

Abstract
Apoptotic cell death has been proposed to play a role in the neuronal loss observed following traumatic injury in the CNS and PNS. The present study uses an in vitro tissue culture model to investigate whether free fatty acids (FFAs), at concentrations comparable to those found following traumatic brain injury, trigger cell death. Nerve growth factor (NGF)-differentiated PC12 cells exposed to oleic and arachidonic acids (2 : 1 ratio FFA/BSA) showed normal cell survival. However, when cells were exposed to stearic and palmitic acids, there was a dramatic loss of cell viability after 24 h of treatment. The cell death induced by stearic acid and palmitic acid was apoptotic as assessed by morphological analysis, and activation of caspase-8 and caspase-3-like activities. Western blotting showed that differentiated PC12 cells exposed to stearic and palmitic acids exhibited the signature apoptotic cleavage fragment of poly (ADP-ribose) polymerase (PARP). Interestingly, blockade of caspase activities with the pan-caspase inhibitor z-VAD-fmk failed to prevent the cell death observed induced by palmitic or stearic acid. RT-PCR and RNA blot experiments showed an up-regulation of the Fas receptor and ligand mRNA. These findings are consistent with our hypothesis that FFAs may play a role in the cell death associated with trauma in the CNS and PNS.
Keywords: caspases, Fas ligand, Fas receptor, fatty acid, neuronal injury
Traumatic and hypoxic-ischemic injury to the CNS is a major source of morbidity and mortality. In both forms of injury, cell death occurs in two distinct phases (Pulsinelli et al. 1982; Bramlett et al. 1997). The first phase is coincident with injury and is essentially necrotic (Hicks et al. 1996), attributed mainly to the dysfunctional ion homeostasis and excitotoxicity characteristic of trauma (Faden et al. 1987; Nilsson et al. 1990) and hypoxia-ischemia (Lekieffre et al. 1992). The second or delayed phase of cell death develops progressively following experimental (Smith et al. 1995) and clinical head injury (Gale et al. 1995), and hypoxia-ischemia (MacManus et al. 1994; Du et al. 1996; Vexler et al. 1997). This prolonged phase of cell death can continue for up to a year (Smith et al. 1995; Conti et al. 1998), causing further tissue degeneration and contributing to post-injury neurobehavioral complications (Gualtieri and Cox 1991).
Although little is known about the mechanisms underlying secondary CNS injury, recent studies have established a role for apoptosis in delayed cell loss (Ferrer et al. 1994; Beilharz et al. 1995; Du et al. 1996; Liu et al. 1997; Conti et al. 1998; Lou et al. 1998; Matsushita et al. 2000). Studies of the molecular mechanisms of this apoptotic cell death have shown activation of the effector caspase-3 (Yakovlev et al. 1997; Northington et al. 2001) and the initiator caspase-8 (Velier et al. 1999; Matsushita et al. 2000) and caspase-11 (Kang et al. 2000). Altered balance of pro- and anti-apoptotic members of the Bcl-2 family proteins has also been demonstrated in response to trauma (Clark et al. 1997; O'Dell et al. 2000) and hypoxia-ischemia (Northington et al. 2001). Fas has been implicated as a possible apoptosis trigger in studies showing up-regulation of Fas receptor and ligand following brain trauma (Beer et al. 2000; Matsushita et al. 2000) and hypoxia-ischemia (Herdegen et al. 1998; Martin-Villalba et al. 1999; Velier et al. 1999; Felderhoff-Mueser et al. 2000; Matsushita et al. 2000).
An important neurochemical event initiated at the onset of traumatic and hypoxic-ischemic CNS injury is the degradation of membrane phospholipids with release of FFAs (Bazan 1970; Rehncrona et al. 1982; Zhang and Sun 1995; Dhillon et al. 1997; Homayoun et al. 1997). FFAs are liberated from membrane lipid by intracellular lipases (Abe et al. 1987; Faden et al. 1987) and may mediate much of the secondary damage (White et al. 2000). Arachidonic acid, which forms toxic metabolites, has received much attention as a possible mediator of FFA-induced injury. However, oleic, stearic and palmitic acids are also released following injury (Lukacova et al. 1998).
In this study we tested the hypothesis that FFAs, in concentrations comparable to those found following CNS injury, are toxic to neurons. Using NGF-differentiated pheochromocytoma 12 (PC12) cells as an in vitro neuronal model, we examined the effects of stearic, palmitic, oleic and arachidonic acids. Our study showed that stearic and palmitic acids are toxic while oleic and arachidonic acids are not. Cell death induced by stearic and palmitic acid was associated with caspase-8 and -3 activation, specific cleavage of PARP, and an increase in Fas receptor and ligand mRNA. In the presence of pan-caspase inhibition, these fatty acids were still capable of inducing cell death. These results are consistent with the possibility that injury-induced elevation of palmitic and stearic acids may induce apoptosis following traumatic and hypoxic-ischemic brain injury.
Materials and methods
Cell culture
Undifferentiated PC12 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% horse serum, 5% fetal bovine serum (FBS), 2 mm l-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin (Mediatech, Herndon, VA, USA) at 37°C with 95% air–5% CO2. Culture medium was replaced every 2–3 days. PC12 cells were differentiated by exposure to 50 ng/mL 2.5S (grade II) nerve growth factor (Alomone Laboratories, Jerusalem, Israel) for 10–14 days in DMEM supplemented with 1% FBS, penicillin/streptomycin and l-glutamine 24–36 h prior to treatment. NGF-differentiated PC12 cells were re-plated at 12 000 cells/cm2.
Preparation of fatty acid-methyl-β-cyclodextrin complexes
We used methyl-β-cyclodextrin (MβCD) as a delivery system to have optimal conditions for fatty acids remaining in solution in their monomeric form (Dansen et al. 1999). Preparation of fatty acid– MβCD (Sigma, St. Louis, MO, USA) inclusion complexes was performed with slight modifications to a previously described procedure (Klein et al. 1995). Briefly, 50 mm ethanol solutions of each fatty acid (all from Sigma) were converted to their sodium salts by addition of 5 mm Na2CO3. MβCD was dissolved in water to yield a 0.2 g/mL solution. Fatty acids were added at 65.2 mg/g MβCD. The resulting solutions of arachidonic and oleic acids were then incubated at 37°C for 1 h, and solutions of stearic and palmitic acids were incubated at 60°C for 15 min with occasional shaking until a clear solution was obtained. Ethanol in the solutions was then evaporated under a constant flow of argon at 60°C, and the remaining volume aliquoted and lyophilized in a vacuum concentrator. Inclusion complexes were stored at − 20°C and used within 2 months. MβCD was present in culture medium at a final concentration of 0.12% (w/v). Fatty acid-free BSA (Calbiochem, San Diego, CA, USA) was also used to buffer the concentration of fatty acid in the culture medium. Treatment of differentiated PC12 cells with vehicle alone (0.12% MβCD and 150 μm BSA) for 24 h resulted in no significant loss of viability with 94.7% ± 1.8% (n = 7) remaining viable.
Fatty acid treatment
NGF-differentiated PC12 cells were treated with 300 μm fatty acid (complexed with MβCD) and 150 μm BSAin low-serum medium (1% FBS). To prepare medium, fatty acid and BSA were added to DMEM containing 1% FBS and incubated at 50°C with occasional shaking for ∼ 12 h. Prior to treatment, fatty acid-containing medium was cooled to 37°C, 2 mm l-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin were added, and the medium was sterilized using a 0.22 μm filter.
Cell viability
Cell viability was determined by trypan blue exclusion. PC12 cells were harvested and incubated in 0.2% trypan blue (Sigma) for 10 min at 37°C, washed in phosphate-buffered saline (PBS), and the cells counted under phase-contrast microscopy. In this assay, blue cells were considered to have lost membrane integrity and were scored as non-viable. A minimum of 1000 cells was counted per sample and the number of trypan blue-excluding cells was expressed as a percentage of the total counted. Counts were performed in triplicate.
Morphologic determination of apoptotic nuclei
Apoptosis was measured by quantifying the number of cells exhibiting apoptotic nuclear morphology. Nuclear morphology was visualized by harvesting and fixing/staining cells in methanol containing 4′,6′-diamidino-2-phenylindole (DAPI, Sigma) at 5 μg/mL. Cells were then washed twice in PBS and examined under fluorescent microscopy at 400× magnification. Nuclei with condensed and/or fragmented morphology were scored as apoptotic and expressed as a percentage of the total nuclei counted. Counts were performed in triplicate with at least 1000 nuclei counted per sample.
Caspase activity assay
Caspase enzymatic activity was assessed by cleavage of the site-selective tetrapeptide chromogenic reporter substrates with the specificity of Asp-Glu-Val-Asp (DEVD; caspase-3 and -7) (Alexis, San Diego, CA, USA), Ile-Glu-Thr-Asp (IETD; caspase-8) and Leu-Glu-His-Asp (LEHD; caspase-9) (Calbiochem). Cells were lyzed at 4°C in HEPES–KOH 50 mm, pH 7.4, EDTA 1 mm buffer containing NaCl 75 mm, 1% Triton X-100, dithiothreitol (DTT) 1 mm, PMSF 1 mm, 10 μg/mL pepstatin A and 10 μg/mL aprotinin. Cells were then spun at 15 000 g for 20 min at 4°C and the supernatant fluid recovered and stored immediately at − 80°C until use. Enzymatic reactions (100 μL final volume) were performed at 37°C with 50 μg cell lysate and 100 μm chromogenic reporter substrate in HEPES–KOH 50 mm, pH 7.4 buffer containing NaCl 75 mm, DTT 2 mm, and (3-[(3-cholamidopropyl)dimethylammonio]-propane-1-sulfonate (CHAPS) 0.1%. Caspase-catalysed release of the chromophore p-nitroanilide was monitored spectrophotometrically at 405 nm. Optical density reading was corrected for background and normalized to lysate from cells treated for equal periods with vehicle only (0.12% MβCD/150 μm BSA).
Western blot analysis
Cells were lysed in sample buffer [200 mm Tris-HCl, 5% sodium dodecyl sulfate (SDS) and 10% glycerol] containing a complete protease inhibitor cocktail (Roche, Indianapolis, IN, USA). A 50 μg aliquot of total cellular protein was electrophoresed on 12% SDS– polyacrylamide gel (Bio-Rad, Hercules, CA, USA). Proteins were transferred to nitrocellulose membrane (Osmonics, Westerborough, MA, USA) and blocked in 5% skim milk in TPBS (PBS with 0.05 %Tween) for 1 h. Blots were then probed with an antibody to PARP (C2-10; Pharmingen, San Diego, CA, USA) followed by a secondary antibody which was horseradish peroxidase-conjugated. Antigen– antibody complexes were visualized with enhanced chemiluminescence (Amersham International, Biosciences, Pistcataway, NJ, USA).
Caspase inhibition experiments
Caspase-8 inhibitor benzyoxycarbonyl-Ile-Glu-Thr-Asp-fluoromethyl ketone (z-IETD-fmk) was purchased from Calbiochem. The pan-caspase inhibitor benzyoxycarbonyl-Val-Ala-Asp (Ome)-fluoromethylketone (z-VAD-fmk) was purchased from Enzyme Systems (Livermore, CA, USA). Stock solutions of caspase inhibitors were made in dimethyl sulfoxide (DMSO). For experiments employing caspase inhibitors, cells were pre-treated for 4 h prior to exposure to fatty acid.
Experiments were also undertaken to assess the ability of IETD-fmk to inhibit purified active caspase-3. Caspase activity assays were carried out (as above, without cell lysate) with 25 ng/mL of purified active caspase-3 (Pharmingen). Various concentrations of IETD-fmk and z-VAD-fmk were added to these reactions to determine a concentration of IETD-fmk at which the activity of caspase-3 is not inhibited.
RNA isolation
Total RNA was extracted using TRI-Reagent (Molecular Research Center, Cincinnati, OH, USA) as described (Chomczynski 1993). RNA was quantified by measuring the optical density at 260 nm. RNA samples were stored at − 80°C until use. All RNA samples were used within 1 week of extraction.
Semi-quantitative RT-PCR
RT-PCR was performed as a one-step reaction in 50 μL with the GeneAmp® Gold PCR kit (PE Applied Biosystems, Foster City, CA, USA). First-strand cDNA was prepared by reverse transcription of 1 μg of RNA with sequence-specific antisense primers, followed by PCR amplification with both sense and antisense primers. The primers used were as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense, 5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′ and antisense, 5′-CATGTGGGCCATGAGGTCCACCAC-3′ (983 bp fragment); Fas death receptor (Fas) sense, 5′-CAAGGGACTGATAGCATCTTTGAG-3′ and antisense, 5′-TCCAGATTCAGGGTCACAGGTTG-3′ (568 bp fragment); Fas ligand (FasL) sense, 5′-GTCCTGTCCTTGACACTTCAGTCTCC-3′ and antisense, 5′-TCAACTTCTTCTCCTCCATTAGCACC-3′ (750 bp fragment). The initial amount of mRNA and reaction conditions were optimized to obtain linearity for GAPDH in control cells. RT-PCR reactions for GAPDH, Fas and FasL were carried out under the same conditions throughout the incubation. RT-PCR cycling conditions were as follows: 12 min at 42°C, 10 min at 95°C, 60 cycles of 30 s at 94°C, 1 min at 60°C, 1 min 30 s at 72°C and 10 min at 72°C. The RT-PCR products (50 μL) were separated on a 2% agarose gel, stained with ethidium bromide and visualized. GAPDH, Fas and FasL RT-PCR reactions each generated one product that was subsequently purified and sequenced. The nucleotide sequences of each product were in full agreement with those expected in each respective reaction. Similar results were obtained from two independent preparations of RNA.
RNA blot analysis
RNA blots were done as previously described (De Leon et al. 1996). RNA (30 μg) was separated by electrophoresis on a 1.5% formaldehyde agarose gel and transferred onto Hybond-N membrane (Amersham). The membrane was subsequently dried under vacuum at 80°C for 30 min and the RNA immobilized by UV cross-linking. The templates for GAPDH and Fas RNA blots were derived from the respective RT-PCR fragments generated above. Probes were labeled with [α–32P]dCTP using the Prime-a-Gene® labeling system (Promega). RNA blot pre-hybridization and hybridization were performed with hybridization buffer containing 25 mm KPO4 (pH 7.4), 50% formamide, 5× Denhardt's solution, 50 μg/mL salmon sperm DNA and 5× SSPE (0.75 m NaCl, 50 mm NaH2PO4, 5 mm EDTA). Hybridization was carried out at 42°C with radiolabeled probes for ∼ 16 h. The membranes were washed twice with 2× SSPE/0.1% SDS for 10 min at room temperature, once for 15 min with 1× SSPE/0.1% SDS at 55°C, and once for 10 min with 1× SSPE/0.1% SDS at 55°C. Autoradiography was performed using Hyperfilm ECL (Amersham Bioscience) at − 80°C.
Results
Incubation of NGF-differentiated PC12 cells with stearic or palmitic acids results in loss of viability
The species of FFAs that are most notably elevated after CNS trauma and hypoxia-ischemia, arachidonic, oleic, stearic and palmitic acids, were tested for toxic effects (Homayoun et al. 1997). Our aim was to use a concentration of fatty acid comparable to that found following neuronal injury. Following traumatic brain injury (TBI), arachidonic acid increases from ∼30 to 300 μm, oleic acid from ∼40 to 100 μm, palmitic acid from ∼60 to 180 μm and stearic acid from ∼50 to 350 μm (Lipton 1999). Consequently, NGF-differentiated PC12 cells were incubated with 300 μm of the respective fatty acid complexed with MβCD in low-serum (1% FBS) medium containing 150 μm fatty acid-free BSA to emulate physiological conditions.
Cell membrane integrity was assessed by the ability of cells to exclude trypan blue. Stearic and palmitic acids were shown to be toxic, with dead cells first appearing after 6 h of incubation, and with 85 ± 1.4% of stearic acid-treated and 89.1 ± 2.9% palmitic acid-treated cells remaining viable after 6 h (mean viability ± SD, n = 9; Fig. 1a). By 24 h of incubation, only 8.81 ± 2.3% of stearic acid-treated and 16.6 ± 3.2% of palmitic acid-treated cells remained viable (mean viability ± SD, n = 9; Fig. 1a). Neither arachidonic nor oleic acids at 300 μm showed toxicity during the 24 h incubation (Fig. 1a).
Fig. 1.

Stearic and palmitic acid treatments result in loss of viability and increased apoptosis. (a) Stearic and palmitic acid treatments (300 μm) result in loss of viability of NGF-differentiated PC12 cells. Differentiated PC12 cells were treated for up to 24 h with either stearic, palmitic, oleic or arachidonic acid complexed with BSA (2 : 1 ratio). Viability was assessed by trypan blue exclusion during the course of fatty acid treatment. (b) Stearic and palmitic acid treatments increase the percentage of nuclei exhibiting apoptotic morphology. Cells were fixed and stained with 4′,6′-diamidino-2-phenylindole (DAPI) during the course of stearic and palmitic acid treatments. Nuclei were visualized under fluorescent microscopy and nuclei exhibiting condensed and/or fragmented morphology were scored as apoptotic. A minimum of 1000 cells were counted per sample for viability and apoptosis assays. Data represent the mean ± SD of three independent experiments performed in triplicate.
Stearic and palmitic acid treatment produce apoptotic cellular and nuclear morphology with fragmentation of DNA
Cell shrinkage with membrane blebbing, chromatin condensation and fragmentation, and cleavage of DNA into internucleosomal (180–200 bp) fragments, are commonly used criteria for distinguishing apoptosis from other forms of cell death (Kerr 1969; Kerr et al. 1972; Wyllie et al. 1980). In most systems, including neuronal cells, the morphologic changes that define apoptosis begin to appear before cell death is evident (Estus et al. 1994; Jacobson et al. 1997). Accordingly, the changes in nuclear morphology occurring in response to treatment with palmitic and stearic acid were documented (Fig. 1b). The portion of cells exhibiting apoptotic morphology (nuclear condensation/fragmentation) was clearly elevated after 12 h, with 33.9 ± 9.53% of stearic acid-treated and 41.9 ± 6.85% of palmitic acid-treated cells undergoing apoptosis (mean percent condensed/fragmented nuclei ± SD, n = 9; Fig. 1b). By 24 h of incubation, 84.4 ± 2.8% of stearic acid-treated and 80.8 ± 5.4% of palmitic acid-treated cells exhibited nuclear apoptotic morphology (mean percent condensed/ fragmented nuclei ± SD, n = 9; Fig. 1b). The morphological and nuclear changes induced by treatment with stearic and palmitic acids are illustrated in Fig. 2. After 24 h of stearic and palmitic acid treatment, nearly all cells were shrunken and exhibited blebbing into apoptotic bodies and nuclear fragmentation (Fig. 2, upper two panels). Consistent with the data presented in Fig. 1(a), treatment with either oleic or arachidonic acids for 24 h did not result in any of the morphological or nuclear changes evident in stearic and palmitic acid-treated cells (lower four panels, Fig. 2).
Fig. 2.
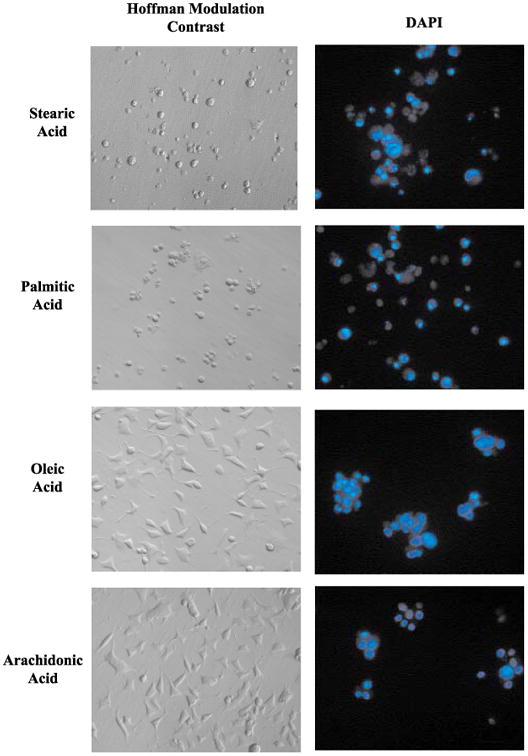
Hoffmann modulation contrast and fluorescent micrographs of NGF-differentiated PC12 cells exposed to selected fatty acids complexed with BSA (2 : 1). Images show NGF-differentiated PC12 cells exposed to the indicated fatty acid for 24 h. Fluorescent nuclear staining with DAPI was performed as described in Materials and methods. Panels on the left show Hoffman modulation contrast images of cells before fixation. Panels on the right show nuclear staining with DAPI. The fields shown on the left panels are different from the fields shown on the right panels.
Stearic and palmitic acids activate caspase-8- and caspase-3-like activity
Caspases have been implicated in models of neuronal apoptotic cell death and their activation occurs before the relatively late morphological changes characterizing apoptosis (Schwartz and Milligan 1996; Armstrong et al. 1997). The morphological characteristics of stearic and palmitic acid-induced toxicity suggest an apoptotic mode of cell death. Accordingly, DEVD-pNA, the colorimetric substrate for effector caspases-3 and -7, was used to assess the activity of these caspases. Both stearic and palmitic acid treatment resulted in activation of DEVDase activity by 6 h of treatment (Figs 3a and b). The peak of DEVDase activity was achieved for palmitic acid at 12 h (∼ 12-fold; Fig. 3a) and for stearic activity at 15 h (∼ 10-fold; Fig. 3b). Caspases-9 and-8aretwo, well-characterized initiator caspases, and their activity was also assessed with the colorimetric substrates LEDH-pNA and IETD-pNA, respectively. Activity of caspase-9 was not detectable during the 24 h fatty acid treatment (Figs 3a and b). The activity of caspase-8, on the other hand, was significantly elevated as early as 6 h, synchronous with the rise in caspase-3-like activity (Figs 3a and b, p < 0.05, both compared to the activity at 3 h). Caspase-8 activity continued to rise after 6 h, but did not achieve the high fold induction seen in caspase-3-like activity.
Fig. 3.

Induction of caspases-3 and -8, but not caspase-9, activity in cellular extracts of differentiated PC12 cells exposed to either palmitic or stearic acid complexed (2 : 1) with BSA. (a) Palmitic acid. (b) Stearic acid. Data represent the mean ± SD of three independent experiments performed in triplicate. # indicates significance of p < 0.05 in a two-sample t-test comparing caspase-8 activity at 3 h with that at 6 h. Data passed a test for normality and equality of variance.
PARP cleavage in NGF-differentiated PC12 cells exposed to stearic and palmitic fatty acids
Cleavage of PARP into fragments distinctive for apoptosis and necrosis has been described in Jurkat T-cell lines (Casiano et al. 1998; Gobeil et al. 2001; Wu et al. 2001). Consequently, initial verification of the reported cleavage patterns of PARP in NGF-stimulated PC12 cells was carried out by exposure of cells to 2 μm staurosporine (apoptosis) and 40 μm HgCl2 (necrosis), followed by the assessment of PARP cleavage by western blot analysis. Figure 4a shows that full-length (110 kDa) PARP is cleaved into an 85 kDa fragment in response to exposure to staurosporine for 24 h. Likewise, full-length PARP was cleaved into an 85 kDa and 50 kDa fragments upon exposure to HgCl2 for 24 h. These results confirm that the apoptosis- and necrosis-specific cleavage patterns of PARP obtained in NGF-stimulated PC12 cells are the same as those reported for Jurkat cells.
Fig. 4.
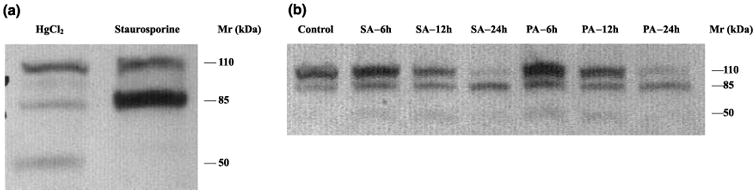
Cleavage pattern of PARP in cell lysates of NGF-differentiated PC-12 cells exposed to palmitic and stearic fatty acids. PC12 cells were differentiated with NGF for 14 days and exposed to the different treatments as shown in the Figure. Western blots using cell lysates were performed as described in Materials and methods. (a) Control NGF-differentiated PC12 cells treated with staurosporine or HgCl2. NGF-differentiated PC12 cells were treated with 40 μm HgCl2 or 2 μm staurosporine to verify the response of this cell line to conditions where a necrotic (HgCl2) or apoptotic (staurosporine) PARP cleavage pattern is induced. (b) PARP cleavage in NGF-differentiated PC12 cells treated with FFAs. Both palmitic and stearic acid induced the appearance of the signature apoptotic 85 kDa PARP but not the necrosis-associated 50 kDa fragment. SA, stearic acid; PA, palmitic acid; h, hour after initial exposure to either stearic or palmitic acid.
Cleavage of PARP into the 85 kDa fragment generated during apoptotic cell death was evident by 6 h incubation with either stearic or palmitic acids (Fig. 4b). Cleavage is more pronounced by 12 h and nearly complete by 24 h of exposure, with no visible full-length 110 kDa PARP remaining at this time point (Fig. 4b). The cleavage of PARP into the 85 kDa fragment correlates with DEVDase activity induced by fatty acid treatment (Fig. 3).
Activation of caspase-3-like activity is caspase-8 dependent
The kinetics of stearic and palmitic acid-induced activation of caspases did not clearly place the activation of caspase-8 ahead of caspase-3-like activation. Caspase-8 functions as an upstream caspase capable of initiating a cascade of caspase activation upon signaling at either the Fas death-inducing signaling complex (DISC) or tumor necrosis factor receptor (Boldin et al. 1996; Nagata 1997). However, activation of caspase-8 may also occur downstream of mitochondrial dysfunction as a result of caspase-3-like activation and independent from signaling at the death-inducing signaling complex (DISC) (Fulda et al. 1997).
To investigate the sequential activation of caspase-8 and caspase-3-like activity we used a selective inhibitor of caspase-8, IETD-fmk. Because the selective inhibition of caspase-8 by IETD-fmk is more pronounced at lower concentrations, an initial study with purified active caspase-3 was performed to determine a concentration of IETD-fmk at which caspase-3 is not inhibited. Figure 5a shows the results of this experiment. Z-VAD-fmk at 100 μm was included as a positive control for inhibition of caspase-3. IETD-fmk at 50 and 100 μm inhibited caspase-3 activity to an extent comparable with 100 μm z-VAD-fmk. However, at lower concentrations, IETD-fmk did not produce significant caspase-3 inhibition, with 5 μm being the highest tested concentration not demonstrating an inhibitory effect. Consequently, NGF-differentiated PC12 cells were pre-treated for 2 h with 5 μm IETD-fmk and then treated, in the presence of 5 μm IETD-fmk, with either stearic or palmitic acid. Figure 5(b and c) illustrates the inhibitory effects of IETD-fmk toward caspase-3-like activity in palmitic or stearic acid-treated cells, respectively. These findings show that caspase-8 activation is required for caspase-3-like activity in response to palmitic and stearic acid treatments.
Fig. 5.

Activation of caspase-3-like activity in cellular extracts of differentiated PC12 cells exposed to palmitic or stearic acid complexed with BSA (2 : 1) is caspase-8-dependent. (a) Inhibition of purified active caspase-3 with z-IETD-fmk (2–100 μm) and z-VAD-fmk (100 μm). The activity of purified active caspase-3 was assessed in vitro alone and in the presence of z-IETD-fmk or z-VAD-fmk. The activity of purified active caspase-3 in the presence of caspase inhibitors was expressed as a percentage of the activity in the absence of any inhibitor. Data represent the mean ± SD of two independent experiments performed in duplicate. (b) Inhibition of palmitic acid-induced caspase-3-like activation by z-IETD-fmk (5 μm). (c) Inhibition of stearic acid-induced caspase-3-like activation by z-IETD-fmk (5 μm). Data in (b) and (c) represent the mean ± SD of three independent experiments performed in duplicate. Control reactions (stearic and palmitic acid-induced caspase-3-like activity) were assessed in parallel with z-IETD-fmk reactions.
Stearic and palmitic acid-induced cell death is not blocked by pan-caspase inhibition
Apoptotic cell death is a caspase-dependent process in many systems that have been studied (Schwartz et al. 1996; Nicholson and Thornberry 1997). Caspase-independent cell death pathways, however, have been described in several systems (Miller et al. 1997; Vercammen et al. 1998; Matsumura et al. 2000). Z-VAD-fmk has been used widely to study the caspase dependence of apoptotic processes due to its ability to inhibit irreversibly a broad spectrum of the known caspases (Nicholson et al. 1995; Garcia-Calvo et al. 1998). This inhibitor was used to assess the caspase dependence of stearic and palmitic acid-induced apoptosis.
After 4 h of pre-treatment with 100 μm z-VAD-fmk, NGF-differentiated PC12 cells were exposed to stearic or palmitic acid in the presence of z-VAD-fmk. Caspase inhibition did not protect or delay the course of cell death as measured at 12 and 24 h of incubation with stearic or palmitic acids (Fig. 6a), although caspase-3-like activity was completely abolished (Fig. 6b). This result, along with previous evidence for caspase activation (Fig. 4a,b), indicates that stearic and palmitic acids are capable of activating both caspase-dependent and caspase-independent processes. The caspase-independent process becomes evident when caspase activity is blocked by z-VAD-fmk.
Fig. 6.

Inhibition of caspase activity with z-VAD-fmk does not prevent loss of cell viability in PC12 cultures exposed to either palmitic or stearic fatty acid complexed with BSA (2 : 1). (a) Cell viability was assessed after 12 and 24 h of exposure to either stearic and palmitic acid in the presence and absence of 100 μm z-VAD-fmk as indicated. Data represent the mean ± SD from three independent experiments performed in triplicate. (b) Caspase-3-like activity in cellular extracts of PC12 cells exposed to stearic and palmitic in the presence and absence of z-VAD-fmk (100 μm). Time points for stearic (15 h) and palmitic (12 h) acid were chosen to correspond to the peak of induced caspase-3-like activity. Data represent the mean ± SD for two independent experiments.
Stearic and palmitic acids induce up-regulation of Fas Death receptor and ligand mRNA
The apoptotic death occurring after both traumatic and hypoxic-ischemic brain injury has been associated with an up-regulation of Fas receptor and ligand mRNA (Martin-Villalba et al. 1999; Beer et al. 2000; Matsushita et al. 2000). Stearic and palmitic acid activate caspase-3-like activity in a caspase-8-dependent manner (Figs 5b and c), suggesting activation of a death receptor pathway. We were unable to detect the Fas ligand mRNA using northern blots (data not shown), suggesting that this mRNA is present at very low abundance. To explore this possibility further, the expression of mRNA for the Fas receptor and ligand were examined with RNA blot analysis and semi-quantitative RT-PCR.
Figure 7a shows that Fas receptor mRNA is up-regulated in NGF-differentiated PC12 cells exposed either to stearic or palmitic acid. The up-regulation was observed as early as 2 h after the initial exposure. Following probing with the Fas probe, the RNA blot was stripped and re-probed with GAPDH to verify even loading and transfer of RNA in each lane.
Fig. 7.
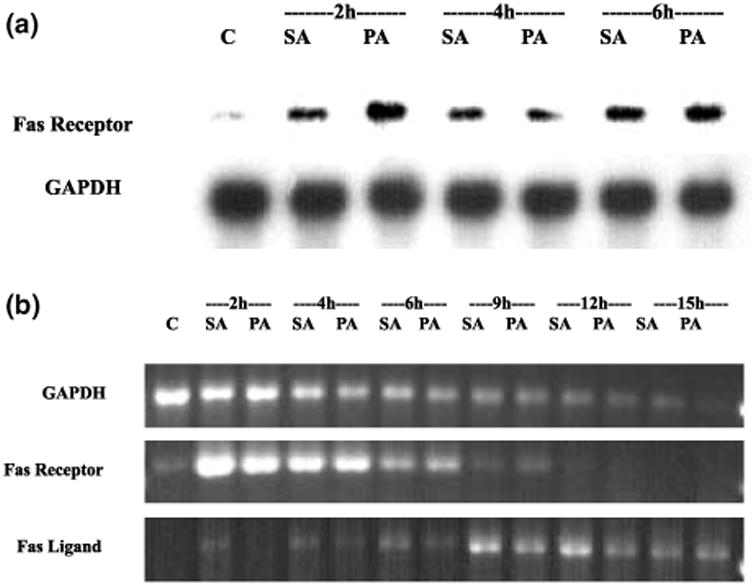
Induction of Fas receptor and Fas ligand mRNA in cellular extract of NGF-differentiated PC12 cells exposed to either stearic or palmitic acid complexed with BSA (2 : 1). (a) RNA blot showing the induction of Fas receptor mRNA during the course of exposure to either stearic or palmitic fatty acid. Each lane was loaded with 30 μg of RNA extracted from differentiated PC12 cells after 2, 4 and 6 h exposure to stearic or palmitic acid. Representative blot of two independent experiments. (b) RT-PCR analysis performed on total RNA extracted from cells treated for 2, 6, 9, 12 and 15 h with either palmitic or stearic acid complexed with BSA (2 : 1). The PCR products shown are representative of the results obtained from two independent experiments. C, control; SA, stearic acid; PA, palmitic acid; h, hours after initial exposure to fatty acids.
Semi-quantitative RT-PCR has been used to show that neuronal apoptotic cell death triggers a significant reduction of most mRNA species (Estus et al. 1994; Freeman et al. 1994; Miller et al. 1997). To confirm the RNA blot data, we extracted total RNA at various time points during stearic or palmitic acid-induced death (2, 4, 6, 9, 12 and 15 h) and 1 μg was used in a one-step RT-PCR reaction as described in Materials and methods. The primer sets used to amplify GAPDH, Fas receptor and Fas ligand each generated only one product of the predicted size. Sequence analysis of the receptor and ligand RT-PCR products confirmed that the amplified fragment corresponded to the intended product. As expected, GAPDH mRNA declined during the course of fatty acid-induced apoptosis (Fig. 7b). This finding is consistent with the degradation of GAPDH mRNA demonstrated in other models of neuronal apoptosis (Estus et al. 1997). Fas death receptor mRNA, on the other hand, increased significantly early (within 2 h) after treatment with stearic or palmitic acid. Expression of Fas ligand mRNA was also increased, although to a lesser degree, and was not apparent until 4 h of incubation (Fig. 7b). The up-regulation of Fas ligand is remarkable because it occurs at a time when mRNA species in general, as represented by GAPDH, are declining.
Discussion
The findings in this study show that stearic and palmitic acid induce apoptotic cell death in NGF-differentiated PC12 cells. This cell death was characterized by: (i) an early (within 2 h) increase in Fas receptor expression; (ii) subsequent cell shrinkage and membrane blebbing; (iii) nuclear condensation and fragmentation; (iv) activation of caspase-3 in a caspase-8-dependent manner; (v) late up-regulation of Fas ligand.
These findings support the conclusion that palmitic and stearic acid induce Fas-mediated apoptosis. Interestingly, broad spectrum caspase inhibition did not alter the kinetics of cell death, indicating that caspases are seemingly dispensable for stearic and palmitic acid-induced death.
Maximal tissue damage does not occur at the time of traumatic or hypoxic-ischemic injury (Cooper 1985; Soblosky et al. 1996; Dhillon et al. 1997) but tissue degeneration continues at the site of initial lesion, as well as in distant selectively-vulnerable regions (Furlan et al. 1996; Liu et al. 1997; Northington et al. 2001). Apoptosis has been shown to play an important role in delayed cell loss (Du et al. 1996; Conti et al. 1998; Newcomb et al. 1999; Nakajima et al. 2000). The molecular cascade of hypoxic-ischemia and trauma-induced apoptosis are similarly characterized by increased expression of Fas death receptor and activation of caspases-8 and -3 (Yakovlev et al. 1997; Martin-Villalba et al. 1999; Velier et al. 1999; Beer et al. 2000; Matsushita et al. 2000; Northington et al. 2001). These similarities raise the possibility that apoptosis in both injury settings has activator(s) that may regulate the development of the secondary to injury.
Traumatic, hypoxic-ischemic brain injury initiates cascades of injury events that can contribute to tissue damage, including loss of ion homeostasis, release of neurotransmitters, increased lactic acid and degradation of membrane phospholipid (Cooper 1985; White et al. 2000). Breakdown of membrane lipids with resultant accumulation of FFAs begins very rapidly following both types of injury and may be a factor in tissue damage (Bazan 1970; Dhillon et al. 1994, 1995, 1997). FFAs plateau at approximately 8–10-fold basal levels within the first hour after injury (Yoshida et al. 1984; Dhillon et al. 1997). The species of FFAs elevated in response to traumatic and hypoxic-ischemic injury are similar, with stearic, palmitic, oleic and arachidonic acids accounting for most of the accumulation (Rehncrona et al. 1982; Dhillon and Prasad 1999). The few studies that have examined phospholipid metabolism late after trauma (Homayoun et al. 1997) and hypoxia-ischemia (Abe et al. 1991) have demonstrated persistent elevation of FFAs during the days and weeks following injury. Additionally, a correlation exists between the level of FFAs accumulation and the severity of tissue injury (Bazan et al. 1971; Zhang and Sun 1995; Dhillon et al. 1999). For instance, the rate of lipolysis during ischemia is significantly greater in selective vulnerable zones, areas that notably exhibit large numbers of apoptotic cells (Beilharz et al. 1995), than in other regions of the brain (Umemura 1990). The rate of lipolysis and the subsequent magnitude of accumulation of FFAs following brain trauma and hypoxia-ischemia then correlate with selective vulnerability and the extent of tissue damage and may, consequently, be an important instigator of secondary damage.
Loss of fatty acid from the plasma membrane may alter its structure and function by changing membrane fluidity and signaling capacity (Siejo and Katsura 1992). Arachidonic acid in particular is a suspected mediator of tissue damage following both traumatic and hypoxic-ischemic neuronal death (Malecki et al. 2000). Arachidonic acid exerts its toxic effects through metabolism into eicosaniods (i.e. prostaglandins, thromboxanes and leukotrienes) (Katsuki and Okuda 1995) which increase production of free radicals, causing lipid peroxidation and oxidative damage to proteins (Krause et al. 1988). Arachidonic acid has also been shown to cause formation of cellular and vasogenic edema (Chan et al. 1983). In neuronal cell cultures, 10 μm arachidonic acid in serum-free medium without BSAhas been shown to be toxic (Toborek et al. 1999; Garrido et al. 2000, 2001). Arachidonic acid was not toxic in our experiments even though we tested a higher concentration (300 μm). This disparity may be explained by the presence of BSA (150 μm) in our system, which binds long-chain fatty acids with high affinity thereby reducing their free concentration (Bojesen and Bojesen 1994).
Neurons in the CNS can be exposed to albumin and other blood proteins during normal development and a number of pathological conditions including traumatic injury. This increase in albumin in the CSF occurs as a result of a breakdown of the blood–brain barrier (Cavanagh and Wareen 1985; Skultetyova et al. 1993; Fekuda et al. 1995). The presence of albumin in these situations can serve neuroprotective and neurotrophic functions, or have other effects associated with its interactions with fatty acids (Tabernero et al. 2002; Guajardo et al. 2002). For instance, albumin forms high affinity complexes with fatty acids that facilitate their solubility and transport in aqueous solution. The quantity of fatty acids bound to albumin is determined by the degree of saturation and the length of the carbon chain of the fatty acids involved. As a result of this, albumin binds to fully saturated fatty acids with higher affinity than to unsaturated fatty acids (Spector 1975); the dissociation constant for the complex is higher for saturated than unsaturated fatty acids and it decreases as the length of carbon backbone increases (Demant et al. 2002). These observations suggest, having all other conditions equal, that there should be higher levels of saturated stearic and palmitic acids bound to BSAcompared to unsaturated oleic and arachinodic fatty acids. These BSA-bound fatty acids are more efficiently transported to the cell membrane and into the cell than unbound unsaturated fatty acids. Interestingly, neuronal cell line HN2-5 undergoing apoptosis exhibits an increased percentage of saturated fatty acids, such as palmitic and stearic acids, incorporated into cell membrane phospholipids (Singh et al. 1996). We speculate that an increase in the percentage of stearic and palmitic acids in the cell membrane phospholipids will alter the immediate local lipid environment of receptor proteins, such as the Fas receptor, and may serve as a trigger for apoptosis.
Another way that albumin influences fatty acids is by preventing their exposure to free radicals that can result in the formation of hydroperoxides. Albumin as an antioxidant can reduce lipid peroxidation and increase cell viability (Lozinsky et al. 2001; Guajardo et al. 2002). Unsaturated fatty acids, such as oleic and arachidonic acid, are more vulnerable to oxidation than saturated stearic and palmitic acids. Our experimental design was developed taking into consideration the potential antioxidant effects of BSA, and we treated the cell cultures with the same high concentration of albumin to maximize the antioxidant effect. The absence of apoptosis in the cell culture exposed to oleic and arachinodic acid suggests that albumin could indeed be playing such a role.
Although a well-defined mechanism of toxicity exists for polyunsaturated fatty acids (arachidonic and docosahexinoic acids), they may not be crucial mediators of secondary damage given that they are notably absent during the second or prolonged phase of FFA release following hypoxia-ischemia (Zhang and Sun 1995). This observation is confirmed and partially explained by the finding that the rate at which arachidonic acid is re-incorporated into brain phospholipid is selectively accelerated after hypoxia-ischemia (Rabin et al. 1998). Selective re-acylation of arachidonic acid into lysophospholipid and re-incorporating into the membrane potentially reduces the generation of toxic cyclooxygenase and lipoxygenase metabolites. Although the relative rate of arachidonic acid re-acylation has not been measured in the context of trauma, saturated (stearic) and monounsaturated (oleic) fatty acids account for most of the FFA accumulation 24 h after traumatic injury (Dhillon et al. 1997; Homayoun et al. 1997).
A major obstacle in attempting to reproduce the conditions of FFAs accumulation following trauma and hypoxia-ischemia is presenting cells in culture with large amounts of monomeric fatty acid. Long-chain free fatty acids are extremely hydrophobic and, as such, are highly insoluble in the aqueous phase. In the blood, most FFA is bound to albumin with only a small amount existing in solution monomerically (Spector 1975; Bojesen and Bojesen 1992; Richieri and Kleinfeld 1995). Further complicating this scenario are the differences in monomer solubility arising from variation in fatty acid chain length and degree of saturation (Richieri et al. 1993). It is the water-phase shuttle of fatty acid monomers that mediates the exchange of fatty acids between albumin and cell membranes; consequently, differences in solubility may result in differing rates of transfer. Furthermore, fully-saturated fatty acids, such as stearic and palmitic, which have less soluble than mono- and polyunsaturated fatty acids, tend to form dimers and/or higher aggregates when solubilized in organic solvent and then diluted in aqueous medium.
Consequently, methyl-β-cyclodextrin was employed to increase the solubility of monomeric fatty acid molecules. A question that arises is whether the method of solubilizing fatty acid is physiologically important. This question has been addressed in studies using MβCD to form water-soluble complexes of lipophilic substances. By forming inclusion complexes with lipophilic molecules, methyl-β-cyclodextrin is able to function as a non-toxic pharmaceutical vehicle to solubilize and deliver water-insoluble substances to cells in culture (Pitha and Pitha 1985; Vicanova et al. 1999). A study analysing carotenoid uptake by cells in culture demonstrated that solubilization of β-carotene with MβCD results in increased stability and enhanced cellular uptake of β-carotene as compared to solubilization in organic solvents (Pftizer et al. 2000). Furthermore, it has been demonstrated that binding of fatty acid to a non-specific lipid transfer protein is substantial only when fatty acids are presented in their monomeric form complexed with MβCD (Dansen et al. 1999). These results suggest that presentation of fatty acids monomerically can be essential for their cellular uptake and protein-binding capabilities.
Injury induces release of fatty acid monomers from the inner layer of the plasma membrane of the cell body by the action of intracellular phospholipases (Abe et al. 1987). Released fatty acid monomers are then free to interact with intracellular proteins including transport proteins, such as fatty acid binding proteins (FABPs) and peroxisome proliferator-activated receptors (PPARs), whose binding sites accommodate monomolecular fatty acids (Storch and Thumser 2000). In addition to fatty acid transport, a partnership has been demonstrated between FABPs and PPARs in the transduction of long-chain fatty acid-mediated changes in gene expression (Wolfrum et al. 2001). Injury-induced released of monomeric fatty acids could conceivably alter patterns of gene expression upon their interaction with FABPs and, subsequently, PPARs. For instance, epidermal fatty acid binding protein (E-FABP), a neuronal injury-associated FABP, exhibits a robust increase in neurons following nerve injury (De Leon et al. 1996).
We found that PARP is cleaved into the signature 85 kDa apoptotic fragment upon treatment with stearic or palmitic acid. This provided further support for the hypothesis that these fatty acids induce apoptotic cell death in NGF-stimulated PC12 cells.
An additional question raised by these experiments is the temporal relationship between the up-regulation of Fas receptor and Fas ligand mRNA. A previous study examining immunohistologically the pattern of Fas receptor and Fas ligand expression found that while Fas receptor was up-regulated within 15 min of traumatic injury, Fas ligand up-regulation was not detected until 72 h after injury (Beer et al. 2000). These results are similar to those of our study in that we found Fas receptor to be up-regulated within 2 h of fatty acid exposure while Fas ligand up-regulation was not detected until 4 h (Fig. 7b). The biological significance of this observation is not clear. Studies are underway to clarify whether differences in the affinities of the probes used to amplify Fas receptor and ligand are responsible for the delayed detection of the up-regulation of Fas ligand mRNA or indeed there are sufficient basal levels of Fas ligand to mediate Fas receptor induction during fatty acid cytotoxicity.
The cell death induced by stearic and palmitic acid described in this study was not inhibited by the pan-caspase inhibitor z-VAD-fmk. Inhibition of caspases with z-VAD-fmk effectively blocks apoptosis in several models of neuronal cell death (Deshmukh et al. 1996, 2000; Armstrong et al. 1997). Conversely, many models of apoptotic cell death that are not blocked by caspase inhibition have been described (Miller and Johnson 1996; Miller et al. 1997). Apoptotic signaling through the Fas death receptor serves as a well-characterized example of this type of model. The Fas receptor, under certain circumstances, has been shown to transduce both caspase-dependent and caspase-independent cell death signals (Vercammen et al. 1999; Matsumura et al. 2000). The caspase-dependent death pathway apparently dominates the caspase-independent pathway when caspases are functional (Matsumura et al. 2000). Yet when caspases are inhibited, the caspase-independent pathway ensures that cell death occurs in the absence of caspase activation and has prominent features of necrosis. We hypothesize that a similar phenomenon is occurring in stearic and palmitic acid-induced death. Current studies in our laboratory are being directed at exploring the mechanisms underlying this caspase-inde-pendent pathway.
In conclusion, this study demonstrates that stearic and palmitic acids induce apoptosis in NGF-differentiated PC12 cells, and provides evidence for the involvement of the Fas receptor and ligand in this process. Because of the similarities in mechanism, these results suggest a possible role for elevated intracellular levels of stearic and palmitic acids in the acute and delayed apoptotic death occurring after traumatic and hypoxic-ischemic injury. Although it is certain that the Fas receptor/ligand pathway is not the only apoptotic program activated following injury, it may serve as an important therapeutic target for improving functional recovery following brain injury.
Acknowledgments
This work was supported by grants from the National Science Foundation (IBN-9728662) and the Montgomery Street Foundation. We would like to acknowledge Drs Sandy Hilliker and Jo-Wen Liu for their critical reading of the manuscript.
Abbreviations used
- BSA
bovine serum albumin
- CHAPS
(3-[(3-cholamidopropyl)dimethylammonio]-propane-1-sulfonate
- DAPI
4′,6′-diamidino-2-phenylindole
- DEVD
Asp-Glu-Val-Asp
- DISC
death-inducing signaling complex
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- FBS
fetal bovine serum
- FFA
free fatty acid
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IETD
Ile-Glu-Thr-Asp
- z-IETD-fmk
benzyoxycarbonyl-Ile-Glu-Thr-Asp-fluoromethyl ketone
- LEHD
Leu-Glu-His-Asp
- MBCD
methyl-β-cyclodextrin
- NGF
nerve growth factor
- PARP
poly-(ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PMSF
phenylmethylsulfonyl fluoride
- SDS
sodium dodecyl sulfate
- TBI
traumatic brain injury
- z-VAD-fmk
benzyoxycarbonyl-Val-Ala-Asp (OMe)-fluoromethylketone
References
- Abe K, Kogure K, Yamamoto H, Imazawa M, Miyamoto K. Mechanism of arachidonic acid liberation during ischemia in gerbil cerebral cortex. J Neurochem. 1987;48:503–509. doi: 10.1111/j.1471-4159.1987.tb04121.x. [DOI] [PubMed] [Google Scholar]
- Abe K, Yoshidomi M, Kogure K. Arachidonic acid metabolism in ischemic neuronal damage. Ann NY Acad Sci. 1991;559:259–268. doi: 10.1111/j.1749-6632.1989.tb22614.x. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karenewsky DS, Fritz LC, Tomaselli KJ. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. Effects of ischemia and electroconvulsive shock on free fatty acid pool in the brain. Biochim Biophys Acta. 1970;218:1–10. doi: 10.1016/0005-2760(70)90086-x. [DOI] [PubMed] [Google Scholar]
- Bazan NG, De Bazan HEP, Kennedy WG, Joel CD. Regional distribution and rate of production of free fatty acids in rat brain. J Neurochem. 1971;18:1487–1393. doi: 10.1111/j.1471-4159.1971.tb00003.x. [DOI] [PubMed] [Google Scholar]
- Beer R, Franz G, Schopf M, Reindl M, Zelger B, Schmutzhard E, Poewe W, Kampfl A. Expression of Fas and Fas ligand after experimental traumatic brain injury in the rat. J Cereb Blood Flow Metab. 2000;20:669–677. doi: 10.1097/00004647-200004000-00004. [DOI] [PubMed] [Google Scholar]
- Beilharz EJ, Williams CE, Dragunow M, Sirimanne ES, Gluckman PD. Mechanisms of delayed cell death following hypoxic-ischemic injury in the immature rat: evidence for apoptosis during selective neuronal loss. Mol Brain Res. 1995;29:1–14. doi: 10.1016/0169-328x(94)00217-3. [DOI] [PubMed] [Google Scholar]
- Bojesen IN, Bojesen E. Water-phase palmitate concentrations in equilibrium with albumin-bound palmitate in a biological system. J Lipid Res. 1992;33:1327–1334. [PubMed] [Google Scholar]
- Bojesen IN, Bojesen E. Binding of arachi11/27/02donate and oleate to bovine serum albumin. J Lipid Res. 1994;35:770–778. [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;84:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD, Green EJ, Busto R. Chronic histopathological consequences of fluid-percussion brain injury in rats: effects of post-traumatic hypothermia. Acta Neuropathol. 1997;93:190–199. doi: 10.1007/s004010050602. [DOI] [PubMed] [Google Scholar]
- Casiano CA, Ochs RL, Tan EM. Distinct cleavage products of nuclear proteins in apoptosis and necrosis revealed by autoantibody probes. Cell Death Differ. 1998;1998:183–190. doi: 10.1038/sj.cdd.4400336. [DOI] [PubMed] [Google Scholar]
- Cavanagh ME, Wareen A. The distribution of native and foreign albumin injected into lateral ventricles of prenatal and neonatal rat ventricles of prenatal and neonatal rat forebrains. Anat Embryol. 1985;172:345–351. doi: 10.1007/BF00318983. [DOI] [PubMed] [Google Scholar]
- Chan PH, Fishman RA, Caronna J, Schimdley JW, Prioleau G, Lee J. Induction of brain oedema following intracerebral injection of arachidonic acid. Ann Neurol. 1983;13:625–632. doi: 10.1002/ana.410130608. [DOI] [PubMed] [Google Scholar]
- Chomczynski P. Areagent for the single-step simultaneous isolation of RNA, DNA and protein from cell and tissue samples. Biotechniques. 1993;15:532–537. [PubMed] [Google Scholar]
- Clark RSB, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA, Loeffert JE, Graham SH. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci. 1997;17:9172–9182. doi: 10.1523/JNEUROSCI.17-23-09172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper PR. Delayed brain injury: Secondary insults. In: Becker PD, Povlishock JT, editors. Central Nervous System Trauma Status Report. National Institute of Neurological and Communicative Disorders and Stroke, National Institutes of Health; Bethesda, MD: 1985. pp. 217–228. [Google Scholar]
- Dansen TB, Westerman J, Wouters FS, Wanders RJA, van Hoek A, Gadella TWJ, Wirtz KWA. High-affinity binding of very-long-chain fatty acyl-CoA esters to the peroxisomal non-specific lipid-transfer protein (sterol carrier protein-2) Biochem J. 1999;339:193–199. [PMC free article] [PubMed] [Google Scholar]
- De Leon M, Welcher AA, Nahin RH, Liu Y, Ruda MA, Shooter EM, Molina CA. Fatty acid binding protein is induced in neurons of the dorsal root ganglia after peripheral nerve injury. J Neurosci Res. 1996;44:283–292. doi: 10.1002/(SICI)1097-4547(19960501)44:3<283::AID-JNR9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Demant EJF, Richieri GV, Kleinfeld AM. Stopped-flow kinetic analysis of long-chain fatty acid dissociation from bovine serum albumin. Biochem J. 2002;363:809–815. doi: 10.1042/0264-6021:3630809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Kuida K, Johnson EM. Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon HS, Prasad RM. Kynurenate attenuates the accumulation of diacylglycerol and free fatty acids after experimental brain injury in the rat. Brain Res. 1999;832:7–12. doi: 10.1016/s0006-8993(99)01437-7. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Donaldson D, Dempsey RJ, Prasad MR. Regional levels of free fatty acids and Evans Blue extravasation after experimental brain injury. J Neurotrauma. 1994;11:405–415. doi: 10.1089/neu.1994.11.405. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Carbary T, Dose J, Dempsey RJ, Prasad MR. Activation of phosphatidylinositol biphosphate signal transduction pathway after experimental brain injury: a lipid study. Brain Res. 1995;698:100–106. doi: 10.1016/0006-8993(95)00840-m. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Dose JM, Scheff SW, Prasad MR. Time course of changes in lactate and free fatty acids after experimental brain injury and relationship to morphologic damage. Exp Neurol. 1997;146:240–249. doi: 10.1006/exnr.1997.6524. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Carman HM, Zhang D, Scheff SW, Prasad MR. Severity of experimental brain injury on lactate and free fatty acid accumulation and Evans blue extravasation in the rat cortex and hippocampus. J Neurotrauma. 1999;16:455–469. doi: 10.1089/neu.1999.16.455. [DOI] [PubMed] [Google Scholar]
- Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- Estus S, Zaks WJ, Freeman RS, Gruba M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estus S, Tucker MH, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. Aggregated amyloid-P protein induces cortical neuronal apoptosis and concomitant ‘apoptotic’ pattern of gene induction. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI, Chan PH, Longar S. Alterations in lipid metabolism, Na+, K+-ATPase activity, and tissue water content of spinal cord following experimental traumatic injury. J Neurochem. 1987;48:1809–1816. doi: 10.1111/j.1471-4159.1987.tb05740.x. [DOI] [PubMed] [Google Scholar]
- Fekuda K, Tanno H, Okimura Y, Nakamura M, Yamaura A. The blood-brain barrier disruption to circulating proteins in the early period after fluid percussion brain injury in rats. J Neurotrauma. 1995;12:315–324. doi: 10.1089/neu.1995.12.315. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Taylor DL, Greenwood K, Kozma M, Stilbenz D, Joashi UC, Edwards AD, Mehmet H. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic-ischemic injury to the developing rat brain. Brain Pathol. 2000;10:17–29. doi: 10.1111/j.1750-3639.2000.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Tortosa A, Macaya A, Sierra A, Moreno D, Munell F, Blanco R, Squier W. Evidence of nuclear DNA fragmentation following hypoxia-ischemia in the infant rat brain, and transient forebrain ischemia in the adult gerbil. Brain Pathol. 1994;4:115–122. doi: 10.1111/j.1750-3639.1994.tb00821.x. [DOI] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Horigome K, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- Fulda D, Sieverts H, Friesen C, Herr I, Debatin KM. The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 1997;57:3822–3829. [PubMed] [Google Scholar]
- Furlan M, Marchal G, Viader F, Derlon JM, Baron JC. Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann Neurol. 1996;40:216–226. doi: 10.1002/ana.410400213. [DOI] [PubMed] [Google Scholar]
- Gale SD, Johnson SC, Bigler ED, Blatter DD. Trauma-induced degenerative changes in brain injury: a morphometric analysis of three patients with preinjury and post-injury MR scans. J Neurotrauma. 1995;12:151–158. doi: 10.1089/neu.1995.12.151. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Garrido R, Malecki A, Hennig B, Toborek M. Nicotine attenuates arachidonic acid-induced neurotoxicity in cultured spinal cord neurons. Brain Res. 2000;861:59–68. doi: 10.1016/s0006-8993(00)01977-6. [DOI] [PubMed] [Google Scholar]
- Garrido R, Mattson MP, Hennig B, Toborek M. Nicotine protects against arachidonic-acid-induced caspase activation, cytochrome c release and apoptosis of cultured spinal cord neurons. J Neurochem. 2001;76:1395–1403. doi: 10.1046/j.1471-4159.2001.00135.x. [DOI] [PubMed] [Google Scholar]
- Gobeil S, Boucher CC, Nadeau D, Poirier GG. Characterization of the necrotic cleavage of poly (ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001;8:588–594. doi: 10.1038/sj.cdd.4400851. [DOI] [PubMed] [Google Scholar]
- Guajardo MH, Terrasa AM, Catalá A. Retinal fatty acid binding protein reduces lipid peroxidation stimulated by long-chain fatty acid hydroperoxides on rod outer segments. Biochim Biophys Acta. 2002;1581:65–74. doi: 10.1016/s1388-1981(02)00121-x. [DOI] [PubMed] [Google Scholar]
- Gualtieri T, Cox DR. The delayed neurobehavioral sequelae of traumatic brain injury. Brain Inj. 1991;5:219–232. doi: 10.3109/02699059109008093. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks RR, Soares HD, Smith DH, McIntosh TK. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neurochir. 1996;51(Suppl):331–333. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- Homayoun P, Rodriguez de Turco EB, Parkins NE, Lane DC, Soblosky J, Carey ME, Bazan NG. Delayed phospholipid degradation in rat brain after traumatic brain injury. J Neurochem. 1997;69:199–205. doi: 10.1046/j.1471-4159.1997.69010199.x. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, Huang Z, Srinivasan S, Tomaselli KJ, Thornberry NA, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol. 2000;149:613–622. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol. 1995;46:607–636. doi: 10.1016/0301-0082(95)00016-o. [DOI] [PubMed] [Google Scholar]
- Kerr JFR. Shrinkage necrosis: a distinct mode of cellular death. J Pathol. 1969;105:13–20. doi: 10.1002/path.1711050103. [DOI] [PubMed] [Google Scholar]
- Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with β-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- Krause GS, White BC, Aust SD, Nayini NR, Kumar K. Brain cell death following ischemia and reperfusion: a proposed biochemical sequence. Crit Care Med. 1988;16:714–726. doi: 10.1097/00003246-198807000-00015. [DOI] [PubMed] [Google Scholar]
- Lekieffre D, Callebert J, Plotkine M, Boulu RG. Concomitant increases in the extracellular concentrations of excitatory and inhibitory amino acids in the rat hippocampus during forebrain ischemia. J Neurosci Lett. 1992;137:78–82. doi: 10.1016/0304-3940(92)90303-o. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J, Lenke LG, Ludwig FJ, O'Brien MF. Apoptosis as a mechanism of neuronal cell death following acute experimental spinal cord injury. Spinal Cord. 1998;39:683–690. doi: 10.1038/sj.sc.3100632. [DOI] [PubMed] [Google Scholar]
- Lozinsky E, Novoselsky A, Shames AI, Saphier O, Meyerstein GI, Meyerstein D. Effect of albumin on the kinetics of ascorbate oxidation. Biochim Biophys Acta. 2001;1526:53–60. doi: 10.1016/s0304-4165(01)00100-3. [DOI] [PubMed] [Google Scholar]
- Lukacova N, Jalc P, Marsala J. Phospholipid composition in spinal cord regions after ischemia/reperfusion. Neurochem Res. 1998;23:1069–1077. doi: 10.1023/a:1020708102702. [DOI] [PubMed] [Google Scholar]
- MacManus JP, Hill IE, Huang ZG, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient focal ischemic neurocortex. Neuroreport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- Malecki A, Garrido R, Mattson MP, Hennig B, Toborek M. 4-Hydroxynonenal induces oxidative stress and death of cultured spinal cord neurons. J Neurochem. 2000;74:2278–2287. doi: 10.1046/j.1471-4159.2000.0742278.x. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J. CD95 (Fas-L/APO-1L) and tumor necrosis factor related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151:1247–1256. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Wu Y, Qiu J, Lang-Lazdunski L, Hirt L, Waeber C, Hyman BT, Yuan J, Moskowitz MA. Fas receptor and neuronal cell death after spinal cord ischemia. J Neurosci. 2000;20:6879–6887. doi: 10.1523/JNEUROSCI.20-18-06879.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Johnson EM., Jr Metabolic and genetic analysis of apoptosis in rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nakajima W, Ishida A, Lange MS, Grabielson KL, Wilson MA, Martin LJ, Blue ME, Johnston MV. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in newborn rats. J Neurosci. 2000;20:7994–8004. doi: 10.1523/JNEUROSCI.20-21-07994.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb JK, Zhao X, Pike BR, Hayes RL. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp Neurol. 1999;158:76–88. doi: 10.1006/exnr.1999.7071. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Hillered L, Ponten U, Ungerstedt U. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab. 1990;10:631–637. doi: 10.1038/jcbfm.1990.115. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Flock DL, Martin LJ. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J Neurosci. 2001;21:1931–1938. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell DM, Raghupathi R, Crino PB, Eberwine JH, McIntosh TK. Traumatic brain injury alters the molecular fingerprint of TUNEL-positive cortical neurons in vivo: a single-cell analysis. J Neurosci. 2000;20:4821–4828. doi: 10.1523/JNEUROSCI.20-13-04821.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pftizer I, Francz PI, Biesalski HK. Carotenoid: methyl-β-cyclodextrin formulations: an improved method for supplementation of cultured cells. Biochim Biophys Acta. 2000;1474:163–168. doi: 10.1016/s0304-4165(00)00014-3. [DOI] [PubMed] [Google Scholar]
- Pitha J, Pitha J. Amorphous water-soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients. J Pharm Sci. 1985;74:987–990. doi: 10.1002/jps.2600740916. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Rabin O, Chang MCJ, Grange E, Bell J, Rapoport SI, Deutsch J, Purdon AD. Selective acceleration of arachidonic acid reincorporation into brain membrane phospholipid following transient ischemia in awake gerbil. J Neurochem. 1998;70:325–334. doi: 10.1046/j.1471-4159.1998.70010325.x. [DOI] [PubMed] [Google Scholar]
- Rehncrona S, Westerberg E, Akesson B, Siesjo BK. Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. J Neurochem. 1982;56:84–93. doi: 10.1111/j.1471-4159.1982.tb10857.x. [DOI] [PubMed] [Google Scholar]
- Richieri GV, Kleinfeld AM. Unbound free fatty acid levels in human serum. J Lipid Res. 1995;36:229–240. [PubMed] [Google Scholar]
- Richieri GV, Anel A, Kleinfeld AM. Interactions of long-chain fatty acids and albumin: determination of free fatty acid levels using the fluorescent probe ADIFAB. Biochemistry. 1993;32:7574–7589. doi: 10.1021/bi00080a032. [DOI] [PubMed] [Google Scholar]
- Schwartz LM, Milligan CE. Cold thoughts of death—the role of ICE proteases in neuronal cell death. Trends Neurosci. 1996;19:555–562. doi: 10.1016/s0166-2236(96)10067-9. [DOI] [PubMed] [Google Scholar]
- Siejo BK, Katsura K. Ischemic brain damage: focus on lipids and lipid mediators. Adv Exp Med Biol. 1992;318:41–56. doi: 10.1007/978-1-4615-3426-6_5. [DOI] [PubMed] [Google Scholar]
- Singh JK, Dasgupta A, Adayev T, Shahmehdi SA, Hammond D, Banerjee P. Apoptosis is associated with an increase in saturated fatty acid containing phospholipids in the neuronal cell line, HN2-5. Biochim Biophys Acta. 1996;1304:171–178. doi: 10.1016/s0005-2760(96)00134-8. [DOI] [PubMed] [Google Scholar]
- Skultetyova I, Tokarev DI, Jezova D. Albumin content in the developing rat brain in relation to the blood brain barrier. Endocr Regul. 1993;27:209–213. [PubMed] [Google Scholar]
- Smith DH, Chen XH, Pierce JES, Wolf JA, Trojanowski JQ, Graham KI, McIntosh L. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1995;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- Soblosky JS, Matthews MA, Davidson JF, Tabor SL, Carey ME. Traumatic brain injury of the forelimb and hindlimb sensorimotor areas in the rat: physiological, histological and behavioral correlates. Behav Brain Res. 1996;79:79–82. doi: 10.1016/0166-4328(95)00264-2. [DOI] [PubMed] [Google Scholar]
- Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16:165–179. [PubMed] [Google Scholar]
- Storch J, Thumser AEA. The fatty acid transport function of fatty acid-binding proteins. Biochim Biophys Acta. 2000;1486:28–44. doi: 10.1016/s1388-1981(00)00046-9. [DOI] [PubMed] [Google Scholar]
- Tabernero A, Velasco A, Granda B, Lavado EM, Medina JM. Trancytosis of albumin in astrocytes activates the sterol regulatory element binding protein 1 which promotes the synthesis of the neurotrophic factor oleic acid. J Biol Chem. 2002;277:4240–4246. doi: 10.1074/jbc.M108760200. [DOI] [PubMed] [Google Scholar]
- Toborek M, Malecki A, Garrido MP, Mattson B, Hennig B, Young B. Arachidonic acid-induced oxidative injury to cultured spinal cord neurons. J Neurochem. 1999;73:684–692. doi: 10.1046/j.1471-4159.1999.0730684.x. [DOI] [PubMed] [Google Scholar]
- Umemura A. Regional difference in free fatty acids release and the action of phospholipase during ischemia in rat brain. No To Shinkei. 1990;42:979–986. [PubMed] [Google Scholar]
- Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC, Feuerstein GZ. Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J Neurosci. 1999;19:5932–5941. doi: 10.1523/JNEUROSCI.19-14-05932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vexler ZS, Roberts TP, Bollen AW, Derugin N, Arieff AI. Transient cerebral ischemia. Association of apoptosis induction with hypoperfusion. J Clin Invest. 1997:1543–1549. doi: 10.1172/JCI119304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicanova J, Weerheim AM, Kempenaar JA, Ponec M. Incorporation of linoleic acid by cultured human keratinocytes. Arch Dermatol Res. 1999;29:405–412. doi: 10.1007/s004030050430. [DOI] [PubMed] [Google Scholar]
- White BC, Sullivan JM, DeGarcia DJ, O'Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. doi: 10.1016/s0022-510x(00)00386-5. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Borrman CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors a- and c-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci USA. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Molinaro C, Johnson N, Casiano CA. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunity. Arthr Rheum. 2001;11:2642–2652. doi: 10.1002/1529-0131(200111)44:11<2642::aid-art444>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Harik SI, Busto R, Santiso M, Martinez E, Ginsberg MD. Free fatty acids and energy metabolites in ischemic cerebral cortex with noradrenaline depletion. J Neurochem. 1984;42:711–717. doi: 10.1111/j.1471-4159.1984.tb02741.x. [DOI] [PubMed] [Google Scholar]
- Zhang JP, Sun GY. Free fatty acids, neutral glycerides, and phosphoglycerides in transient focal cerebral ischemia. J Neurochem. 1995;64:1688–1695. doi: 10.1046/j.1471-4159.1995.64041688.x. [DOI] [PubMed] [Google Scholar]