

Abstract
Activation of RhoA, a low molecular-weight G-protein, plays an important role in protecting the heart against ischemic stress. Studies using non-cardiac cells demonstrate that the expression and subsequent secretion of the matricellular protein CCN1 is induced by GPCR agonists that activate RhoA. In this study we determined whether and how CCN1 is induced by GPCR agonists in cardiomyocytes and examined the role of CCN1 in ischemic cardioprotection in cardiomyocytes and the isolated perfused heart.
Methods and results
In neonatal rat ventricular myocytes (NRVMs), S1P, lysophosphatidic acid (LPA) and endothelin-1 induced robust increases in CCN1 expression while phenylephrine, isoproterenol and carbachol had little or no effect. The ability of agonists to activate the small G-protein RhoA correlated with their ability to induce CCN1. CCN1 induction by S1P was blocked when RhoA function was inhibited with C3 exoenzyme or a pharmacological RhoA inhibitor. Conversely overexpression of RhoA was sufficient to induce CCN1 expression. To delineate the signals downstream of RhoA we tested the role of MRTF-A (MKL1), a co-activator of SRF, in S1P-mediated CCN1 expression. S1P increased the nuclear accumulation of MRTF-A and this was inhibited by functional inactivation of RhoA. In addition, pharmacological inhibitors of MRTF-A or knockdown of MRTF-A significantly diminished S1P-mediated CCN1 expression, indicating a requirement for RhoA/MRTF-A signaling. We also present data indicating that CCN1 is secreted following agonist treatment and RhoA activation, and binds to cells where it can serve an autocrine function To determine the functional significance of CCN1 expression and signaling, simulated ischemia/reperfusion(sI/R)-induced apoptosis was assessed in NRVMs. The ability of S1P to protect against sI/R was significantly reduced by inhibition of RhoA, ROCK or MRTF-A or by CCN1 knockdown. We also demonstrate that ischemia/reperfusion induces CCN1 expression in the isolated perfused heart and that this functions as a cardioprotective mechanism, evidenced by the significant increase in infarct development in response to I/R in the cardiac specific CCN1 KO relative to control mice.
Conclusion
Our findings implicate CCN1 as a mediator of cardioprotection induced by GPCR agonists that activate RhoA/MRTF-A signaling.
Keywords: CCN1, RhoA, MRTF-A, GPCRs, cardioprotection
1. Introduction
CCN1 (also known as Cyr61) was first identified as an immediate early gene upregulated by growth factor stimulation and subsequently shown to be induced in response to a wide range of extracellular stimuli [1, 2]. CCN1, classified as a matricellular protein, is secreted from cells and serves to regulate diverse responses including cell migration, proliferation, angiogenesis, senescence and cell survival. CCN1 is a multi-domain protein which includes a number of distinct integrin binding sites [2, 3]. CCN1 binding to integrins mediates the majority of its diverse, and at times opposing cellular effects [2, 4-7]. We have demonstrated that activation of GPCRs by lysophospholipids (S1P and LPA) or thrombin leads to robust induction of CCN1 expression in glioblastoma cells and that this is mediated through activation of RhoA [6, 8]. RhoA involvement in S1P induced CCN1 induction has also been demonstrated in other glioma cells lines [9, 10] and in stretch-induced responses of smooth muscle cells [11, 12].
RhoA is best recognized as transducer of signals for actin cytoskeletal rearrangement. A critical, albeit less appreciated role, for RhoA is in transcriptional regulation, as first discovered through effects of RhoA activation on serum response factor (SRF) target gene expression [13] SRF, a widely expressed member of the MADS (MCM-1, Agamous, and Deficients, SRF) box superfamily, is constitutively localized to the nucleus and bound to SRE sequences [14, 15]. Transcriptional activity of SRF is regulated through its association with other transcriptional co-activators which provide combinatorial control of SRF target genes [16, 17]. To date, two major families of coactivators are known to activate SRF, the ternary complex factors (TCFs) and the myocardin-related transcription factors (MRTFs; also known as MAL or MKL). The effect of RhoA on SRF dependent genes is mediated through a TCF-independent mechanism [15]. Recent seminal studies demonstrated that the myocardin family proteins MRTF-A/B provide the link between RhoA-dependent cytoskeletal regulation and SRF-dependent gene expression [16, 18-20]. Mechanistically, MRTF-A associates with G-actin and is thus sequestrated in the cytoplasm under resting conditions. Serum stimulation and signals that activate RhoA to promote actin polymerization lead to MRTF-A dissociation from G-actin, whereupon it translocates into the nucleus and triggers activation of SRF target genes. MRTF-A activation was recently implicated in the ability of mechanical stretch to induce RhoA-mediated CCN1 gene expression in smooth muscle cells [11]. In the heart, deletion of MRTF-A has been shown to decrease cardiac hypertrophic responses induced by pressure overload or angiotensin II (Ang-II) [21], consistent with our early findings on RhoA involvement in hypertrophic ANF gene expression in cardiomyocytes [22, 23].
Relatively little is known about the regulation or functional role of CCN1 in cardiomyocytes. Global CCN1 gene deletion results in embryonic lethality associated with altered cardiac development [24] and Drexler's laboratory reported that CCN1 expression is highly upregulated in the myocardium of patients with heart failure or ischemic myopathy [25]. CCN1 appears to serve as a survival signal for cardiomyocytes by activating kinases such as Akt and ERK that protect against oxidative stress [7]. Conversely CCN1 has been shown to sensitize to apoptosis induced by TNFα or Fas ligand [4, 5] but this depends on specific integrin binding sites [4, 5] and is context dependent [2, 5, 7, 26].
We and others have demonstrated that S1P and RhoA signaling confers cardioprotection [27-30]. It is not known whether CCN1 signaling contributes to this response. In the present study we demonstrate that CCN1 is induced in cardiomyocytes by S1P and other agonists that activate RhoA, that this occurs through MRTF-A signaling, and that CCN1 confers cardioprotection against ischemia/reperfusion injury both in cardiomyocytes and the isolated perfused heart based on findings using mice in which cardiac CCN1 is genetically deleted.
2. Materials and Methods
2.1. Materials
S1P and LPA were purchased from Avanti Polar Lipids (Alabaster, AL, USA), prepared according to manufactory instruction. C3 exoenzyme was obtained from Cytoskeleton, Inc. Antibodies against CCN-1, RhoA, MRTF-A and α-actinin were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-RhoGDI and anti-Lamin A/C were purchased from Cell Signaling Technology (Danvers, MA, USA). CCG-1423 and CCG-203,971 were kindly provided by Dr. Scott Larsen (University of Michigan). All other chemicals and reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated.
2.2. Cell culture
Neonatal rat ventricular myocytes (NRVMs) were prepared from 1 to 3 day old Sprague-Dawley rat pups as described previously [31]. All procedures were performed in accordance with NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee. Cardiomyocytes were plated at a density of 1.0 × 106 per 6 cm dish or 3.5 × 106 per 10 cm dish in 15% fetal bovine serum containing Dulbecco-modified Eagle's medium (DMEM) overnight. The cells were washed and the medium replaced with serum-free DMEM. All experiments were performed after 24 hours of serum starvation.
2.3. Quantitative RT-PCR for CCN1 mRNA
Quantitative RT-PCR was carried out as described previously[32]. Briefly, RNA was extracted using Trizol (Ambion), cDNA synthesis was carried out with the Verso cDNA synthesis kit (Thermo Scientific) and qRT-PCR was carried out using standard Taman primers and TaqMan Universal Mastermix II (Applied Biosystems) on a 7500 Fast Real-Time PCR system (Applied Biosystems). Fold difference was calculated according to the comparative CT (2−ΔΔCt) method using GAPDH as a control.
2.4. Whole cell lysate preparation and Western blot analysis
Whole cell lysate was prepared and Western blot analysis was performed using Invitrogen NuPage system, as described previously [31]. All primary antibodies were diluted 1:1000 in 5% BSA and secondary immunoglobulin G –horseradish peroxidase at 1:3500 in 5% non-fat milk. Data was processed and quantitated using gel documentation software, AlphaEaseFC (Alpha Innotech Corp, CA, USA).
2.5. Nuclear protein extraction
Nuclear protein was isolated from NRVMs as described previously [31]. Briefly, the cells were lysed in ice-cold Buffer C, containing 10 mM HEPES (pH7.6), 10 mM NaCl, 1.5 mM MgCl2, 10% glycerol, 0.1% NP-40, 1 mM phosphatase and protease inhibitors. The samples were kept on ice for 15min and by centrifugation at 2600 rcf for 5 min. The pellet was washed twice then lysed in high salt RIPA buffer. The samples were centrifuged at 21,000 rcf for 15 min and the supernatant containing extracted nuclear protein were collected.
2.6. Rho activation assay
The assay for activated RhoA was carried out as described previously [33]. Briefly, cell lysate was incubated with Rho binding domain of rhotekin and then subjected to series of washes and centrifugations. 4X Laemmli buffer was added and boiled for 5 min prior to SDS-PAGE analysis. Activated GTP-bound RhoA was detected by Western blotting for RhoA and normalized to total RhoA in lysate.
2.7. siRNA knockdown
Pre-designed CCN1 ON-TARGETplus siRNA for rat (catalog number; L-099437-01), MRTF-A ON-TARGETplus siRNA for rat (catalog number; L-081405-00) and control siRNA (catalog number; D-001810-02) were purchased from Thermo Scientific. NRVMs were transfected with siRNA using DharmaFECT-I transfection reagent (Thermo Scientific) as previously described [31]. After overnight incubation, cells were washed and cultured for another 48 hours in serum free DMEM.
2.8. Immunofluorescence
Myocytes were fixed in 4 % formaldehyde, permeabilized in 0.1% Triton-X 100, and blocked in 3% bovine serum albumin. Cells were incubated with a primary antibody against MRTF-A (Santa Cruz. sc-32909) overnight at 4 °C, followed by Alexa Fluor 488-conjugated secondary antibody for 1 hour at room temperature. DAPI was also added to visualize myocyte nuclei. Images were visualized by confocal microscopy (Olympus FluoView FV1000 confocal microscope).
2.9. Simulated Ischemia/Reperfusion (sI/R) and Apoptosis assay
Myocytes were incubated with simulated ischemia solution, which contained (mM/L) NaCl 140, KCl 12, MgCl2 1, HEPES 10 and CaCl2 2, (pH 6.5 and saturated with 95% N2 and 5% CO2) for 4 hours, washed with DMEM, and cultured for 20 hours. DNA fragmentation, an indicative of apoptosis, was assayed using the cell death detection ELISAPLUS (Roche Applied Science; catalog number/11 774 425 001) as previously described [34].
2.10. Generation of cardiac specific CCN1 knockout mice
Ccn1flox/+ mice were backcrossed to C57BL/6 mice >10 times [35]. The floxed mice were crossed with αMHC-Cre mice to generate cardiomyocyte-specific CCN1 knockout mice (CCN1fl/fl, αMHC-Cre). Offspring were born with expected Mendelian frequency and showed no overt cardiac abnormalities or echocardiographic differences from WT or control (CCN1fl/fl) mice for up to 2 months of age.
2.11 Ischemia/reperfusion in the isolated perfused mouse heart
Hearts were rapidly excised, washed in ice-cold modified Krebs-Henseleit solution (118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 25 mM NaHCO3, 0.5 mM EDTA, 1.2 mM MgSO4, 11 mM glucose, 1.5 mM Na-Pyruvate, and 2 mM CaCl2), mounted on a Langendorff apparatus and perfused with oxygenated Krebs-Henseleit buffer at 37 °C at a constant pressure of 80 mmHg. Hearts were perfused for 20 min to allow for equilibration and subjected to no-flow ischemia for 30 min followed by reperfusion for 60 min. To measure infarct size, the ventricles were then frozen and cut transversely into 5 slices of equal thickness. The slices were then incubated in 1% 2,3,5-triphenyltetrazolium chloride (TTC) in PBS and fixed in 10% formalin-PBS for 24 hours. Fixed slices were then scanned, and ImageJ was used to measure and calculate the size of the infarct size and the total area.
2.12. Statistical analysis
Results are reported as averages ± SEM. Statistical significance was determined using ANOVA followed by the Tukey post hoc test. Comparisons of two groups were accomplished using unpaired Student's t test. P<0.05 was considered statistically significant.
3. Results
3.1. Differential effects of GPCR agonists on CCN1 expression in neonatal rat ventricular myocytes
The cardioprotective lysophospholipid S1P was tested for its ability to induce CCN1 expression in neonatal rat ventricular myocytes (NRVMs). CCN1 mRNA assessed by quantitative RT-PCR increased within 15 min after S1P treatment, reached a peak at 30 min and declined to basal levels by 2 hours (Fig. 1A). S1P treatment also lead to robust increases in CCN1 protein in whole cell lysates which were significant as early as 30 min and sustained over 24 hours (Fig. 1B). We then compared the ability of other GPCR agonists to induce CCN1 expression in cardiomyocytes. Robust increases in CCN1 expression were observed after 1 hour treatment with S1P, LPA and endothelin-1 (ET-1)(Fig. 1C). The effects of another group of agonists shown to activate signaling pathways regulating hypertrophy or cardiac contractility in NRVMs were also tested. This included phenylephrine (PE), isoproterenol (ISO) and carbachol (CRB) which activate α-adrenergic, β-adrenergic and muscarinic cholinergic receptors respectively. Myocytes treated with these agonists showed little to no increase in CCN1 expression at 1 hour (Fig. 1C), or at times up to 24 hours (not shown).
Figure 1.
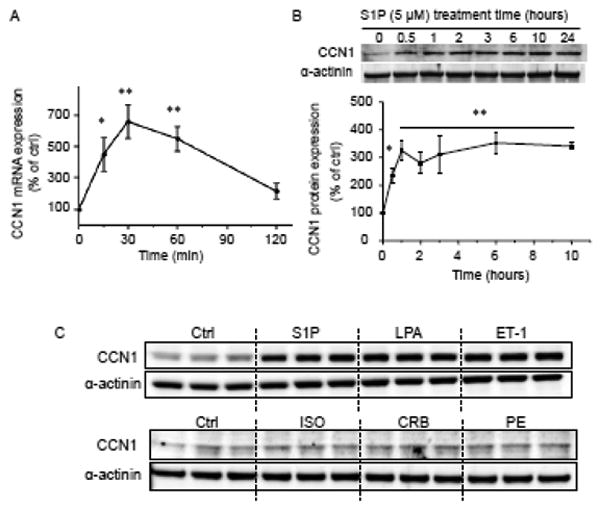
NRVMs were serum-starved for 24 hours prior to agonist stimulation. A) Cells were treated with 5 μM S1P and mRNA was isolated and subjected to quantitative RT-PCR analysis (n=5). *, **; P<0.05, 0.01 vs. control. B) Cells were treated with S1P, lysed at various time points and subjected to SDS-PAGE followed by Western blot for CCN1 or α-actinin. Representative blot and quantitative analysis of S1P-induced CCN1 induction are shown (n=4). C) NRVMs were stimulated with 5 μM S1P, 10 μM LPA, 100 nM ET-1, 1 μM ISO, 100 μM CRB, or 100 nM PE for 1 hour and subjected to Western blot.
3.2. RhoA involvement in agonist-induced CCN1 expression
We next examined the potential importance of RhoA activation in CCN1 expression in NRVMs. RhoA activation was examined by immunoprecipitating the GTP-bound activated form of RhoA using the Rho effector rhotekin. Among the agonists tested, S1P elicited the most robust RhoA activation, followed by LPA and ET-1. In contrast, PE, CRB and ISO showed little or no ability to activate RhoA (Fig. 2A). Thus RhoA activation is closely correlated with the ability of agonists to induce CCN1 expression in NRVMs. The contribution of RhoA activation to CCN1 induction was examined more directly in experiments using the C3 exoenzyme, which ADP ribosylates and functionally inactivates RhoA [36]. The ability of C3 exoenzyme treatment (2 μg/ml) to inhibit Rho activation induced by S1P was first confirmed (Supplemental Fig. 1). Cardiomyocytes were pretreated with C3 exoenzyme, stimulated with agonists for 1 hour and CCN1 expression assessed by Western blotting. Figures 2B and C demonstrate that C3 exoenzyme treatment blocks CCN1 expression in response to S1P, LPA and ET-1 without affecting basal CCN1 expression. CGX0287, a RhoA selective inhibitor [37] was also found to prevent S1P-mediated CCN1 expression in NRVMs (Fig. 2B). ROCK, a downstream effector of RhoA, was also demonstrated to participate in CCN1 induction by S1P since the pharmacological inhibitor Y-27632 blocked S1P-induced CCN1 expression (Fig. 2D), as observed by others [6, 38]. To determine if RhoA activation was sufficient as well as necessary for CCN1 expression, we demonstrated that adenoviral overexpression of RhoA elicits significant increases in CCN1 expression (Fig. 2E). These composite data provide multiple lines of evidence that GPCRs signal through RhoA activation to increase CCN1 expression in NRVMs.
Figure 2.
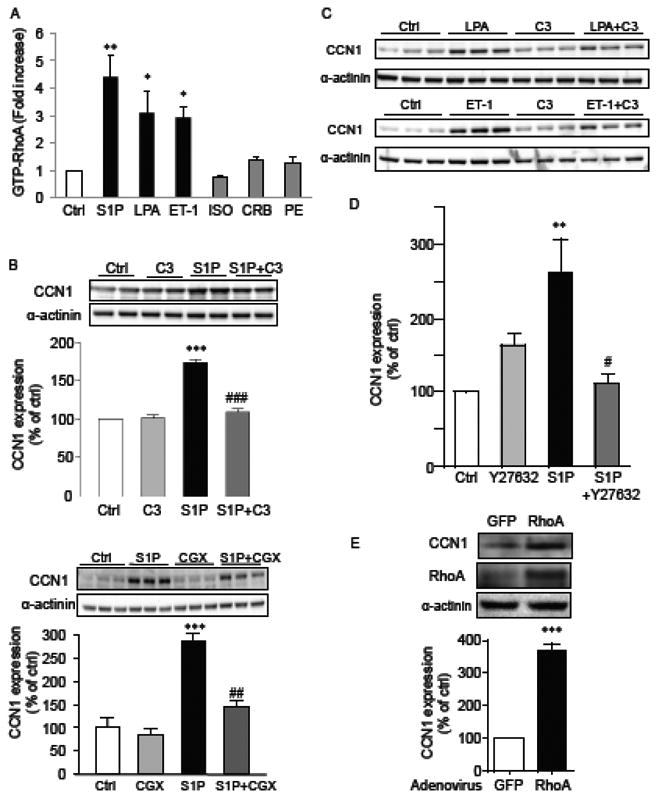
A) NRVMs were stimulated with S1P (5 μM), LPA (10 μM), ET-1 (100 nM), ISO (1 μM), CRB (100 μM), or PE (100 nM) for 5 min. RhoA activity was assessed by RBD pull down assay as described in Materials and Methods. *,**; P<0.05, 0.01 vs. control (n=4). B) Blocking RhoA function decreases CCN1 induction by S1P. Cells were treated with 2 μg/ml C3 exoenzyme overnight or with 30 μM CGX 0287 for 30 min prior to the addition of 5 μM S1P. After 1 hour of S1P treatment, cells were harvested and cell lysates were subjected to CCN1 Western blotting. Bar graph shows quantitated results from 3 independent experiments (n=5-7). *** P<0.001 vs. control. ##, ### P<0.01, P<0.001 vs. S1P alone. C) Cells were treated with 2 μg/ml C3 exoenzyme overnight, stimulated with 10 μM LPA or 100 nM ET-1 for 1 hour, lysed and analyzed by Western blotting for CCN1 expression. D) NRVMs were treated with 5 μM Y-27632 for 30 min prior to S1P treatment and CCN1 expression was assessed by Western blotting (n=5). **; P<0.01 vs. control, #; P<0.05 vs. S1P alone. E) NRVMs were infected with L63RhoA adenovirus or control adenovirus (AdCMV). Cell lysates were prepared 24 hours after adenovirus washout and CCN1 levels were assessed by Western blotting. *** P<0.001 vs. control (GFP), n=4.
3.3. S1P induces MRTF-A nuclear accumulation through RhoA
MRTF-A, a transcriptional co-activator for SRF, is activated through RhoA signaling and now recognized to mediate RhoA effects on transcription of various genes. To determine whether MRTF-A plays a role in CCN1 induction in cardiomyocytes we first asked whether S1P treatment causes nuclear accumulation of MRTF-A. NRVMs were stimulated with S1P and the nuclear fraction isolated and subjected to Western blotting. As shown in Figure 3A, MRTF-A increased in the nuclear fraction in a time dependent manner following S1P treatment. Increased nuclear MRTF-A was evident by 30 min and significant by 60 min, and the increase was prevented by C3 exoenzyme pretreatment (Fig. 3B) indicating the RhoA dependence of nuclear MRTF-A accumulation. Immunohistochemical analysis also demonstrated that MRTF-A retention in cytosol was decreased by S1P treatment in a C3 exoenzyme-dependent manner (Fig. 3C).
Figure 3.
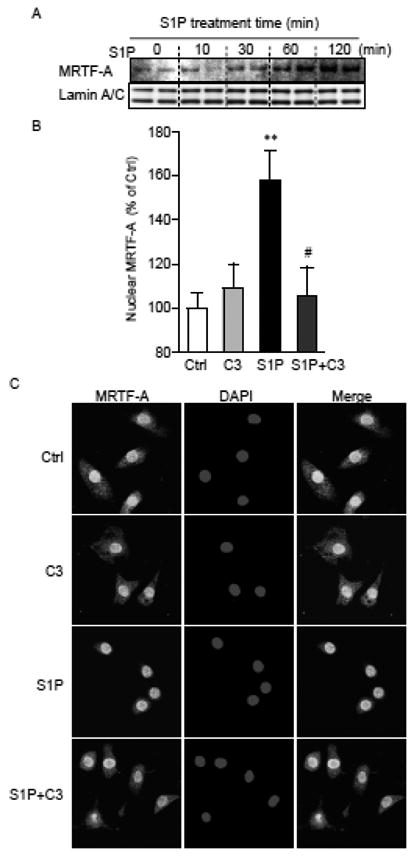
A) NRVMs were treated with 5 μM S1P for various times, lysed, fractionated and nuclear proteins extracted for Western blotting for MRTF-A and Lamin A/C (loading control). B) Cells were treated with 2 μg/ml C3 exoenzyme overnight followed by stimulation with 5 μM S1P. After 1 hour incubation, nuclear proteins were extracted and analyzed for MRTF-A by Western blotting (n=5-6). **; P<0.01 vs. control. #; P<0.05 vs. S1P alone. C) After 1 hour treatment with S1P, cells were fixed, permeabilized, and subject to nuclear (DAPI) and MRTF-A staining. Representative confocal images from 3 independent experiments are shown.
3.4. Inhibition of MRTF-A attenuates S1P-induced CCN1 expression
We next determined whether MRTF-A activation is responsible for agonist-induced CCN1 expression in NRVMs. CCG-1423 is a small molecule inhibitor discovered through a transcription-based high-throughput SRE-luciferase screening assay [39]. This inhibitor was recently shown to prevent Rho-mediated increases in SRF mediated transcription by blocking MRTF-A activation [39-42]. In cells treated with 30 μM CCG-1423 for 30 min prior to S1P stimulation, the S1P-induced increase in nuclear MRTF-A (Fig. 4A) and decrease in cytosolic sequestration of MRTF-A (Fig. 4B) were inhibited. CCG-203,971, a derivative of CCG-1423 [40], had a similar effect (Fig. 4B). Treatment with CCG-1423 reduced CCN1 expression in a dose-dependent manner (Fig. 4C) and CCG-203,971 was an even more potent and effective inhibitor of CCN1 induction by S1P (Fig. 4D). To further demonstrate the contribution of MRTF-A to CCN1 induction, we pretreated cells for 48 hrs with control or MRTF-A siRNA, resulting in a greater than 50% decrease in MRTF-A expression. MRTF-A knockdown significantly decreased CCN1 expression upon S1P stimulation (Fig. 4E), supporting the conclusion that MRTF-A is required for S1P/RhoA-induced CCN1 expression.
Figure 4.
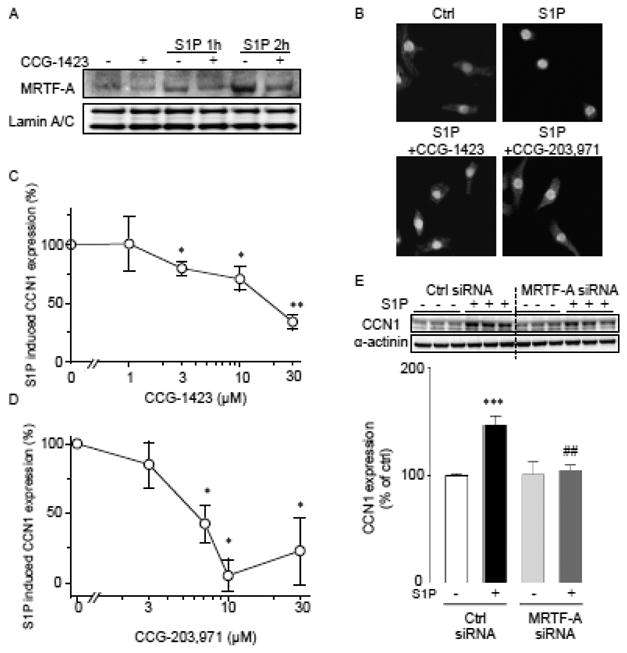
A) Cells were treated with 30 μM CCG1423 for 30 min prior to 5 μM S1P stimulation. Cells were fractionated and nuclear proteins extracted for analysis of MRTF-A levels. Lamin A/C was used as nuclear loading control. B) Confocal images of cells stained for MRTF-A immunolocalization. Cells were pre-treated with CCG-1423 (30 μM) or CCG-203,971 (10 μM) for 30 min prior to S1P treatment. C, D) Dose-dependent effects of CCG-1423 and CCG-203,971 on S1P-induced CCN1 expression. Treatment with CCG compounds was for 30 min prior to S1P addition (n=5). *,** P<0.05, P<0.01 vs. control. E) Effect of siRNA-mediated MRTF-A knockdown on CCN1 induction by S1P. Cells were transfected with MRTF-A siRNA or control siRNA for 48 hours prior to treatment with 5 μM S1P for 1 hour (n=5). ***; P<0.001 vs. control, ##; P<0.01 vs. S1P alone.
3.5. CCN1 is secreted and binds to the plasma membrane
CCN1 is a heparin-binding matricellular protein. To detect CCN1 accumulated outside the cells in response to S1P treatment, cells were exposed to S1P for 4 hours and soluble heparin plus agarose bead-bound heparin were added to the medium for an additional hour. As shown previously heparin competes for CCN1 bound to the cell surface and the CCN1 accumulated outside the cell can be collected by using heparin agarose beads [6, 43]. CCN1 bound to the beads was collected and subject to Western blot analysis (Fig. 5). S1P treatment lead to robust increases in CCN1 in the media. CCN1 remaining in the whole cell lysate was undetectable after heparin and heparin-beads treatment, suggesting that nearly all of the CCN1 generated in response to S1P exits from the cells. The accumulation of CCN1 in the extracellular space following S1P treatment was significantly diminished by inhibition of Rho with C3 (Fig. 5A), ROCK with Y-27632 or MRTF-A with CCG-203,971 (Fig. 5B).
Figure 5.
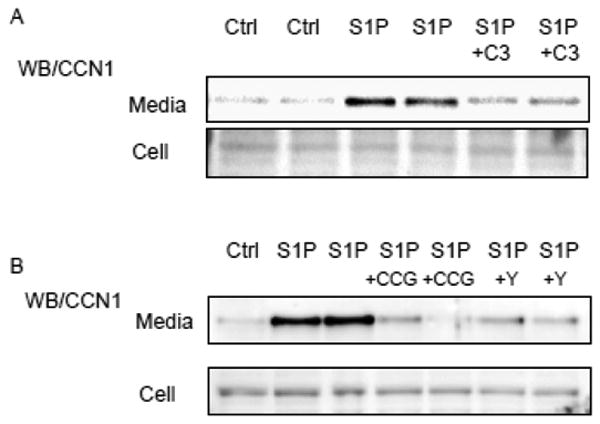
NRVMs were treated with S1P for 4 hours with or without inhibitors, and 5 U/ml heparin and 50 μl of heparin beads were subsequently added to the media for an additional hour. The beads were collected from media, resuspended in LDS buffer and boiled to remove bound CCN1 from the beads. Cells were also harvested in RIPA buffer from the dishes. Proteins bound to beads and in whole cell lysates were resolved by SDS-PAGE and Western blot using an anti-CCN1 antibody. Some cells were treated with C3 (2 μg/ml ), Y-27632 (Y)(5 μM ) or CCG-203,971 (CCG)(10 μM) to S1P treatment.
3.6. CCN1 contributes to the protective effect of S1P in NRVMs
Our group previously demonstrated a cardioprotective role for S1P in the heart [28, 30]. The hypothesis that RhoA-mediated MRTF-A activation and CCN1 expression serve as mediators of the cardioprotective effect of S1P was therefore examined. NRVMs treated with CCN1 siRNA showed decreased basal CCN1 and no significant S1P-stimulated increase in CCN1 expression (Fig. 6A). These cells, along with cells in which RhoA, ROCK and MRTF-A activation were pharmacologically inhibited (with CGX0287, Y-27632 and CCG-203,971, respectively) were subjected to simulated ischemia/reperfusion (sI/R) and apoptosis assessed (Fig. 6B). The effect of sI/R was significantly attenuated by S1P treatment (white bar), confirming that S1P is cardioprotective. This protection was partially but significantly blocked by CCN1 knockdown. Blocking RhoA, ROCK or MRTF-A activation also diminished the protective effect of S1P. These results indicate that RhoA/ROCK-dependent MRTF-A activation and CCN1 induction contribute to S1P-mediated cardioprotection.
Figure 6.
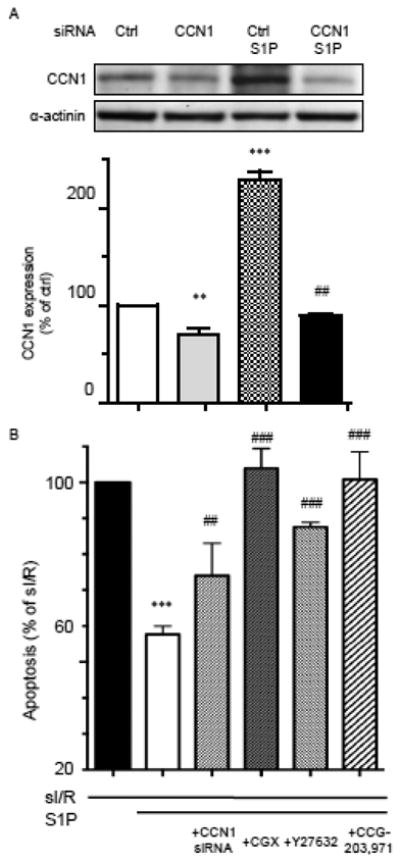
A) NRVMs were transfected with control or CCN1 siRNA, incubated for 48 hours and treated with 5 μM S1P for 1 hour. Inhibition of S1P-induced CCN1 expression is shown. **, ***; P<0.01, P<0.001 vs siCtrl, ##; P<0.01 vs. siCtrl+S1P (n=5). B) NRVMs treated with CCN1 siRNA for 48 hours, with CGX 0287 (30 μM) for 30 min, with Y-27632 (5 μM ) for 30 min or with CCG-203,971 (5 μM) for 30 min were subjected to simulated ischemia for 4 hours and reperfusion for 20 hours with or without addition of 5 μM S1P. The ability of S1P to decrease apoptosis was assessed by an ELISA-based DNA fragmentation assay. Inhibition of CCN1 decreases S1P-mediated cardioprotection against simulated ischemia/reperfusion (sI/R) (n=4). ***; P<0.001 vs. control (no S1P), ##, ###; P<0.01, 0.001 vs. S1P alone.
3.7. CCN1 expression is induced by ischemia/reperfusion and provides cardioprotection in the perfused hearts
To determine the role of CCN1 in the heart, we generated cardiac-specific CCN1 knockout mice (CCN1fl/fl, αMHC-Cre). The level of CCN1 mRNA in the CCN1 KO mouse heart was significantly decreased as assessed by quantitative RT-PCR (Fig. 7A). The remaining 20% expression likely reflects CCN1 present in non-cardiomyocytes in the heart e.g. vascular smooth muscle cells and cardiac fibroblasts. We previously reported that RhoA is activated in response to ex vivo ischemia/reperfusion (I/R) [29] and therefore hypothesized that CCN1 would be induced in the heart subjected to I/R. Remarkably in control (CCN1fl/fl) hearts, CCN1 mRNA was increased 7.5 fold after 30 min ischemia and 60 min reperfusion. As expected there was no significant increase in CCN1 KO mouse hearts (Fig. 7B). To determine whether the increase in CCN1 expression could play a regulatory role in cardioprotection against I/R, infarct size at 60 min of reperfusion was assessed by TTC staining. The infarct area was more than twice as large in CCN1 KO hearts compared to control hearts (Fig. 7C). Western blotting for phosphorylated Akt (P-Akt), a pro-survival kinase, revealed that P-Akt increased following I/R in control mice, as reported [28, 31, 44-46] and that this response was significantly reduced in KO heart (Fig. 7D), suggesting that Akt activation occurs through CCN1 signaling.
Figure 7.
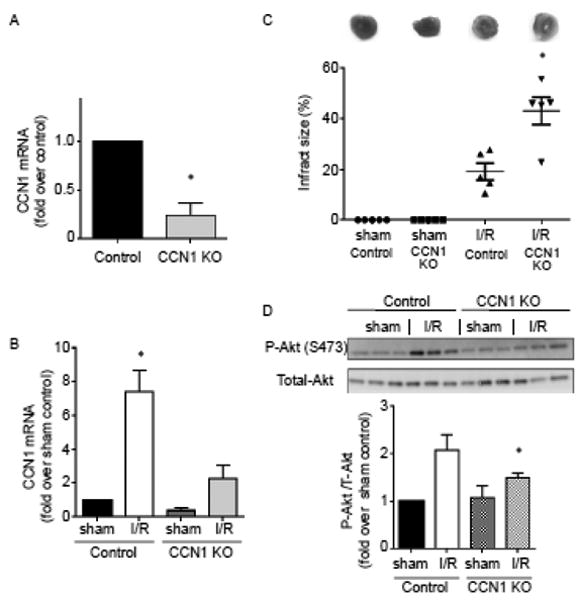
I/R injury is exacerbated in cardiac specific CCN1 KO mouse hearts. A, Quantitative RT-PCR analysis of CCN1 mRNA expression levels in the ventricles from control (CCN1fl/fl) and CCN1 KO (CCN1fl/fl, αMHC-Cre) mouse hearts. *, P<0.05 vs. control (n=6). B, Ischemia/reperfusion (I/R: 30/60 min) in ex vivo perfused hearts leads to increase in CCN1 mRNA expression in control group but not in KO group. *, P<0.05 vs. sham control (n=5). C, Infarct size was determined by TTC staining after I/R. *, P<0.05 vs. I/R in control (n=5). D, Western blotting for phosphorylation of Akt at S473 in the whole heart homogenates. *, P<0.05 vs. I/R in control (n=6-7).
4. Discussion
CCN1 is a pleiotropic molecule, expression of which is highly induced in response to diverse stimuli. Cellular functions including cell migration, proliferation, differentiation, survival/apoptosis and senescence can be regulated through CCN1 signaling [2]. CCN1 is highly expressed in the myocardium of patients with heart failure or ischemic myopathy, and it has also been shown to be increased in the mouse heart in response to pressure overload and myocardial infarction [25]. The molecular mechanisms by which CCN1 expression is regulated in the heart have not been elucidated nor has its role been fully determined. In this study we provide several lines of evidence that GPCRs and interventions that increase RhoA signaling are efficacious inducers of CCN1 expression, that this occurs through the transcription co-factor MRTF-A, and that CCN1 contributes to GPCR agonist-mediated cardioprotection.
4.1. Activation of GPCRs that couple to RhoA induces CCN1 in cardiomyocytes
A previous publication reported that PE and Ang-II increase CCN1 expression in cardiomyocytes [25]. Interestingly our studies demonstrated that CCN1 induction by PE was very modest compared to that by S1P, LPA or ET-1. We suggest that the disparate efficacy of these agonists reflects differences in G-protein coupling. Specifically several S1P and LPA receptor subtypes couple to G12/13 [47-50]. The endothelin receptor ETA has also been reported to couple to G12 in addition to its established coupling to Gq [51, 52]. G12/13 proteins regulate guanine nucleotide exchange factors (GEFs) for RhoA [6, 27, 53, 54] and indeed we also show robust activation of RhoA by these ligands (Fig. 2A). In contrast the α- and β- adrenergic receptors stimulated by PE and ISO in cardiomyocytes couple to Gq and Gs respectively, and the cardiomyocyte M2 muscarinic receptor stimulated by carbachol couples mainly to Gi. Work presented here shows that activation of these receptors does not lead to significant RhoA activation, nor does it induce CCN1 induction in cardiomyocytes. Studies in which we block RhoA signaling with C3 exoenzyme or CGX0287 (Figs. 2B and 2C), or inhibit its downstream effector, Rho kinase (Fig. 2D), further demonstrate the critical role of RhoA activation in GPCR-induced CCN1 expression in cardiomyocytes. We also demonstrate that adenoviral overexpression of RhoA is sufficient to increase CCN1 expression (Fig. 2E), and that CCN1 expression is robustly increased in the heart subjected to I/R (Fig. 7B), a condition which we have previously shown to lead to activation of RhoA [29].
4.2. MRTF-A mediates RhoA induced CCN1 induction
RhoA regulates transcriptional activity and immediate-early genes through its effects on SRF. The CCN1 protein falls into the category of immediate-early genes since increases in CCN1 mRNA and protein can be observed within 15 min to 30 min of S1P treatment (Fig. 1A and 1B). The ability of SRF to transactivate its target genes is regulated by transcriptional co-activators [16, 17] one of which is MRTF-A [20, 55]. Using two pharmacological inhibitors and siRNA-mediated knockdown, we demonstrate that MRTF-A is required for CCN1 induction in cardiomyocytes (Fig. 4). MRTF-A is translocated to the nucleus in response to S1P through RhoA signaling, as evidenced by inhibition with C3 exoenzyme (Fig. 3). This supports the critical role of RhoA signaling in MRTF-A activity previously reported in fibroblasts stimulated with serum and in smooth muscle cells subjected to strain [11, 16, 20]. CCN1 is a matricellular protein and our data indicate that it is increased in response to transcriptional activation in cardiomyocytes and secreted into the extracellular space (Fig. 5), as shown previously by us and others for non-cardiac cells [2, 6, 56]. The increase in CCN1 in the media reflects the increase in CCN1 mRNA and protein in the cell as it is also prevented by RhoA inhibition with C3, ROCK inhibition with Y-27632 or MRTF-A inhibition with CCG-203,971. Thus MRTF-A-mediated CCN1 expression could integrate GPCR signaling with responses mediated through the extracellular matrix and integrins, the receptors through which CCN1 affects cellular functions (Fig. 8).
Figure 8.
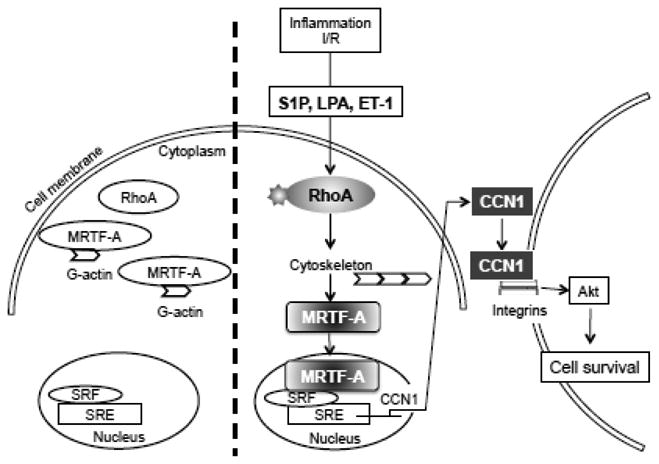
Schematic depicting the mechanism by which RhoA and MRTF-A participate in agonist-induced CCN1 expression and cardioprotection.
4.3. CCN1 plays a protective role in the heart against I/R
The diverse effects of CCN1 are suggested to be due to its binding to various cell surface integrins [2]. Integrin activation can have cardioprotective effects, as established by the observation that heterozygous knockout of β1 integrin increases cardiac dysfunction after infarction [57], and that conditional and complete deletion of integrin β1 leads to development of cardiac fibrosis and cardiac failure [58]. Activation of α7β1 integrin was also recently demonstrated to be cardioprotective against I/R [59]. Recombinant CCN1 protein has been shown to bind to β1 integrin and activate survival kinases such as Akt and ERK in cardiomyocytes [7]. Our findings using CCN1 gene knockdown or gene deletion confirm that CCN1 expression, induced by RhoA, agonist, or I/R, provides cardioprotection both in cardiomyocytes and in the ex vivo perfused heart. It has been reported that CCN1 signaling through some of its integrin binding sites can facilitate apoptosis [4, 5, 26] although this is context dependent [2, 4, 5, 7, 26]. Mutation of two of the CCN1 integrin binding sites implicated in death receptor mediated apoptosis [4] decreases isoproterenol toxicity in the mouse heart suggesting that CCN1 contributes to this toxicity [5]. Wild-type CCN1 protein does not, however, induce apoptosis in isolated cardiomyocytes [5, 7], indeed it protects against H2O2 induced cell death [7]. These data and the observations presented here support a predominantly salutary effect of CCN1 induction and integrin activation on cardiac responses to injury.
While induction of CCN1 in response to S1P is a remarkably rapid response, leading to increased protein expression within 30 min, it may not be fast enough to protect against the earliest events leading to reperfusion injury. S1P can also activate protective protein kinase signaling pathways including Akt and this response, which occurs within minutes, has been shown to contribute to cardioprotection [28, 30, 60]. CCN1 induction may, however, provide a second phase of protective signaling activation to insure cardiomyocyte survival. Cardiomyocytes express S1P1, S1P2 and S1P3 receptors and we have suggested that it is the S1P2 and S1P3 receptors that are responsible for the initial Akt activation [28].. Thus it could be clinically advantageous to selectively activate S1P receptors and to recruit both immediate post-transcriptional- and later transcriptional- salvage pathways to protect hearts.
While this study focuses on CCN1, another CCN family protein CCN2 (connective tissue growth factor: CTGF) , has also been shown to be elevated in hypertrophied and failing hearts[61, 62]. The role of CCN2 in the heart has been examined in transgenic mice models, leading to the conclusion that large increases in CCN2 can confer cardioprotection against I/R[63], suggesting functional similarity in CCN1 and CCN2.. It will be of considerable importance to examine the role of CCN2 as well as CCN1 in future in vivo experiments stressing the heart through interventions such as in vivo I/R, pressure overload and myocardial infarction.
4.4. Conclusion
Our findings demonstrated that CCN1 expression in cardiomyocytes is highly induced by a select subset of GPCR agonists. This does not occur through the ligands or GPCRs involved in acute physiological regulation of the heart (e.g., β-AdR and Gs; α1-AdR and Gq; m2AChR and Gi) but rather through agonists such as S1P and LPA which act on receptors that couple to G12/13 and RhoA. Ligands such as S1P and LPA are known to be generated under conditions of inflammation and at sites of cell injury [64], as well as during ischemia reperfusion in the heart [60, 65, 66]. Based on the observations reported here the actions of these endogenous cardioprotective signals may depend on and would be diminished in the absence of CCN1 . These findings suggest more broadly that other G12/13 and RhoA coupled receptor ligands released at sites of ischemia in the heart could activate RhoA and MRTF-A, induce CCN1 expression, and protect cardiomyocytes against ischemic injury. Early activation of RhoA and CCN1 during reperfusion could provide a therapeutic avenue for protecting cardiomyocytes and limiting further development of heart failure and cardiac remodeling.
Supplementary Material
Highlights.
RhoA coupled GPCR ligands induce CCN1 expression in cardiomyocytes
MRTF-A is activated by RhoA and regulates CCN1 expression
CCN1 plays a protective role in the heart against ischemic stress
Acknowledgments
We thank Dr. Scott Larsen for kindly providing CCG-1423 and CCG-203,971 and Dr. David Roberts for excellent technical assistance.
Funding: This work was supported by National Institutes of Health grant HL028143 to Joan Heller Brown and SM by HL097037 to Shigeki Miyamoto. Sunny Xiang was supported by an AHA Postdoctoral Fellowship (11POST7580130) and Olivia Yu by Pharmacological Sciences Training Grant (T32-GM007752).
Footnotes
Conflict of interest: none declared.
Disclosure: none
References
- 1.O'Brien TP, Yang GP, Sanders L, Lau LF. Expression of cyr61, a growth factor-inducible immediate-early gene. Mol Cell Biol. 1990 Jul;10(7):3569–77. doi: 10.1128/mcb.10.7.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lau LF. CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol Life Sci. 2011 Oct;68(19):3149–63. doi: 10.1007/s00018-011-0778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011 Dec;10(12):945–63. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CC, Young JL, Monzon RI, Chen N, Todorovic V, Lau LF. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J. 2007 Mar 7;26(5):1257–67. doi: 10.1038/sj.emboj.7601596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu PL, Su BC, Kuok QY, Mo FE. Extracellular matrix protein CCN1 regulates cardiomyocyte apoptosis in mice with stress-induced cardiac injury. Cardiovasc Res. 2013 Apr 1;98(1):64–72. doi: 10.1093/cvr/cvt001. [DOI] [PubMed] [Google Scholar]
- 6.Walsh CT, Radeff-Huang J, Matteo R, Hsiao A, Subramaniam S, Stupack D, et al. Thrombin receptor and RhoA mediate cell proliferation through integrins and cysteine-rich protein 61. FASEB J. 2008 Nov;22(11):4011–21. doi: 10.1096/fj.08-113266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida Y, Togi K, Matsumae H, Nakashima Y, Kojima Y, Yamamoto H, et al. CCN1 protects cardiac myocytes from oxidative stress via beta1 integrin-Akt pathway. Biochem Biophys Res Commun. 2007 Apr 13;355(3):611–8. doi: 10.1016/j.bbrc.2007.01.195. [DOI] [PubMed] [Google Scholar]
- 8.Walsh CT, Stupack D, Brown JH. G protein-coupled receptors go extracellular: RhoA integrates the integrins. Mol Interv. 2008 Aug;8(4):165–73. doi: 10.1124/mi.8.4.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young N, Pearl DK, Van Brocklyn JR. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol Cancer Res. 2009 Jan;7(1):23–32. doi: 10.1158/1541-7786.MCR-08-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young N, Van Brocklyn JR. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp Cell Res. 2007 May 1;313(8):1615–27. doi: 10.1016/j.yexcr.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanna M, Liu H, Amir J, Sun Y, Morris SW, Siddiqui MA, et al. Mechanical regulation of the proangiogenic factor CCN1/CYR61 gene requires the combined activities of MRTF-A and CREB-binding protein histone acetyltransferase. J Biol Chem. 2009 Aug 21;284(34):23125–36. doi: 10.1074/jbc.M109.019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura I, Rosenbloom J, Macarak E, Chaqour B. Regulation of Cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am J Physiol Cell Physiol. 2001 Nov;281(5):C1524–32. doi: 10.1152/ajpcell.2001.281.5.C1524. [DOI] [PubMed] [Google Scholar]
- 13.Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995 Jun 30;81(7):1159–70. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 14.Gauthier-Rouviere C, Cavadore JC, Blanchard JM, Lamb NJ, Fernandez A. p67SRF is a constitutive nuclear protein implicated in the modulation of genes required throughout the G1 period. Cell Regul. 1991 Jul;2(7):575–88. doi: 10.1091/mbc.2.7.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995 Jan 27;80(2):199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 16.Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science. 2007 Jun 22;316(5832):1749–52. doi: 10.1126/science.1141084. [DOI] [PubMed] [Google Scholar]
- 17.Shore P, Sharrocks AD. The MADS-box family of transcription factors. Eur J Biochem. 1995 Apr 1;229(1):1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- 18.Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, et al. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci U S A. 2002 Nov 12;99(23):14855–60. doi: 10.1073/pnas.222561499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lockman K, Hinson JS, Medlin MD, Morris D, Taylor JM, Mack CP. Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J Biol Chem. 2004 Oct 8;279(41):42422–30. doi: 10.1074/jbc.M405432200. [DOI] [PubMed] [Google Scholar]
- 20.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003 May 2;113(3):329–42. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 21.Kuwahara K, Kinoshita H, Kuwabara Y, Nakagawa Y, Usami S, Minami T, et al. Myocardin-related transcription factor A is a common mediator of mechanical stress-and neurohumoral stimulation-induced cardiac hypertrophic signaling leading to activation of brain natriuretic peptide gene expression. Mol Cell Biol. 2010 Sep;30(17):4134–48. doi: 10.1128/MCB.00154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morissette MR, Sah VP, Glembotski CC, Brown JH. The Rho effector, PKN, regulates ANF gene transcription in cardiomyocytes through a serum response element. Am J Physiol Heart Circ Physiol. 2000 Jun;278(6):H1769–74. doi: 10.1152/ajpheart.2000.278.6.H1769. [DOI] [PubMed] [Google Scholar]
- 23.Sah VP, Hoshijima M, Chien KR, Brown JH. Rho is required for Galphaq and alpha1-adrenergic receptor signaling in cardiomyocytes. Dissociation of Ras and Rho pathways. J Biol Chem. 1996 Dec 6;271(49):31185–90. doi: 10.1074/jbc.271.49.31185. [DOI] [PubMed] [Google Scholar]
- 24.Mo FE, Lau LF. The matricellular protein CCN1 is essential for cardiac development. Circ Res. 2006 Oct 27;99(9):961–9. doi: 10.1161/01.RES.0000248426.35019.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, et al. Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation. 2004 May 11;109(18):2227–33. doi: 10.1161/01.CIR.0000127952.90508.9D. [DOI] [PubMed] [Google Scholar]
- 26.Todorovic V, Chen CC, Hay N, Lau LF. The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol. 2005 Nov 7;171(3):559–68. doi: 10.1083/jcb.200504015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008 Dec 19;283(51):35622–9. doi: 10.1074/jbc.M804036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Means CK, Xiao CY, Li Z, Zhang T, Omens JH, Ishii I, et al. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007 Jun;292(6):H2944–51. doi: 10.1152/ajpheart.01331.2006. [DOI] [PubMed] [Google Scholar]
- 29.Xiang SY, Vanhoutte D, Del Re DP, Purcell NH, Ling H, Banerjee I, et al. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest. 2011 Aug;121(8):3269–76. doi: 10.1172/JCI44371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang SY, Ouyang K, Yung BS, Miyamoto S, Smrcka AV, Chen J, et al. PLCepsilon, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci Signal. 2013 Dec;17:6. doi: 10.1126/scisignal.2004405. (306): ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K, et al. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ Res. 2010 Aug 20;107(4):476–84. doi: 10.1161/CIRCRESAHA.109.215020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013 Jun 15;27(12):1365–77. doi: 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagi SA, Seasholtz TM, Kobiashvili M, Wilson BA, Toksoz D, Brown JH. Physical and functional interactions of Galphaq with Rho and its exchange factors. J Biol Chem. 2001 May 4;276(18):15445–52. doi: 10.1074/jbc.M008961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howes AL, Miyamoto S, Adams JW, Woodcock EA, Brown JH. Galphaq expression activates EGFR and induces Akt mediated cardiomyocyte survival: dissociation from Galphaq mediated hypertrophy. J Mol Cell Cardiol. 2006 May;40(5):597–604. doi: 10.1016/j.yjmcc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Kim KH, Chen CC, Monzon RI, Lau LF. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol. 2013 May;33(10):2078–90. doi: 10.1128/MCB.00049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aktories K, Just I. In vitro ADP-ribosylation of Rho by bacterial ADP-ribosyltransferases. Methods Enzymol. 1995;256:184–95. doi: 10.1016/0076-6879(95)56023-8. [DOI] [PubMed] [Google Scholar]
- 37.Carles M, Lafargue M, Goolaerts A, Roux J, Song Y, Howard M, et al. Critical role of the small GTPase RhoA in the development of pulmonary edema induced by Pseudomonas aeruginosa in mice. Anesthesiology. 2010 Nov;113(5):1134–43. doi: 10.1097/ALN.0b013e3181f4171b. [DOI] [PubMed] [Google Scholar]
- 38.Han JS, Macarak E, Rosenbloom J, Chung KC, Chaqour B. Regulation of Cyr61/CCN1 gene expression through RhoA GTPase and p38MAPK signaling pathways. Eur J Biochem. 2003 Aug;270(16):3408–21. doi: 10.1046/j.1432-1033.2003.03723.x. [DOI] [PubMed] [Google Scholar]
- 39.Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, et al. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007 Aug;6(8):2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 40.Evelyn CR, Bell JL, Ryu JG, Wade SM, Kocab A, Harzdorf NL, et al. Design, synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG-1423. Bioorg Med Chem Lett. 2010 Jan 15;20(2):665–72. doi: 10.1016/j.bmcl.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PD. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014 Jan;20(1):154–65. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lundquist MR, Storaska AJ, Liu TC, Larsen SD, Evans T, Neubig RR, et al. Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell. 2014 Jan 30;156(3):563–76. doi: 10.1016/j.cell.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang GP, Lau LF. Cyr61, product of a growth factor-inducible immediate early gene, is associated with the extracellular matrix and the cell surface. Cell Growth Differ. 1991 Jul;2(7):351–7. [PubMed] [Google Scholar]
- 44.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008 Apr;88(2):581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006 Mar;38(3):414–9. doi: 10.1016/j.biocel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 46.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004 Feb 15;61(3):448–60. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 47.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, et al. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998 Jun 26;280(5372):2109–11. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 48.Windh RT, Lee MJ, Hla T, An S, Barr AJ, Manning DR. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J Biol Chem. 1999 Sep 24;274(39):27351–8. doi: 10.1074/jbc.274.39.27351. [DOI] [PubMed] [Google Scholar]
- 49.Takuwa Y, Takuwa N, Sugimoto N. The Edg family G protein-coupled receptors for lysophospholipids: their signaling properties and biological activities. J Biochem. 2002 Jun;131(6):767–71. doi: 10.1093/oxfordjournals.jbchem.a003163. [DOI] [PubMed] [Google Scholar]
- 50.Rieken S, Herroeder S, Sassmann A, Wallenwein B, Moers A, Offermanns S, et al. Lysophospholipids control integrin-dependent adhesion in splenic B cells through G(i) and G(12)/G(13) family G-proteins but not through G(q)/G(11) J Biol Chem. 2006 Dec 1;281(48):36985–92. doi: 10.1074/jbc.M605287200. [DOI] [PubMed] [Google Scholar]
- 51.Kawanabe Y, Okamoto Y, Nozaki K, Hashimoto N, Miwa S, Masaki T. Molecular mechanism for endothelin-1-induced stress-fiber formation: analysis of G proteins using a mutant endothelin(A) receptor. Mol Pharmacol. 2002 Feb;61(2):277–84. doi: 10.1124/mol.61.2.277. [DOI] [PubMed] [Google Scholar]
- 52.Arai K, Maruyama Y, Nishida M, Tanabe S, Takagahara S, Kozasa T, et al. Differential requirement of G alpha12, G alpha13, G alphaq, and G beta gamma for endothelin-1-induced c-Jun NH2-terminal kinase and extracellular signal-regulated kinase activation. Mol Pharmacol. 2003 Mar;63(3):478–88. doi: 10.1124/mol.63.3.478. [DOI] [PubMed] [Google Scholar]
- 53.Del Re DP, Miyamoto S, Brown JH. RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J Biol Chem. 2007 Mar 16;282(11):8069–78. doi: 10.1074/jbc.M604298200. [DOI] [PubMed] [Google Scholar]
- 54.Hilal-Dandan R, Means CK, Gustafsson AB, Morissette MR, Adams JW, Brunton LL, et al. Lysophosphatidic acid induces hypertrophy of neonatal cardiac myocytes via activation of Gi and Rho. J Mol Cell Cardiol. 2004 Apr;36(4):481–93. doi: 10.1016/j.yjmcc.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 55.Cen B, Selvaraj A, Prywes R. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J Cell Biochem. 2004 Sep 1;93(1):74–82. doi: 10.1002/jcb.20199. [DOI] [PubMed] [Google Scholar]
- 56.Kireeva ML, Mo FE, Yang GP, Lau LF. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol Cell Biol. 1996 Apr;16(4):1326–34. doi: 10.1128/mcb.16.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krishnamurthy P, Subramanian V, Singh M, Singh K. Deficiency of beta1 integrins results in increased myocardial dysfunction after myocardial infarction. Heart. 2006 Sep;92(9):1309–15. doi: 10.1136/hrt.2005.071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, et al. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002 Mar 8;90(4):458–64. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 59.Okada H, Lai NC, Kawaraguchi Y, Liao P, Copps J, Sugano Y, et al. Integrins protect cardiomyocytes from ischemia/reperfusion injury. J Clin Invest. 2013 Oct 1;123(10):4294–308. doi: 10.1172/JCI64216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vessey DA, Li L, Honbo N, Karliner JS. Sphingosine 1-phosphate is an important endogenous cardioprotectant released by ischemic pre- and postconditioning. Am J Physiol Heart Circ Physiol. 2009 Oct;297(4):H1429–35. doi: 10.1152/ajpheart.00358.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsui Y, Sadoshima J. Rapid upregulation of CTGF in cardiac myocytes by hypertrophic stimuli: implication for cardiac fibrosis and hypertrophy. J Mol Cell Cardiol. 2004 Aug;37(2):477–81. doi: 10.1016/j.yjmcc.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 62.Ohnishi H, Oka T, Kusachi S, Nakanishi T, Takeda K, Nakahama M, et al. Increased expression of connective tissue growth factor in the infarct zone of experimentally induced myocardial infarction in rats. J Mol Cell Cardiol. 1998 Nov;30(11):2411–22. doi: 10.1006/jmcc.1998.0799. [DOI] [PubMed] [Google Scholar]
- 63.Ahmed MS, Gravning J, Martinov VN, von Lueder TG, Edvardsen T, Czibik G, et al. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol Apr. 300(4):H1291–302. doi: 10.1152/ajpheart.00604.2010. [DOI] [PubMed] [Google Scholar]
- 64.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003 May;4(5):397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 65.Cordis GA, Yoshida T, Das DK. HPTLC analysis of sphingomylein, ceramide and sphingosine in ischemic/reperfused rat heart. J Pharm Biomed Anal. 1998 Mar;16(7):1189–93. doi: 10.1016/s0731-7085(97)00260-4. [DOI] [PubMed] [Google Scholar]
- 66.Hernandez OM, Discher DJ, Bishopric NH, Webster KA. Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ Res. 2000 Feb 4;86(2):198–204. doi: 10.1161/01.res.86.2.198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.