

Abstract
Pathologic cardiac hypertrophy can lead to heart failure, but the mechanisms involved are poorly understood. SERCA2 is critical for normal cardiac calcium handling and function and SERCA2 mRNA and protein levels are reduced by cardiac hypertrophy. We hypothesized that Extracellular Signal-regulated Kinase (ERK) 1/2 activation during hypertrophy reduced SERCA2 transcription. Using a neonatal rat ventricular myocyte model of hypertrophy, we found that pharmacologic inhibitors of ERK activation preserve SERCA2 mRNA levels during hypertrophy. ERK activation is sufficient to reduce SERCA2 mRNA. We determined that ERK represses SERCA2 transcription via nuclear factor-kappaB (NFkB), and activation of NFkB is sufficient to reduce SERCA2 mRNA in cardiomyocytes. This work establishes novel connections between ERK, NFkB, and SERCA2 repression during cardiac hypertrophy. This mechanism may have implications for the progression of hypertrophy to heart failure.
Keywords: ERK, SERCA2, NF-kappaB, neonatal rat ventricular myocytes, cardiac hypertrophy, cardiovascular disease
1. Introduction
Pathologic cardiac hypertrophy, caused by excessive adrenergic stimulation or pressure overload, can lead to heart failure. Numerous pathways have been implicated, but the mechanisms of the transition from compensated hypertrophy to heart failure are still poorly understood [1]. Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) pumps cytosolic calcium into the sarcoplasmic reticulum, which is critical for normal cardiac calcium handling. SERCA2 mRNA and protein levels are reduced in cardiac hypertrophy, and reduced SERCA2 is thought to have an important role in both systolic and diastolic heart failure pathophysiology [2, 3]. Increasing expression of SERCA2 is a promising strategy to treat heart failure [4, 5].
Neonatal rat ventricular myocytes (NRVM) treated with phenylephrine (an alpha-adrenergic receptor agonist) are a well-established model of cardiac hypertrophy [6]. Phenylephrine induces hypertrophy in NRVM and reduces SERCA2 mRNA and protein levels [7]. Alpha-adrenergic receptor stimulation activates Extracellular Signal-regulated Kinases (ERK) ½, which are serine/threonine kinases with diverse functions in mammalian cells. At baseline, it appears that ERK1/2 has some low level of activity in cardiomyocytes, and ERK activation can protect cultured ventricular myocytes from apoptosis [8]. However, chronic ERK activation appears to be involved in the pathophysiology of hypertrophy and heart failure. Transgenic ERK1/2 activation causes cardiac hypertrophy in vivo [9]. ERK is upregulated in human heart failure from both ischemic and non-ischemic causes [10]. ERK is also activated in the hearts of diet-induced obesity wild-type mice and the LMNA mutant mouse model of cardiomyopathy, and pharmacologic ERK inhibition improves cardiac function in the transgenic model [11, 12]. Further, Erk2T188S (which functions as a dominant negative for both ERK1 and ERK2) transgenic mice have a reduced hypertrophic response [13].
There has been speculation that ERK activation may decrease SERCA2 expression, suggested by the fact that overexpressing Ras (which activates ERK) in NRVM reduces SERCA2 [14]. However, Ras activates other pathways besides ERK. There has been no direct evidence demonstrating that ERK activation decreases cardiac SERCA2 transcription. Furthermore, other cell types may have the opposite relationship between ERK activation and SERCA2 expression. Macrophage from ob/ob mice have less ERK activation and less SERCA2 mRNA, which is rescued by MEK adenovirus-induced activation of ERK [15]. Thus, it is unclear if activating ERK in cardiomyocytes increases or decreases SERCA2 expression. We hypothesized that ERK activation during hypertrophy reduced SERCA transcription and sought to determine the downstream mediators.
2. Materials and Methods
2.1 Animal Care
Animal protocols were approved by the Columbia University Institutional Animal Care and Use Committee and were carried out in accordance with the NIH guidelines for the care and use of laboratory animals. Pregnant Sprague-Dawley rats were purchased from Harlan Laboratories. Neonatal rat ventricular myocytes (NRVM) were isolated from 1–3 day old pups using techniques described previously [16].
2.2 Cell Culture
NRVM were cultured in MEM with 10% FBS overnight, then in MEM without serum plus phenylephrine 100 μmol/L and propranolol 5 μmol/L for 3 days as previously described [17]. Media was changed daily. H9c2 cells were purchased from ATCC.
2.3 Chemicals and Viruses
U0126 and PD98059 were from Cell Signaling and were used at final concentrations 10 μM and 25 μM, respectively. Phenylephrine and propranolol were purchased from Sigma. The MEK adenovirus (constitutively active, ADV-119) was purchased from Cellbiolabs. The IKKBeta adenovirus (#1487) was purchased from Vector Biolabs. The GFP virus was a generous gift from Dr. Konstantinos Drosatos. Viral transduction was performed according to the manufacturers instructions.
2.4 Real-time PCR
Cells were homogenized and RNA was then purified using a Qiagen RNeasy kit (#74104). cDNA was synthesized using the Applied Biosystems high capacity RNA to cDNA kit (#4387406) and diluted to 10 ng/μL for use as a template (20 ng template was used for each 20 μL reaction). Real-time PCR was performed using an Applied Biosystems StepOne Plus Real-Time PCR system with StepOne Software v2.0 and inventoried primers from Applied Biosystems. PCR was performed for 40 cycles with automated detection of crossing threshold; the ΔΔCT method was used for relative quantification. PCR reactions were performed with duplicate wells and with the housekeeping gene ribosomal 18S as a reference.
2.5 Promoter-Luciferase Vectors, Transfection, and Mutagenesis
The mouse SERCA2 proximal promoter (3 kB region upstream from the start of transcription) was digested from a BAC template (bacpac.chori.org) using native Mlu1 and Sma1 restriction sites and ligated into the pGL3 basic vector (Promega). The human SERCA2 proximal promoter was cloned by PCR from a BAC template. Constructs were verified by DNA sequencing. Restriction enzymes and ligase were from New England Biolabs. Taq and dNTPs were from Invitrogen. The NFkB reporter luciferase construct is from Agilent technologies (#219077). The CREB1 expression vector (#22395), ERK1 expression vector (#12656), ERK2 expression vector (#8974), PKD expression vectors (#10808, 10812), NFAT expression vectors (11790, 11788), the MEK expression vector (40809), and the p50 and p65 expression vectors (#21965, #21966) were purchased from Addgene.
Vectors were co-transfected into rat cardiomyocyte H9c2 cells with a beta-gal reporter vector as a transfection control using Lipofectamine (Invitrogen). We transfected 200 nanograms of luciferase construct and 100 nanograms of protein expression vector per well; control transfection wells received empty vector pcDNA3 to equalize the amount of DNA. Standard 12-well tissue culture plates were used. Cells lysates were harvested 36–48 hrs after transfection. After cell lysis, luciferase substrate (Promega, E1501) was added to lysates in a 96-well plate and luminescence was measured with a Tecan Infinite 200 plate reader.
Mutagenesis to eliminate the NFkB binding site in the SERCA2 promoter was performed with the Agilent kit (#210515) following the manufacturer’s protocol. Mutagenesis primers were designed to substitute two base pairs in the middle of binding site to disrupt the NFkB binding sequence, as shown in the supplement.
2.6 Microscopy
Cells on glass coverslips were fixed with 4% paraformaldehyde and were visualized using a phase-contrast microscope (Nikon Eclipse Ti) with a digital camera (Photometrics Coolsnap HQ2). Surface area was measured using ImageJ software.
2.7 Bioinformatics
Transcription factor binding sites were predicted using Matinspector (http://www.genomatix.de/cgi-bin/matinspector).
2.8 Statistical Analysis
Results are presented as mean ± SEM. The unpaired t-test was used for comparisons of means; a 2-tailed value of P < 0.05 was considered statistically significant. For groups of 2 or more ANOVA was used with post-hoc testing (Prism v5, GraphPad Software).
3. Results
3.1 Phenylephrine-induced hypertrophy reduces SERCA2 mRNA and protein
We used a well-established model of hypertrophy, NRVM treated with phenylephrine (PE) [17, 18]. NRVM with PE-induced hypertrophy had larger surface area and greater expression of the hypertrophy marker Nppa (aka atrial natriuretic peptide), indicating that hypertrophy was induced (Figure 1). As previously reported, hypertrophy resulted in a significant decrease in SERCA2 mRNA (Figure 1D) and protein (Figure 1E).
Figure 1. Phenylephrine induces hypertrophy in NRVM.
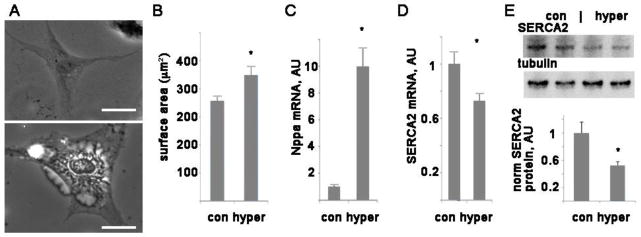
A. Representative images of control cell (top) and hypertrophied cell (bottom), scale bars are 20 μm. B. Graph of mean surface area + SEM, n=20–50 cells. C. Graph of NPPA mRNA levels, mean + SEM, arbitrary units. D. Graph of SERCA2 mRNA levels, arbitrary units. n=3, *= p<0.05 by t-test. E. Representive western blot of SERCA2, with tubulin as a loading control, and graph of protein level normalized to tubulin, average of 3 experiments each done in triplicate.
3.2 Inhibition of ERK activation preserves SERCA2 mRNA, and ERK activation reduces SERCA2 mRNA
PE causes a rapid activation of ERK in NRVM, as indicated by phosphorylation, (fig 2A). ERK phosphorylation status causes a conformational change and has been found to correlate with activity [19, 20]. To determine if ERK activation is responsible for reducing SERCA2 mRNA during hypertrophy, we inhibited MEK, the upstream kinase that activates ERK1/2, with two different pharmacologic inhibitors, PD98059 and U0126 (figure 2B). Both MEK inhibitors were able to preserve SERCA2 mRNA levels during hypertrophy, indicating that ERK1/2 activation is necessary for the reduction in SERCA2 mRNA. In addition, pharmacologic inhibition of MEK preserves SERCA2 protein levels during hypertrophy (figure 2E). In the absence of hypertrophic stimulation, treating NRVM with PD does not change the native SERCA2 mRNA levels, and U0126 by itself causes a decrease (figure 2C). This shows that the ability of these drugs to rescue native SERCA2 mRNA during hypertrophy is a specific effect. The inhibitor PD98059 is relatively specific for MEK1 (whereas U0126 inhibits MEK 1 and 2) suggesting that MEK1 is the critical upstream kinase in this pathway.
Figure 2. Phenylephrine activates ERK, and ERK activation causes a reduction in SERCA2 mRNA.

A. Representative wesern blot of phospho-ERK from NRVM treated with vehicle (control) or phenylephrine for 30 min. Commercially available antibodies recognize both ERK1 and ERK2, giving the characteristic double-band signal at 42 and 44 kDa.
B. Pharmacologic blockage of MEK preserves SERCA2 mRNA in NRVM, mean + SEM, arbitrary units. PD = PD98059 (25 μM), U0126 (10 μM). *= p<0.05 by ANOVA with post-hoc
C. PD98059 and U0126 do not increase SERCA2 mRNA in the absence of hypertrophic stimulation. *= p<0.05 by ANOVA with post-hoc
D. Activating ERK in NRVM by viral transduction with MEK1 decreases SERCA2 mRNA. GFP virus used as a control. n=3, *= p<0.05 by t-test
E. Pharmacologic blockage of MEK preserves SERCA2 protein level in NRVM, arbitrary units. PD = PD98059 (25 μM), *= p<0.05 by ANOVA with post-hoc
To determine if ERK activation is sufficient to reduce SERCA2 mRNA, we transduced NRVM with a constitutively active form of MEK, which is the most selective method for activating ERK. This resulted in a significant reduction in SERCA2 mRNA, to 37% of control viral transduction levels (figure 2D). Thus, pharmacologic inhibition of ERK activation rescues SERCA2 mRNA levels during hypertrophy, and specific activation of ERK is sufficient to reduce SERCA2 mRNA levels in cardiac myocytes.
3.3 ERK represses SERCA2 transcription
To determine if ERK reduces SERCA2 mRNA by repressing transcription, we made promoter-luciferase constructs with the mouse or human genomic DNA up to 3kB upstream from the start of transcription. The transcription factor CREB was used as a positive control since it is known to upregulate SERCA2 transcription [21]. Because NRVM have extremely low transfection efficiency with standard methods, we used the cardiomyocyte cell line H9c2 for transfection experiments. The promoter-luciferase constructs demonstrate that transcription driven by the SERCA2 promoter is significantly downregulated by ERK or MEK cotransfection (figure 3A,B). This effect is reduced by MEK inhibition (figure 3B). Further, PE reduces the activity of the SERCA2 promoter-luciferase construct, and this repression is almost eliminated by pharmacologic inhibition of MEK (figure 3C). Protein kinase D (PKD) also has a prominent role in cardiac hypertrophy [22]. However, cotransfection of WT PKD or a constitutively active mutant form of PKD did not decrease the activity of the SERCA2 promoter (figure 3D). Thus, the decrease in SERCA2 transcription is specific to MEK-ERK activation and does not necessarily occur when other hypertrophic pathways are activated.
Figure 3. ERK decreases transcription of the SERCA2 promoter.

A. Mouse SERCA2 promoter-luciferase construct show that CREB increases the transcriptional activity of the promoter and ERK1/2 decreases the transcription. All panels in this figure are representative experiments using H9c2 in triplicate, mean + SEM.
B. Mouse SERCA2 promoter-luciferase activity is decreased by cotransfection with a mutant active MEK, and this effect is reduced by MEK inhibitors. PD = PD98059, U = U0126.
C. PE significantly decreases transcription of the SERCA2 promoter, and MEK inhibition prevents the decrease.
D. Mouse SERCA2 promoter-luciferase contransfected with WT PKD, or a mutant active PKD does not have a significant change in transcription.
3.4 PE activates NFkb via ERK activation
To determine the mechanism by which ERK activation downregulates SERCA2 transcription, we cotransfected the SERCA2 promoter luciferase construct with expression vectors for the transcription factors Elk1 and fos, both of which are known targets of ERK and are believed to have a role in hypertrophy [23]. Both of these proteins increased SERCA2 promoter transcription significantly (supplemental figure 1) making it unlikely that they are responsible for the decrease in SERCA2 mRNA. NFAT transcription factors have also been implicated in cardiac hypertrophy. We cotransfected NFAT expression vectors to test the interaction of NFATs with the SERCA2-luciferase construct. We used NFAT2 and NFAT4, since these are thought to be the two major NFATs in cardiomyocytes [24]. NFAT2 increases transcription of the mouse SERCA2 promoter significantly, and NFAT4 increases transcription modestly (not significant by ANOVA post-test; supplemental figure 1C).
Next, we turned our attention to transcription factor/repressor nuclear factor-kappaB (NFkB), which has also been postulated to play a role in cardiac hypertrophy [25–27]. There is evidence from skeletal muscle that ERK activates NFkb [28]. Using an NFkb reporter-luciferase construct, we demonstrate that PE causes a significant increase in NFkb activity, which occurs by 4 hours and is sustained after overnight exposure to PE (figure 4A). MEK inhibitors blunt the increase in NFkb activity, showing that ERK activation is responsible for the PE-induced increase in NFkb activity (figure 4B). Furthermore, cotransfecting a constitutively active MEK with the NFkB reporter luciferase construct shows that MEK-ERK activation increases NFkb activity (figure 4C). This effect is reduced, but not eliminated completely, by the MEK inhibitors. The MEK inhibitors have no effect on their own.
Figure 4. PE activates NFkb via ERK activation, and NFkB represses SERCA2 transcription.

A. NFkb reporter-luciferase construct show that PE increases the transcriptional activity of the NFkb after 4 hrs or overnight. Representative experiment in H9c2 done with triplicates, mean + SEM. The means are significantly different by ANOVA; *= sig different from control by post-hoc.
B. NFkb reporter-luciferase treated with PE and MEK inhibitors for 4 hrs, PD = PD98059, U = U0126. Representative experiment in H9c2 done with triplicates, mean + SEM. The means are significantly different by ANOVA; *= sig different from control by post-hoc.
C. NFkb reporter-luciferase cotransfected with MEK, with or without MEK inhibitors. Representative experiment in H9c2 done with triplicates, mean + SEM. The means are significantly different by ANOVA; *= sig different from control by post-hoc.
D. Mouse SERCA2 promoter-luciferase cotransfected with either NFkb components p50 or p65 has a significant decrease in the transcription. Representative experiment in H9c2 done with triplicates, mean + SEM. The means are significantly different by ANOVA; *= sig different from control by post-hoc.
E. NFkB site mutant mouse SERCA2 promoter-luciferase treated with PE has an increase in transcription that is dependent on MEK/ERK activation. The means are significantly different by ANOVA; *= p<0.05 by post-hoc test.
F. IKK virus, which activates endogenous NFkB, reduces native SERCA2 mRNA in NRVM. n=3, * p< 0.05
3.5 NFkb represses SERCA2 transcription and binds to the SERCA2 promoter
To determine if NFkb could have a direct interaction with the SERCA2 promoter, we analyzed promoters with transcription-factor binding prediction software. The mouse, rat, and human SERCA2 promoters all have several predicted NFkB binding sites, determined by using Matinspector software.
To determine if activation of the NFkB protein complex can reduce SERCA2 transcription, we cotransfected the mouse SERCA2 promoter-luciferase construct with the components of NFkB. Transfection of either of the two main subunits of NFkB, p50 and p65, significantly reduces transcription of the SERCA2 promoter (figure 4D). Mutating a single predicted NFkB site in the SERCA2 promoter-luciferase vector (by changing 2 bases) eliminates the reduction in transcription with PE treatment. In fact, PE causes an increase in transcription with this mutated promoter in a MEK/ERK dependent manner (figure 4E). MEK cotransfection also causes an increase in transcription with this mutated promoter in a MEK/ERK dependent manner (sup figure 3). This indicates that this NFkB site is required for ERK to repress SERCA2 transcription, and without NFkB binding, PE and MEK have the opposite effect, increasing transcription from the SERCA2 promoter. Mutating the other predicted NFkB binding site in the core promoter, which is closer to the start codon, did not change the response to PE (supplement figure 2). This predicted site is probably a false-positive result from the algorithm. Perhaps subtleties of the DNA sequence adjacent to the predicted binding site, or methylation status, prevent transcription factor binding at this predicted site.
Selective activation of endogenous NFkB in NRVM by viral expression of IkappaB kinase (IKKB, which specifically activates the NFkB canonical pathway) causes a significant reduction in native SERCA2 mRNA (figure 4F). This shows that activating NFkB is sufficient to repress SERCA2 transcription in ventricular myocytes.
To generalize our findings beyond rodent models, we performed luciferase experiments with the human SERCA2 promoter. Treating the human SERCA2 promoter construct with PE gives results that are very similar to the mouse promoter (fig 5A). This demonstrates that the downregulation of SERCA2 transcription by ERK activation is conserved in mammalian evolution. A cartoon of the proposed pathway is depicted in figure 5B.
Figure 5. PE significantly decreases transcription of the human SERCA2 promoter and diagram of proposed pathway.
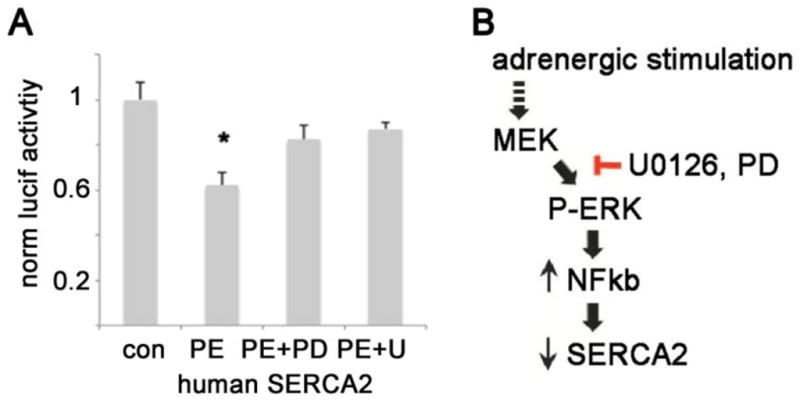
A. PE significantly decreases transcription of the human SERCA2 promoter, and MEK inhibition prevents the decrease. The means are significantly different by ANOVA; *= p<0.05 by post-hoc test. PD = PD98059, U = U0126
B. Diagram of proposed pathway, PD = PD98059
4. Discussion
Despite the importance of reductions in SERCA2 for the pathophysiology of cardiac hypertrophy and heart failure, the mechanisms responsible for reducing SERCA2 expression have not been identified. We present evidence that ERK is required for the hypertrophy-induced reduction of SERCA2 expression, and ERK activation is sufficient to reduce SERCA2 expression in cardiac myocytes. This is in contrast to other cell types, where ERK activation has been shown to increase SERCA [15].
Since NFkB has been found to have a role in cardiac hypertrophy, we tested whether ERK could be working through NFkB to repress transcription of SERCA2. We show that ERK downregulates SERCA2 expression through NFkB activation, and NFkB activation is sufficient to reduce SERCA2 expression in cardiomyocytes. This establishes novel connections between ERK, NFkB, and SERCA2 expression, improving our understanding of the pathways involved in cardiac hypertrophy. Further, we show that ERK activation has multiple downstream effects, many of which increase SERCA2 transcription, but the repression effect predominates in cardiomyocytes. By mutating a single NFkB binding site in the SERCA2 promoter, alpha-adrenergic stimulation changes from causing repression to the opposite, increasing SERCA2 transcription. This NFkB binding site in the SERCA2 promoter can be seen as a transcriptional switch. The NFkB site makes alpha-adrenergic stimulation inhibitory despite the fact that ERK activates other transcription factors that increase SERCA2 transcription. Different cell types could have different transcriptional responses to adrenergic stimulation and ERK activation depending on the relative abundance of NFkB and the other transcription factors that downstream of ERK.
The decrease in SERCA2 expression mediated by ERK and NFkB may have an important role in the transition from cardiac hypertrophy to heart failure. Although NFkB is mostly thought of in term of inflammatory pathways and apoptosis, it has other roles as well. In the neuroscience literature, NFkB has been shown to have a role in synaptic plasticity and long-term memory [29, 30]. The potential for NFkB activation to cause electrophysiologic remodeling of ion channels in cardiomyocytes has only recently been appreciated [17, 31]. Thus, inhibition of NFkB may have multiple beneficial effects in cardiac tissue.
Besides hypertrophy, the repression of SERCA2 transcription by NFkB may be involved in other forms of heart disease. NFkB is activated in human heart failure from diverse etiologies [32]. Genetic inhibition of NFkB improves survival and systolic function after myocardial infarction in a mouse model [33]. Although the benefit was attributed to reduced inflammation and apoptosis in the transgenic mouse heart, SERCA2 expression was not examined.
Limitations
It is technically challenging to maintain adult cardiomyocytes in culture without rapid decreases in viability. NRVM have the advantage of being relatively easy to culture and have been used as a model system of hypertrophy for decades. However, they may differ from adult cardiomyocytes in important ways, including signal transduction. Transcriptional regulation is fundamental to most proteins, but essential proteins such as SERCA2 are often regulated at multiple levels. Another group has recently published that SERCA2 is regulated by a microRNA, for example [34]. This project is focused on transcriptional regulation of SERCA2, but the fact that the SERCA2 protein level is decreased more than the mRNA level may indicate that protein stability and/or regulation of translation are involved as well.
In conclusion, we demonstrate that ERK activation represses SERCA2 transcription during cardiac hypertrophy via NFkB activation. Identification of the pathway involved in the downregulation of SERCA2 transcription may allow for targeted small-molecule therapy to improve SERCA2 expression in patients with heart disease, which would be expected to improve systolic function.
Supplementary Material
Highlights.
Cardiac hypertrophy can lead to heart failure; the mechanisms involved are poorly understood.
Extracellular Signal-regulated Kinase (ERK) is activated during hypertrophy.
ERK activation is sufficient to reduce SERCA2 mRNA.
ERK represses SERCA2 transcription via nuclear factor-kappaB (NFkB)
Activation of NFkB is sufficient to reduce SERCA2 mRNA in cardiomyocytes
Acknowledgments
JPM is supported by NIH grant 1K08HL105801 and the Lewis Katz Prize in Cardiovascular Research. EBT is supported by 1R01HL122309.
Footnotes
Disclosures: The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 2.Andersson KB, Birkeland JA, Finsen AV, Louch WE, Sjaastad I, Wang Y, et al. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J Mol Cell Cardiol. 2009;47:180–7. doi: 10.1016/j.yjmcc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–5. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling--targets for heart failure therapy. Nat Rev Cardiol. 2012;9:717–33. doi: 10.1038/nrcardio.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suarez J, Scott B, Dillmann WH. Conditional increase in SERCA2a protein is able to reverse contractile dysfunction and abnormal calcium flux in established diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1439–45. doi: 10.1152/ajpregu.00736.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glembotski CC. Classic studies of cultured cardiac myocyte hypertrophy: interview with a transformer. Circ Res. 2013;113:1112–6. doi: 10.1161/CIRCRESAHA.113.302490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prasad AM, Ma H, Sumbilla C, Lee DI, Klein MG, Inesi G. Phenylephrine hypertrophy, Ca2+-ATPase (SERCA2), and Ca2+ signaling in neonatal rat cardiac myocytes. Am J Physiol Cell Physiol. 2007;292:C2269–75. doi: 10.1152/ajpcell.00441.2006. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, et al. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation. 2007;115:763–72. doi: 10.1161/CIRCULATIONAHA.106.664862. [DOI] [PubMed] [Google Scholar]
- 9.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–50. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nediani C, Borchi E, Giordano C, Baruzzo S, Ponziani V, Sebastiani M, et al. NADPH oxidase-dependent redox signaling in human heart failure: relationship between the left and right ventricle. J Mol Cell Cardiol. 2007;42:826–34. doi: 10.1016/j.yjmcc.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Amin V, Gurin M, Wan E, Thorp E, Homma S, et al. Diet-induced obesity causes long QT and reduces transcription of voltage-gated potassium channels. J Mol Cell Cardiol. 2013;59C:151–8. doi: 10.1016/j.yjmcc.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–7. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidal M, Wieland T, Lohse MJ, Lorenz K. beta-Adrenergic receptor stimulation causes cardiac hypertrophy via a Gbetagamma/Erk-dependent pathway. Cardiovasc Res. 2012;96:255–64. doi: 10.1093/cvr/cvs249. [DOI] [PubMed] [Google Scholar]
- 14.Ho PD, Zechner DK, He H, Dillmann WH, Glembotski CC, McDonough PM. The Raf-MEK-ERK cascade represents a common pathway for alteration of intracellular calcium by Ras and protein kinase C in cardiac myocytes. J Biol Chem. 1998;273:21730–5. doi: 10.1074/jbc.273.34.21730. [DOI] [PubMed] [Google Scholar]
- 15.Liang CP, Han S, Li G, Tabas I, Tall AR. Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin-resistant macrophages and are reversed by exenatide treatment. Diabetes. 2012;61:2609–20. doi: 10.2337/db11-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo J, Sabri A, Elouardighi H, Rybin V, Steinberg SF. Alpha1-adrenergic receptors activate AKT via a Pyk2/PDK-1 pathway that is tonically inhibited by novel protein kinase C isoforms in cardiomyocytes. Circ Res. 2006;99:1367–75. doi: 10.1161/01.RES.0000252830.01581.fd. [DOI] [PubMed] [Google Scholar]
- 17.Panama BK, Latour-Villamil D, Farman GP, Zhao D, Bolz SS, Kirshenbaum LA, et al. Nuclear factor kappaB downregulates the transient outward potassium current I(to,f) through control of KChIP2 expression. Circ Res. 2011;108:537–43. doi: 10.1161/CIRCRESAHA.110.229112. [DOI] [PubMed] [Google Scholar]
- 18.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–9. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Zhang F, Ebert D, Cobb MH, Goldsmith EJ. Activity of the MAP kinase ERK2 is controlled by a flexible surface loop. Structure. 1995;3:299–307. doi: 10.1016/s0969-2126(01)00160-5. [DOI] [PubMed] [Google Scholar]
- 20.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–9. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moriscot AS, Sayen MR, Hartong R, Wu P, Dillmann WH. Transcription of the rat sarcoplasmic reticulum Ca2+ adenosine triphosphatase gene is increased by 3,5,3′-triiodothyronine receptor isoform-specific interactions with the myocyte-specific enhancer factor-2a. Endocrinology. 1997;138:26–32. doi: 10.1210/endo.138.1.4857. [DOI] [PubMed] [Google Scholar]
- 22.Fielitz J, Kim MS, Shelton JM, Qi X, Hill JA, Richardson JA, et al. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci U S A. 2008;105:3059–63. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babu GJ, Lalli MJ, Sussman MA, Sadoshima J, Periasamy M. Phosphorylation of elk-1 by MEK/ERK pathway is necessary for c-fos gene activation during cardiac myocyte hypertrophy. J Mol Cell Cardiol. 2000;32:1447–57. doi: 10.1006/jmcc.2000.1185. [DOI] [PubMed] [Google Scholar]
- 24.Bourajjaj M, Armand AS, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, et al. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J Biol Chem. 2008;283:22295–303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 25.Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A. 2001;98:6668–73. doi: 10.1073/pnas.111155798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawano S, Kubota T, Monden Y, Kawamura N, Tsutsui H, Takeshita A, et al. Blockade of NF-kappaB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc Res. 2005;67:689–98. doi: 10.1016/j.cardiores.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–86. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho RC, Hirshman MF, Li Y, Cai D, Farmer JR, Aschenbach WG, et al. Regulation of IkappaB kinase and NF-kappaB in contracting adult rat skeletal muscle. Am J Physiol Cell Physiol. 2005;289:C794–801. doi: 10.1152/ajpcell.00632.2004. [DOI] [PubMed] [Google Scholar]
- 29.Oikawa K, Odero GL, Platt E, Neuendorff M, Hatherell A, Bernstein MJ, et al. NF-kappaB p50 subunit knockout impairs late LTP and alters long term memory in the mouse hippocampus. BMC neuroscience. 2012;13:45. doi: 10.1186/1471-2202-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–54. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, et al. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. 2008;294:C372–9. doi: 10.1152/ajpcell.00186.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong SC, Fukuchi M, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–3. doi: 10.1161/01.cir.98.2.100. [DOI] [PubMed] [Google Scholar]
- 33.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–38. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wahlquist C, Jeong D, Rojas-Munoz A, Kho C, Lee A, Mitsuyama S, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014 doi: 10.1038/nature13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.