4A). Expression of Dlg1AB in Dlg1 knockdown cells was able to rescue Nfatc1 levels, whereas expression of Dlg1AB Y222F or Dlg1B only further attenuated Nfatc1 expression (Fig. 4A). Alterations in Dlg1 expression had no significant effect on the level of Ikbα induction upon TCR stimulation (Fig. 4B). We further interrogated these findings in primary CD8+ T cells where overexpression of Dlg1AB but not Dlg1AB Y222F or Dlg1B was able to enhance TCR-dependent Nfatc1 transcription, whereas Dlg1 expression levels had no significant effect on Ikbα transcription (Fig. 4F, 4G). These findings demonstrate that Dlg1 Y222 phosphorylation is required for the specific activation of NFAT- but not NF-κB–dependent transcription in response to TCR stimulation.
FIGURE 4.
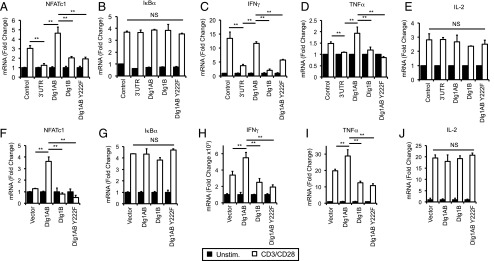
Dlg1 Y222 is required for selective induction of TCR-triggered NFAT- but not NF-κB–dependent genes including proinflammatory cytokines IFN-γ and TNF-α. (A–E) Control, Dlg1-deficient OT-1 hybridomas (3′-UTR) re-expressing Dlg1AB, Dlg1B, or Dlg1AB Y222F (Dlg1Y222F). (F–J) CD8+ T cells were isolated from the spleens of OT-1 mice and expanded on plate-bound anti-CD3 and anti-CD28 Ab for 72 h, followed by overexpression of Dlg1AB, Dlg1B, Dlg1Y222F, or empty vector (control). (A–J) Cells were stimulated with anti-CD3 and anti-CD28 Ab for 2 or 6 h, followed by mRNA isolation, reverse transcription, and qPCR analysis using primers for NFATc1 (A and F), IκBα (B and G), IFN-γ (C and H), TNF-α (D and I), or IL-2 (E and J). All values were normalized to L32 and unstimulated values set to 1.0. Error bars represent SD of triplicates; data are representative of three independent experiments. **p < 0.05.
Dlg1 Y222 is required for optimal NFAT-dependent IFN-γ and TNF-α but not IL-2 production
Dlg1AB-mediated alternative p38 activation has been implicated in selective upregulation of proinflammatory cytokines IFN-γ and TNF-α but not IL-2 in response to TCR stimulation (5, 20, 21, 33, 34) (O. Silva et al., submitted for publication). To examine the requirement of Dlg1 Y222 in TCR-induced cytokine production, CD8+ T cells re-expressing Dlg1AB, Dlg1B, or Dlg1AB Y222F were TCR-stimulated, and their ability to induce cytokine gene expression was measured. Dlg1-deficient CD8+ T cells had impaired induction of IFN-γ and TNF-α but not IL-2 transcription compared with control cells (Fig. 4C–E). Expression of Dlg1AB was able to rescue the defect in IFN-γ and TNF-α whereas Dlg1AB Y222F or Dlg1B was not. In keeping with previous results, expression of Dlg1 had no significant effect on the level of IL-2 transcription (Fig. 4E; O. Silva et al., submitted for publication). In addition, overexpression of Dlg1AB in primary CD8+ CTLs was able to enhance IFN-γ and TNF-α gene expression, whereas expression of Dlg1B or Dlg1AB Y222F led to decreased IFN-γ and TNF-α expression compared with control cells, suggesting that these variants may have a dominant negative effect. Dlg1 expression had no significant effect on IL-2 gene expression (Fig. 4H–J). Taken together, our results support a model where phosphorylation of Dlg1 at Y222 is a key point of control that facilitates selective upregulation of NFAT-dependent gene transcription, including the production of proinflammatory cytokines IFN-γ and TNF-α but not IL-2.
Dlg1 Y222 is not required for Dlg1-mediated target cell killing by CD8+ T cells
Dlg1 is known to regulate cytoskeletal events in epithelial, neuronal, and immunologic systems (4, 7, 10, 35). In particular, Dlg1 association with WASp and ezrin is implicated in TCR-dependent positioning of the microtubule-organizing complex in response to antigenic stimulation (7). Recently, we have demonstrated that both Dlg1AB and Dlg1B can facilitate cytotoxic granule and cytokine release through association with and activation of WASp (O. Silva et al., submitted for publication). To test the requirement of Dlg1 Y222 phosphorylation in release of cytotoxic granules (degranulation), we incubated CD8+ T cells re-expressing Dlg1AB or Dlg1AB Y222F with APCs and tracked the surface expression of degranulation marker CD107a. Total Dlg1 knockdown resulted in significantly impaired Ag-dependent degranulation. Surprisingly, both Dlg1AB and Dlg1Y222F were able to rescue and enhance Ag-dependent degranulation to a similar level (Fig. 5A). Overexpression of Dlg1AB or Dlg1AB Y222F in primary CD8+ CTLs enhanced Ag-dependent degranulation to a similar level compared with control cells (Fig. 5B). Examination of perforin production in primary CD8+ T cells overexpressing Dlg1AB or Dlg1AB Y222F show no significant differences, suggesting that cytotoxic granule components are not affected by loss of Dlg1 Y222 phosphorylation. In the same experiment, Dlg1AB but not Dlg1Y222F was able to enhance NFAT-dependent transcription. Taken together, these data suggest that phosphorylation of Dlg1 Y222 does not control Dlg1-mediated Ag-dependent degranulation but not production of cytotoxic granule components.
FIGURE 5.

Dlg1 Y222 phosphorylation is not required for Ag-dependent CTL degranulation. (A) Control, Dlg1-deficient OT-1 hybridomas (3′-UTR) re-expressing vector only (3′-UTR+Vector), Dlg1AB (3′-UTR+Dlg1AB), or Dlg1AB Y222F (3′-UTR+Dlg1Y222F) (A) or CD8+ T cells were isolated from the spleens of OT-1 mice and expanded on plate-bound anti-CD3 and anti-CD28 Ab for 72 h, followed by overexpression of Dlg1AB, Dlg1B, Dlg1Y222F, or empty vector (control) cells were incubated with EG.7 thymoma target cells at indicated E:T ratios for 2 h in the presence of anti-CD107a Ab. CD107a was assessed by flow cytometry where control cells at E:T of 1:0 was set to 5%. Data are representative of three experiments. (B and C) CD8+ T cells were isolated from the spleens of OT-1 mice and expanded on plate-bound anti-CD3 and anti-CD28 Ab for 72 h, followed by overexpression of Dlg1AB, Dlg1Y222F, or empty vector (control). (B) CTLs were incubated with EG.7 thymoma target cells at indicated E:T ratios for 2 h in the presence of anti-CD107a Ab. CD107a was assessed by flow cytometry where control cells at E:T of 1:0 was set to 5%. (C) CTLs were stimulated with anti-CD3 and anti-CD28 Ab for 6 h, followed by mRNA isolation, reverse transcription, and qPCR analysis using primers for perforin. **p < 0.05.
Acute Dlg1 knockout block TCR-dependent p38 activation, NFAT-dependent transcription and target cell killing in CD8+ T cells
We and others have demonstrated that Dlg1 plays a vital role in T cell development, activation and effector responses utilizing Dlg1 knockdown and overexpression systems (4, 5, 7, 9, 10, 16). However, initial knockout studies that ablate Dlg1 early in T cell development do not show striking functional differences, likely because of compensation from other Dlg1 family members (36, 37). Therefore, we developed an acute Dlg1 knockout system to examine the requirement for Dlg1 in TCR-dependent effector functions. CD8+ T cells isolated from Dlg1flox/flox mice were expanded ex vivo before infection with retroviral cre recombinase (Dlg1KO) or control vector (Dlg1WT) (Fig. 6A). This method yielded CD8+ CTLs with robust and reproducible Dlg1 knockout on both the genomic DNA and protein level (Fig. 6B, 6C). In response to TCR stimulation, Dlg1KO cells showed a significant decrease in total cellular p38 phosphorylation as well as a severe and selective impairment in NFAT-dependent gene expression, including expression of proinflammatory cytokines IFN-γ and TNF-α (Fig. 6D–H). Conversely, there was no decrease in NF-κB–dependent gene expression or IL-2 expression (Fig. 6I). These results were consistent with previous studies using Dlg1 knockdown and overexpression (5, 16, 38) (O. Silva et al. submitted for publication). In addition, we examined the requirement of Dlg1 for TCR-induced release of cytotoxic granules and target cell lysis. We found that Dlg1 knockout significantly diminished Ag-dependent degranulation and target cell killing, but not granzyme B gene expression (Fig. 6J–L). These data further validate our previous findings that Dlg1 is required for the release of cytotoxic granules but not the production of granule elements such as granzyme B (4) (O. Silva et al., submitted for publication). Taken together, our results establish Dlg1 as a vital regulator of TCR-dependent signaling and cytoskeletal events and support a model where Dlg1 Y222 phosphorylation selectively controls Dlg1AB-mediated functions, including proinflammatory cytokine production, while not regulating functions that can be controlled by Dlg1B such as cytotoxic granule release (Fig. 7).
FIGURE 6.

Acute knockout of Dlg1 impairs p38 activation and NFAT-dependent proinflammatory cytokines gene expression and target cell lysis in response to TCR stimulation. (A) Genomic organization of Dlg1flox/flox mice as described previously (28). F and R refer to the location of the forward and reverse primers for gDNA analysis. (B–L) CD8+ T cells were isolated from spleens of OT-1 Dlg1flox/flox mice and expanded on plate-bound anti-CD3 and anti-CD28 Ab for 48–72 h, followed by infection with retroviral cre recombinase or vector control. (B) Genomic DNA was isolated, and PCR analysis was performed. (C) Whole-cell lysates were subjected to SDS-PAGE and blotted with anti-Dlg1 or anti-p38. (D) Cells were stimulated with plate-bound anti-CD3 and anti-CD28 Ab for 15 min, followed by fixation, permeabilization, and staining for p-p38 180/182. Error bars represent SD of three independent experiments. (E–J) Cells were left unstimulated or stimulated with plate-bound anti-CD3 and anti-CD28 for 2 h (E and F) or 6 h (G–J), followed by mRNA isolation, reverse transcription, and qPCR analysis using primers specific for NFATc1(E), IκBα (F), IFN-γ (G), TNF-α (H), IL-2 (I), or granzyme B (J). All values were normalized to L32 and unstimulated values set to 1.0. Error bars represent stand deviation of triplicates; data are representative of four independent experiments. (K and L) Cells were incubated with EG.7 thymoma cells at indicated ratios for 2 h, following surface staining for CD107a (K) or lactate dehydrogenase cytotoxicity assay (L). **p < 0.05.
FIGURE 7.

Model of Dlg1-dependent signaling downstream of the TCR. In response to TCR engagement, Dlg1-bound Lck phosphorylates Dlg1AB at several tyrosine residues including Y222. This phosphorylation event empowers Dlg1 to enhance p38 activation and NFAT-dependent transcription of IFN-γ and TNF-α. Dlg1AB or Dlg1B is able to facilitate Ag-induced degranulation and target cell killing in a phosphorylation-independent manner.
Discussion
CD8+ T cells have the ability to develop a broad range of functionality, with differing capacities to produce and release cytokines and lytic factors in response to TCR stimulation (3). Scaffold protein Dlg1 specifies signaling and function downstream of the TCR by facilitating p38 and NFAT activation leading to the production of proinflammatory cytokines. In addition, Dlg1 coordinates TCR-dependent activation of WASp leading to cytoskeletal reorganization and release of cytotoxic granules (4, 5) (O. Silva et al. submitted for publication). These functions can be differentially regulated by the two Dlg1 splice variants expressed in CD8+ T cells: Dlg1AB and Dlg1B (O. Silva et al., submitted for publication). Both Dlg1AB and Dlg1B facilitate cytotoxicity, whereas only Dlg1AB can coordinate p38 activation and proinflammatory cytokine production. The ability of Dlg1AB to activate p38 has been attributed to its interaction with proximal tyrosine kinase Lck; however, the mechanism by which Dlg1AB but not Dlg1B coordinates p38 activation was unclear. In this study, we identify TCR-induced phosphorylation of Dlg1AB at Y222 as a key point of control required for TCR-induced, Dlg1-mediated p38 activation and expression of proinflammatory cytokines but not cytotoxic granule release.
Dlg1 is rich in tyrosine residues of uncharacterized function and is known to associate with several tyrosine kinases; therefore, we hypothesized that tyrosine phosphorylation may play an important role in regulating Dlg1 function. We found that Dlg1 was tyrosine phosphorylated in response to TCR stimulation in a splice variant dependent manner, where Dlg1AB but not Dlg1B was phosphorylated. We determined that Y222 in the N-terminal linker region of Dlg1 is a phosphorylation site likely targeted by Dlg1-associated Lck. The loss of Y222 phosphorylation impaired TCR-induced p38 activation and NFAT-dependent gene expression of proinflammatory cytokines IFN-γ and TNF-α but not IL-2. Surprisingly, we found that Dlg1 phosphorylation is not required for TCR-mediated degranulation. These data highlight the role of Dlg1 Y222 phosphorylation as a key point of control required for the activation of a subset of effector functions downstream of the TCR. In support of the importance of Dlg1 in regulating T cell function, we demonstrated that acute knockout of Dlg1 in CD8+ CTLs resulted in selective impairment of TCR-induced p38 activation, NFAT-dependent transcription, proinflammatory cytokine gene expression, and Ag-dependent degranulation. Our results indicate that Dlg1 plays an important role in specifying CD8+ T cell function and that Lck-mediated phosphorylation at Y222 is a molecular switch required to authorize a subset of Dlg1-mediated functions.
At least two Dlg1 protein variants, Dlg1AB and Dlg1B, are expressed in CD8+ T cells through alternative splicing that differ in the inclusion or exclusion of the proline-rich i1A region (O. Silva et al., submitted for publication). The inclusion of the i1A region allows Dlg1AB to associate with tyrosine kinase Lck and facilitate p38 activation and NFAT-dependent transcription of proinflammatory cytokine genes in response to TCR stimulation. In contrast, Dlg1AB and Dlg1B are both capable of mediating Ag-dependent degranulation, suggesting that direct association with Lck is not required for coordinating these functions (O. Silva et al., submitted for publication). In this study, we showed that Dlg1AB but not Dlg1B is phosphorylated in response to TCR stimulation and that this phosphorylation is dependent on Dlg1-associated Lck. We also identify Y222 as an Lck-dependent phosphorylation site required for TCR-induced p38 activation and downstream transcriptional activation but not Ag-dependent degranulation. Therefore, we propose that the differential functionality of the two known Dlg1 splice variants is regulated, at least in part, by differential ability to be phosphorylated on tyrosine residues.
Scaffold protein structure, and therefore function, is often regulated through a series of phosphorylation and/or ligand binding events that lead to the gradual unfolding of the scaffold into an active conformation (39–41). These phosphorylation events are hypothesized to provide molecular memory by creating a primed scaffold capable of binding new ligands, endowing the scaffold with novel and/or heightened functionally upon secondary stimulation. Dlg1 forms intramolecular interactions yielding at least two distinct closed conformations hypothesized to impact Dlg1 function by masking ligand binding sites or affecting the juxtaposition of bound ligands (22, 23). However, a role for these intramolecular interactions has not been characterized in the context of Dlg1 ligands in T cells. In this study, we show that tyrosine phosphorylation of Dlg1 in the N-terminal linker region is required for TCR-induced p38 activation. We predict that phosphorylation at Dlg1 Y222 causes a conformational change required for proper association of Dlg1 with ligands such as Zap70. Alternatively, structural changes could affect the positioning of Dlg1-associated proteins relative to each other. Therefore, the closed conformation could disrupt signal propagation on the scaffold even in the presence of all required ligands. Allosteric regulation has been described in other MAGUK family members including CARMA1, which facilitates activation of NF-κB in T and B cells through formation of a signaling complex with Bcl10 and MALT1 (42). Inducible phosphorylation of serine residues in the linker region of CARMA1 regulates its structure, binding partners and function.
Using knockdown methodologies several groups have demonstrated a role for Dlg1 in regulating functionality and differentiation in both CD4+ and CD8+ T cells (4, 5, 7, 16) (O. Silva et al., submitted for publication). Despite growing evidence for Dlg1 in specifying signal transduction downstream of the TCR, recent attempts by numerous groups to extend these studies to knockout models have been inconsistent and largely unavailing (36, 43, 44). The mild phenotype seen in these models is attributed to compensation of other Dlg1 family members; such as PSD-95, which is known to have functional redundancy with Dlg1 in neurons (37). In this study, we have described an acute Dlg1 knockout system where cre recombinase is overexpressed in CD8+ CTLs from Dlg1flox/flox mice, and TCR-triggered functions are assessed 72hr later. In this system, we demonstrate that TCR-triggered p38 activation, NFAT activation, induction of IFN-γ and TNF-α gene expression, and degranulation are impaired in the Dlg1KO cells compared with controls.
The spectrum of effector functions developed by T cells during infection is predicted to optimize T cell responsiveness to specific pathogens (3). Indeed, development of polyfunctional CD8+ T cells is correlated with improved clinical outcomes against HIV and tumors, whereas dysregulation of CD8+ T cell function is implicated in persistence of chronic viral infections and autoimmune disorders (45–49). Therefore, it is essential to understand the molecular mechanisms that regulate TCR-dependent signaling in CD8+ T cells, which together dictate the T cell function. We have recently demonstrated that Dlg1 plays a vital role in CD8+ T cell proinflammatory cytokine production and target cell killing in response to acute viral infection (O. Silva et al., submitted for publication). In this study, we show that TCR-induced Dlg1 phosphorylation is a key point of control capable of uncoupling cytokine production and cytotoxicity in CD8+ T cells. Given these data, it is interesting to imagine that TCR-induced phosphorylation of Dlg1AB acts as a molecular switch turning on p38 dependent functions, including proinflammatory cytokine production, in response to certain Ags, whereas other Ag signals may be sufficient to trigger degranulation.
Our data place Dlg1 Y222 phosphorylation as a vital regulatory step required for activation of p38 and NFAT. In the CD8+ T cells examined in this study, these pathways control the expression of proinflammatory cytokines IFN-γ and TNF-α. Dlg1-mediated p38 and NFAT activation also is thought to play an important role in regulating CD4+ T cell development and function. In CD4+ regulatory T cells, Dlg1 expression and localization to the immune synapse correlates with p38 phosphorylation, NFAT-dependent transcription, and regulatory T cell–suppressive activity (16). In CD4+ effector cells, Dlg1 is thought to play a role in skewing toward expression of Th1 cytokines (36). In addition, Dlg1 also has been shown to influence the development of CD4+ T cell memory (44). Although both Dlg1AB and Dlg1B are expressed in CD4+ T cell subsets, the relative abundance of each isoform and the role of phosphorylation have yet to be explored (O. Silva et al., submitted for publication). Future studies into these cell types are likely to yield important insights into mechanisms of CD4+ T cell development and function.
In summary, Dlg1 is a key signaling and cytoskeletal specifier downstream of the TCR. Our study identifies Lck-mediated Dlg1 phosphorylation as a previously uncharacterized point of control regulating Dlg1-mediated signaling downstream of the TCR. In response to TCR stimulation, Lck phosphorylates the Dlg1AB splice variant at several tyrosine residues, including Y222. Loss of Dlg1 phosphorylation at Y222 through Y222F mutation led to a significant and specific loss of p38 activation and up regulation of IFN-γ and TNF-α gene expression but did not impair cytotoxic granule release. A better understanding of the mechanisms that specifically regulate proinflammatory cytokines should ultimately lead to the discovery and design of more efficient and safe anti-inflammatory treatments for a variety of inflammatory conditions, including autoimmune disorders and severe influenza infection.
Acknowledgments
We thank June L. Round (University of Utah, Salt Lake City, UT) for thoughtful discussion and critical reading of the manuscript. We also thank Ryan O’Connell for providing the microRNA vector and Yisong Wan for providing the murine stem cell virus vector.
This work was supported by National Institutes of Health Grant R01-AI067253-10 (to M.C.M.), a National Institutes of Health research supplement (to O.S.), National Institute of Allergy and Infectious Diseases Grant T32-AI07323 (to O.S. and J.C.), and an Arthritis Foundation postdoctoral fellowship (to L.A.H.).
- Dlg1
- Discs large homolog 1
- GUK
- guanylate kinase
- MAGUK
- membrane-associated GUK
- PDZ
- PSD-95/Dlg/ZO-1
- 3′-UTR
- 3′-untranslated region
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Peng S. L., Gerth A. J., Ranger A. M., Glimcher L. H. 2001. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity 14: 13–20. [DOI] [PubMed] [Google Scholar]
- 2.Rutishauser R. L., Kaech S. M. 2010. Generating diversity: transcriptional regulation of effector and memory CD8 T-cell differentiation. Immunol. Rev. 235: 219–233. [DOI] [PubMed] [Google Scholar]
- 3.Newell E. W., Sigal N., Bendall S. C., Nolan G. P., Davis M. M. 2012. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity 36: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Round J. L., Tomassian T., Zhang M., Patel V., Schoenberger S. P., Miceli M. C. 2005. Dlgh1 coordinates actin polymerization, synaptic T cell receptor and lipid raft aggregation, and effector function in T cells. J. Exp. Med. 201: 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Round J. L., Humphries L. A., Tomassian T., Mittelstadt P., Zhang M., Miceli M. C. 2007. Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF-κB transcription factors. Nat. Immunol. 8: 154–161. [DOI] [PubMed] [Google Scholar]
- 6.Wang D., You Y., Case S. M., McAllister-Lucas L. M., Wang L., DiStefano P. S., Nuñez G., Bertin J., Lin X. 2002. A requirement for CARMA1 in TCR-induced NF-κB activation. Nat. Immunol. 3: 830–835. [DOI] [PubMed] [Google Scholar]
- 7.Lasserre R., Charrin S., Cuche C., Danckaert A., Thoulouze M. I., de Chaumont F., Duong T., Perrault N., Varin-Blank N., Olivo-Marin J. C., et al. 2010. Ezrin tunes T-cell activation by controlling Dlg1 and microtubule positioning at the immunological synapse. EMBO J. 29: 2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanada T., Lin L., Tibaldi E. V., Reinherz E. L., Chishti A. H. 2000. GAKIN, a novel kinesin-like protein associates with the human homologue of the Drosophila discs large tumor suppressor in T lymphocytes. J. Biol. Chem. 275: 28774–28784. [DOI] [PubMed] [Google Scholar]
- 9.Xavier R., Rabizadeh S., Ishiguro K., Andre N., Ortiz J. B., Wachtel H., Morris D. G., Lopez-Ilasaca M., Shaw A. C., Swat W., Seed B. 2004. Discs large (Dlg1) complexes in lymphocyte activation. J. Cell Biol. 166: 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lue R. A., Brandin E., Chan E. P., Branton D. 1996. Two independent domains of hDlg are sufficient for subcellular targeting: the PDZ1-2 conformational unit and an alternatively spliced domain. J. Cell Biol. 135: 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLaughlin M., Hale R., Ellston D., Gaudet S., Lue R. A., Viel A. 2002. The distribution and function of alternatively spliced insertions in hDlg. J. Biol. Chem. 277: 6406–6412. [DOI] [PubMed] [Google Scholar]
- 12.Cavatorta A. L., Facciuto F., Valdano M. B., Marziali F., Giri A. A., Banks L., Gardiol D. 2011. Regulation of translational efficiency by different splice variants of the Disc large 1 oncosuppressor 5′-UTR. FEBS J. 278: 2596–2608. [DOI] [PubMed] [Google Scholar]
- 13.Martinez N. M., Pan Q., Cole B. S., Yarosh C. A., Babcock G. A., Heyd F., Zhu W., Ajith S., Blencowe B. J., Lynch K. W. 2012. Alternative splicing networks regulated by signaling in human T cells. RNA 18: 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lue R. A., Marfatia S. M., Branton D., Chishti A. H. 1994. Cloning and characterization of hdlg: the human homologue of the Drosophila discs large tumor suppressor binds to protein 4.1. Proc. Natl. Acad. Sci. USA 91: 9818–9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Godreau D., Vranckx R., Maguy A., Goyenvalle C., Hatem S. N. 2003. Different isoforms of synapse-associated protein, SAP97, are expressed in the heart and have distinct effects on the voltage-gated K+ channel Kv1.5. J. Biol. Chem. 278: 47046–47052. [DOI] [PubMed] [Google Scholar]
- 16.Zanin-Zhorov A., Lin J., Scher J., Kumari S., Blair D., Hippen K. L., Blazar B. R., Abramson S. B., Lafaille J. J., Dustin M. L. 2012. Scaffold protein Disc large homolog 1 is required for T-cell receptor-induced activation of regulatory T-cell function. Proc. Natl. Acad. Sci. USA 109: 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salvador J. M., Mittelstadt P. R., Guszczynski T., Copeland T. D., Yamaguchi H., Appella E., Fornace A. J., Jr., Ashwell J. D. 2005. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 6: 390–395. [DOI] [PubMed] [Google Scholar]
- 18.Mittelstadt P. R., Yamaguchi H., Appella E., Ashwell J. D. 2009. T cell receptor-mediated activation of p38α by mono-phosphorylation of the activation loop results in altered substrate specificity. J. Biol. Chem. 284: 15469–15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashwell J. D. 2006. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 6: 532–540. [DOI] [PubMed] [Google Scholar]
- 20.Kaminuma O., Kitamura F., Kitamura N., Hiroi T., Miyoshi H., Miyawaki A., Miyatake S. 2008. Differential contribution of NFATc2 and NFATc1 to TNF-α gene expression in T cells. J. Immunol. 180: 319–326. [DOI] [PubMed] [Google Scholar]
- 21.Macian F. 2005. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5: 472–484. [DOI] [PubMed] [Google Scholar]
- 22.Tully M. D., Grossmann J. G., Phelan M., Pandelaneni S., Leyland M., Lian L. Y. 2012. Conformational characterization of synapse-associated protein 97 by nuclear magnetic resonance and small-angle X-ray scattering shows compact and elongated forms. Biochemistry 51: 899–908. [DOI] [PubMed] [Google Scholar]
- 23.Wu H., Reissner C., Kuhlendahl S., Coblentz B., Reuver S., Kindler S., Gundelfinger E. D., Garner C. C. 2000. Intramolecular interactions regulate SAP97 binding to GKAP. EMBO J. 19: 5740–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanada T., Lin L., Chandy K. G., Oh S. S., Chishti A. H. 1997. Human homologue of the Drosophila discs large tumor suppressor binds to p56lck tyrosine kinase and Shaker type Kv1.3 potassium channel in T lymphocytes. J. Biol. Chem. 272: 26899–26904. [DOI] [PubMed] [Google Scholar]
- 25.Sabio G., Arthur J. S., Kuma Y., Peggie M., Carr J., Murray-Tait V., Centeno F., Goedert M., Morrice N. A., Cuenda A. 2005. p38γ regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 24: 1134–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcette J., Hood I. V., Johnston C. A., Doe C. Q., Prehoda K. E. 2009. Allosteric control of regulated scaffolding in membrane-associated guanylate kinases. Biochemistry 48: 10014–10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw A. S., Filbert E. L. 2009. Scaffold proteins and immune-cell signalling. Nat. Rev. Immunol. 9: 47–56. [DOI] [PubMed] [Google Scholar]
- 28.Zhou W., Zhang L., Guoxiang X., Mojsilovic-Petrovic J., Takamaya K., Sattler R., Huganir R., Kalb R. 2008. GluR1 controls dendrite growth through its binding partner, SAP97. J. Neurosci. 28: 10220‑10233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Connell R. M., Chaudhuri A. A., Rao D. S., Baltimore D. 2009. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 106: 7113–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obenauer J. C., Cantley L. C., Yaffe M. B. 2003. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31: 3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Granum S., Andersen T. C., Sørlie M., Jørgensen M., Koll L., Berge T., Lea T., Fleckenstein B., Spurkland A., Sundvold-Gjerstad V. 2008. Modulation of Lck function through multisite docking to T cell-specific adapter protein. J. Biol. Chem. 283: 21909–21919. [DOI] [PubMed] [Google Scholar]
- 32.Isakov N., Wange R. L., Burgess W. H., Watts J. D., Aebersold R., Samelson L. E. 1995. ZAP-70 binding specificity to T cell receptor tyrosine-based activation motifs: the tandem SH2 domains of ZAP-70 bind distinct tyrosine-based activation motifs with varying affinity. J. Exp. Med. 181: 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jirmanova L., Sarma D. N., Jankovic D., Mittelstadt P. R., Ashwell J. D. 2009. Genetic disruption of p38alpha Tyr323 phosphorylation prevents T-cell receptor-mediated p38α activation and impairs interferon-gamma production. Blood 113: 2229–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jirmanova L., Giardino Torchia M. L., Sarma N. D., Mittelstadt P. R., Ashwell J. D. 2011. Lack of the T cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood 118: 3280–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Funke L., Dakoji S., Bredt D. S. 2005. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu. Rev. Biochem. 74: 219–245. [DOI] [PubMed] [Google Scholar]
- 36.Humphries L. A., Shaffer M. H., Sacirbegovic F., Tomassian T., McMahon K. A., Humbert P. O., Silva O., Round J. L., Takamiya K., Huganir R. L., et al. 2012. Characterization of in vivo Dlg1 deletion on T cell development and function. PLoS One 7: e45276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlüter O. M., Xu W., Malenka R. C. 2006. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron 51: 99–111. [DOI] [PubMed] [Google Scholar]
- 38.Adachi K., Davis M. M. 2011. T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T cells. Proc. Natl. Acad. Sci. USA 108: 1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torres E., Rosen M. K. 2003. Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol. Cell 11: 1215–1227. [DOI] [PubMed] [Google Scholar]
- 40.Sommer K., Guo B., Pomerantz J. L., Bandaranayake A. D., Moreno-García M. E., Ovechkina Y. L., Rawlings D. J. 2005. Phosphorylation of the CARMA1 linker controls NF-κB activation. Immunity 23: 561–574. [DOI] [PubMed] [Google Scholar]
- 41.Brenner D., Brechmann M., Röhling S., Tapernoux M., Mock T., Winter D., Lehmann W. D., Kiefer F., Thome M., Krammer P. H., Arnold R. 2009. Phosphorylation of CARMA1 by HPK1 is critical for NF-κB activation in T cells. Proc. Natl. Acad. Sci. USA 106: 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blonska M., Lin X. 2009. CARMA1-mediated NF-κB and JNK activation in lymphocytes. Immunol. Rev. 228: 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephenson L. M., Sammut B., Graham D. B., Chan-Wang J., Brim K. L., Huett A. S., Miletic A. V., Kloeppel T., Landry A., Xavier R., Swat W. 2007. DLGH1 is a negative regulator of T-lymphocyte proliferation. Mol. Cell. Biol. 27: 7574–7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gmyrek G. B., Graham D. B., Sandoval G. J., Blaufuss G. S., Akilesh H. M., Fujikawa K., Xavier R. J., Swat W. 2013. Polarity gene discs large homolog 1 regulates the generation of memory T cells. Eur. J. Immunol. 43: 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walter U., Santamaria P. 2005. CD8+ T cells in autoimmunity. Curr. Opin. Immunol. 17: 624–631. [DOI] [PubMed] [Google Scholar]
- 46.Shin H., Wherry E. J. 2007. CD8 T cell dysfunction during chronic viral infection. Curr. Opin. Immunol. 19: 408–415. [DOI] [PubMed] [Google Scholar]
- 47.Appay V., Nixon D. F., Donahoe S. M., Gillespie G. M., Dong T., King A., Ogg G. S., Spiegel H. M., Conlon C., Spina C. A., et al. 2000. HIV-specific CD8+ T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 192: 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodnow C. C. 2007. Multistep pathogenesis of autoimmune disease. Cell 130: 25–35. [DOI] [PubMed] [Google Scholar]
- 49.Pandolfi F., Cianci R., Pagliari D., Casciano F., Bagalà C., Astone A., Landolfi R., Barone C. 2011. The immune response to tumors as a tool toward immunotherapy. Clin. Dev. Immunol. 2011: 894704. [DOI] [PMC free article] [PubMed] [Google Scholar]