

Abstract
The BM2 protein of influenza B virus functions as an ion channel, which is suggested to be important for virus uncoating in endosomes of virus-infected cells. Because direct support for this function is lacking, whether BM2 plays an essential role in the viral life cycle remains unknown. We therefore attempted to generate BM2 knockout viruses by reverse genetics. Mutant viruses possessing M segments with the mutated initiation codon of BM2 protein at the stop-start pentanucleotide were viable and still expressed BM2. The introduction of multiple stop codons and a one-nucleotide deletion downstream of the stop-start pentanucleotide, in addition to disablement of the BM2 initiation codon, failed to generate viable mutant viruses, but the mutant M segments still expressed proteins that reacted with the BM2 peptide antiserum. To completely abolish BM2 expression, we generated a mutant M gene whose BM2 open reading frame was deleted. Although this mutant was not able to replicate in normal MDCK cells, it did replicate in a cell line that we established which constitutively expresses BM2. Furthermore, a virus possessing the mutant M gene lacking the BM2 open reading frame and a mutant NA gene containing the BM2 open reading frame instead of the NA open reading frame underwent multiple cycles of replication in MDCK cells, with exogenous sialidase used to supplement the deleted viral sialidase activity. These findings demonstrate that the BM2 protein is essential for influenza B virus replication.
The genome of Influenza B virus, a member of the family Orthomyxoviridae, consists of eight negative-strand RNA segments, which encode 11 proteins (12). Of these, nine are also found in influenza A virus: three RNA-dependent RNA polymerase subunits (PB1, PB2, and PA), hemagglutinin (HA), nucleoprotein (NP), neuraminidase (NA), matrix protein (M1), and two nonstructural proteins (NS1 and NS2). Two proteins, NB and BM2, are unique to influenza B virus. NB is encoded by RNA segment 6, which also encodes NA, while BM2 is encoded by segment 7.
The BM2 protein of influenza B virus is a type III integral membrane protein (23). It is incorporated into virions (22), expressed on the surface of virus-infected cells (23), and phosphorylated in the late phase of infection (12). This protein is translated from a +2 open reading frame and is 109 amino acids long. Its translational strategy is unique in that the initiation codon of BM2 protein overlaps the termination codon of M1 protein (UAAUG, a stop-start pentanucleotide) (9). Thus, unlike the M2 protein of influenza A virus, which is translated from a spliced mRNA, the BM2 protein is translated by the stop-start translational mechanism.
Recently, Paterson et al. (23) demonstrated that this protein forms an oligomer, possesses amino acid residues thought to be critical for ion channel activity at the same relative position as in the M2 protein of influenza A virus, and indeed functions as an ion channel (14). Although Mould et al. (14) demonstrated that BM2 protein prevents intracellularly cleaved HA from adopting its low-pH-induced conformation during transport to the cell surface, as shown with the M2 protein of influenza A virus, it remains unclear whether the BM2 protein is essential for viral replication. We therefore tested the requirement for this protein in the life cycle of influenza B virus by attempting to generate BM2 knockout influenza B viruses by reverse genetics.
MATERIALS AND METHODS
Cells and antibodies.
293T human embryonic kidney cells and Madin-Darby canine kidney (MDCK) cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and in minimal essential medium containing 5% newborn calf serum, respectively. The 293T cell line is a derivative of the 293 cell line into which the gene for the simian virus 40 T antigen was inserted (3). All cells were maintained at 37°C in 5% CO2. A rabbit anti-BM2 peptide serum generated against a synthetic peptide (amino acid residues 95 to 109 of BM2 protein) coupled to keyhole limpet hemocyanin was used in these studies.
Construction of plasmids.
Mutant constructs (see Fig. 1 and 4) were produced as follows. M genes with mutations around the initiation codon of BM2 protein and with deletions of the entire BM2 coding region were amplified by PCR from a plasmid (pPolBLeeM) containing the B/Lee/40 M gene and then digested with BsmBI. The BsmBI-digested fragment was cloned into the BsmBI sites of a PolI plasmid vector, which contains the human RNA polymerase I promoter and the mouse RNA polymerase I terminator, separated by BsmBI sites. pPolBNAMCS was constructed by PCR and linker insertion. It consists of 200 bases of the 3′ end of NA viral RNA, a multiple cloning site, and 344 bases of the 5′ end of NA viral RNA. The initiation codons of the NA and NB proteins in this construct were changed to GCG. pPolBNAGFP was constructed by introducing the cyto-green fluorescent protein (GFP) gene (Clontech) between the EcoRI and BglII sites of pPolBNAMCS, and pPolBNABM2 by introducing the BM2 coding region, with a Kozak sequence (GCCGCCACC) upstream of the initiation codon of BM2 protein, between the EcoRI and XhoI sites of pPolBNAMCS. For construction of the BM2 protein expression vector, the BM2 coding region with a Kozak sequence (GCCGCCACC) upstream of the initiation codon of BM2 protein was cloned into the eukaryotic expression vector pCAGGS/MCS (11, 20), resulting in pCABLeeBM2. All of the constructs were sequenced to ensure that unwanted mutations were not present. The sequences of the primers will be provided upon request.
FIG. 1.

Mutations introduced around the pentanucleotide stop-start region of the M segment. (A) M genes with mutations around the initiation codon of BM2 protein were amplified by PCR and cloned into a PolI plasmid vector. The resulting constructs were designated pPolBLeeBM2stop#1, pPolBLeeBM2stop#2, pPolBLeeBM2stop#3, and pPolBLeeBM2stop#4. Mutations are indicated with asterisks (*), the stop codons generated by these mutations are indicated with dashed lines (- - -), and deletions are indicated with single dashes (—). The numbers shown are nucleotide positions. (B) Amino acid deletions and mutation at the N terminus of the protein expressed from pPolBLeeBM2stop#3. Amino acids are designated in the single-letter code.
FIG. 4.

Schematic diagram of PolI plasmids encoding wild-type and mutant NA and M viral RNA segments. pPolBLeeNA encodes wild-type NA viral RNA. pPolBNAMCS was constructed by PCR and linker insertion and consists of 200 bases of the 3′ end of NA viral RNA, a multiple cloning site, and 344 bases of the 5′ end of NA viral RNA. The initiation codons of the NA and NB proteins in this construct were changed to GCG (shown as X). pPolBNAGFP was constructed by introducing the cyto-GFP gene between the EcoRI and BglII sites of pPolBNAMCS. pPolBNABM2 was constructed by introducing the BM2 coding region with a Kozak sequence (GCCGCCACC) upstream of the initiation codon of BM2 protein, between the EcoRI and XhoI sites of pPolBNAMCS. pPolBLeeM encodes wild-type M viral RNA. pPolBLeeBM2del encodes M viral RNA lacking the entire BM2 coding region. MCS, multiple cloning site; NA, the coding region of B/Lee/40 NA protein; cyto-GFP, the coding region of cyto-GFP; M1, the coding region of B/Lee/40 M1 protein; BM2, the coding region of B/Lee/40 BM2 protein.
Plasmid-based reverse genetics.
Transfectant viruses were generated as reported earlier (7, 18). Briefly, 12 plasmids (eight PolI constructs for eight RNA segments and four protein expression constructs for the polymerase proteins and NP) were mixed with transfection reagent (Trans IT LT-1 [Panvera, Madison, Wis.]), incubated at room temperature for 10 min, and added to 106 293T cells cultured in Opti-MEM (Invitrogen) containing 0.3% bovine serum albumin. Forty-eight hours later, viruses in the supernatant were collected and amplified in MDCK cells for the production of stock viruses. For the generation of BNAGFP virus, protein expression plasmids for HA, NA, M1, and BM2 and for NS1 and NS2 were used in addition to the 12 plasmids mentioned above. For the generation of BNABM2 virus, protein expression plasmids for HA and NA and for NS1 and NS2 were used in addition to the 12 plasmids.
Indirect immunofluorescence assay.
For analysis of protein expression from mutant BM2 constructs, 293T cells were transfected with four protein expression plasmids for the polymerase proteins and NP of B/Lee/40 and the wild-type and mutant BM2 PolI constructs. Forty-eight hours after transfection, the cells were fixed with 3% formaldehyde solution and permeated with 0.1% Triton X-100. Antigens were detected with rabbit anti-BM2 peptide serum as a primary antibody and fluorescein isothiocyanate (FITC)-conjugated anti-rabbit immunoglobulin G (IgG) as a secondary antibody. For detection of BNAGFP virus, MDCK cells were incubated with BNAGFP virus at 102 50% tissue culture infectious doses (TCID50). After 72 h of infection, the cells were fixed with 3% formaldehyde solution and observed under a fluorescence microscope. For detection of BM2 protein of the BNABM2 virus, MDCK cells were infected with the virus and overlaid with 0.6% agarose. After incubation for 72 h, the cells were fixed with 3% formaldehyde solution and permeated with 0.1% Triton X-100. The BM2 protein was detected with rabbit anti-BM2 peptide serum, used as a primary antibody, and FITC-conjugated anti-rabbit IgG as a secondary antibody.
Western blotting.
293T cells transfected with four protein expression plasmids (for polymerase proteins and NP) and the wild-type or mutant BM2 PolI construct were lysed in sample buffer at 48 h posttransfection, incubated at 100°C for 5 min, and separated on a 15% PAGEr Gold Tris-glycine gel (BioWhittaker Molecular Applications). Resolved proteins were transferred to nitrocellulose membranes (Schleicher & Schuell) and blocked overnight at 4°C with 5% skim milk in phosphate-buffered saline (PBS). Blots were incubated in rabbit anti-BM2 peptide serum or goat anti-influenza B virus matrix protein serum (National Institute for Allergy and Infectious Diseases Repository, Virology Branch) for 2 h at room temperature, washed three times with PBST (0.05% Tween 20 [Sigma] in PBS), incubated in biotinylated anti-rabbit or anti-goat secondary antibody for 1 h, washed three times with PBST, and incubated in reagent A and reagent B solution (Vectastain ABC kit; Vector Laboratories, Inc., Burlingame, Calif.) for 30 min. The blots were then washed and incubated in Lumi-Light Western blotting substrate (Roche Biochemica) for 5 min and exposed to X-ray film (Kodak).
Establishment of BM2 protein-expressing cell lines.
MDCK cells were cotransfected with plasmid pRHyg (a derivative of pREP7 [Invitrogen Corporation, Carlsbad, Calif.] from which the EBNA-1 and oriP genes were deleted), possessing the hygromycin resistance gene, and plasmid pCABLeeBM2 and cultured under hygromycin selection (150 ng/ml). Hygromycin-resistant cell clones were tested for the expression of BM2 protein with rabbit anti-BM2 peptide serum in an indirect immunofluorescence assay. One of these clones (designated BM2CK#1-3) was used for the experiments.
Replicative properties of viruses.
MDCK and BM2CK#1-3 cells were infected with 103 TCID50 of viruses and overlaid with minimal essential medium containing 0.5 μg of trypsin per ml. Supernatants were examined for viral titers at different times with BM2CK#1-3 cells.
Immunostaining assay.
MDCK and BM2CK#1-3 cells were infected with 104 TCID50 of virus and overlaid with 1.0% agarose. The cells were fixed at different times with 3% formaldehyde (in PBS) for 1 h at room temperature, followed by treatment with 0.1% Triton X-100, and then processed as described previously (16). Viral antigens in cells were detected with mouse anti-influenza virus type B (nucleoprotein) monoclonal antibody, clone INFLB71 (Research Diagnostics Inc., Flanders, N.J.).
RESULTS
Generation of BM2 protein-knockout viruses.
Recently, we and others (7, 8, 10) established reverse genetics systems for generating influenza B virus entirely from cloned cDNAs, based on the in vivo synthesis of viral RNA by RNA polymerase I. Using this system with influenza B/Lee/40 virus, we attempted to generate mutant viruses that did not express the BM2 protein to assess the importance of BM2 in the influenza B viral life cycle. The first two constructs that we produced expressed the mutant M viral RNA by RNA polymerase I and were designated pPolBLeeBM2stop#1 and pPolBLeeBM2stop#2 (Fig. 1A). To eliminate BM2 expression, we converted the sequence around the stop-start pentanucleotide of the M gene from TAATGc (capital letters represent the stop-start pentanucleotide) to TAACCc, a mutation that eliminated BM2 expression in a plasmid expression system (9), in pPolBLeeBM2stop#1 or to TAATAa in pPolBLeeBM2stop#2. When these plasmids were transfected together with protein expression vectors for polymerase proteins and NP, proteins reactive with a rabbit antiserum raised to the BM2 peptide (amino acid residues 95 to 109 of BM2 protein) were detected (Fig. 2 and 3); however, the amount of BM2 protein expressed from pPolBLeeBM2stop#2 was lower than that from the wild-type M PolI (pPolBLeeM) construct and pPolBLeeBM2stop#1 (Fig. 3). By Western blot analysis, the molecular sizes of these mutant proteins appeared identical to that of the wild-type BM2 protein (Fig. 3).
FIG. 2.

Reactivity of cells expressing proteins from wild-type and mutant M viral RNA segments with anti-BM2 peptide serum in an immunofluorescence assay. 293T cells were transfected with four protein expression plasmids for the polymerase proteins and NP of B/Lee/40 and the wild-type or mutant BM2 PolI construct. Antigens were detected with rabbit anti-BM2 peptide serum as a primary antibody and FITC-conjugated anti-rabbit IgG as a secondary antibody. Wild-type, cells transfected with pPolBLeeM; #1, #2, #3, #4, 4Met−, and del, cells transfected with pPolBLeeBM2stop#1, pPolBLeeBM2stop#2, pPolBLeeBM2stop#3, pPolBLeeBM2stop#4, pPolBLeeBM2stop#1(4Met−), and pPolBLeeBM2del, respectively; mock, cells transfected with only the four protein expression plasmids for polymerase proteins and NP.
FIG. 3.

Detection of BM2 protein in 293T cells by Western blot analysis. The 293T cells transfected with four protein expression plasmids for the polymerase proteins and NP and the wild-type or mutant BM2 PolI construct were lysed in sample buffer and separated on a 15% Tris-glycine gel. Resolved proteins were transferred to nitrocellulose membranes and detected with rabbit anti-BM2 peptide serum (A) or goat anti-influenza B virus matrix protein serum (B). The positions of molecular size markers are indicated.
We then generated viruses possessing these mutant M genes. Upon sequencing the M genes of the generated viruses after five passages in MDCK cells, we found no mutations other than those intended to eliminate BM2 expression. To eliminate the possibility that the translation of BM2 protein originated from ATG codons at a site downstream of the stop-start pentanucleotide in these mutant M segments, we converted four ATG codons at the 21st, 70th, 77th, and 98th codons in the BM2 reading frame of pPolBLeeBM2stop#1 to GCG, designating the resultant mutant pPolBLeeBM2stop#1(4Met−). A protein reactive with the BM2 peptide antiserum was still detected at an expression level and intracellular localization similar to those of the wild-type virus (Fig. 2 and 3), although virus possessing this mutant M gene could not be produced. Thus, the methionines downstream of the pentanucleotide do not seem to serve as an initiation codon, but alteration of one or more of these amino acids may have caused the BM2 protein to malfunction.
Since the suggested open reading frame of BM2 starts at nucleotide position 532 of the M segment, the initiation codon might be located upstream of the pentanucleotide. However, since there is no ATG codon in the reading frame of BM2 upstream of the pentanucleotide, we considered that this protein might be translated from an alternative initiation codon. To examine this possibility, we constructed plasmids with the mutant M segment and a stop codon (TAG or TGA) at codon −5, −27, −46, −74, or −86 in the suggested BM2 reading frame upstream of the pentanucleotide (ATG in the pentanucleotide are designated position 0) without changing the amino acids of the M1 protein. These mutations did not abolish the production of a protein reactive with antiserum against the C terminus of the BM2 protein, indicating that translation does not seem to originate upstream of the pentanucleotide (data not shown).
We next constructed pPolBLeeBM2stop#3 and pPolBLeeBM2stop#4 (Fig. 1A), which possessed stop codons and a one-nucleotide deletion at sites downstream of the mutated initiation codon of the BM2 protein. In pPolBLeeBM2stop#3, the above-described alteration created a new ATG codon, predicted to result in a three-amino-acid deletion and an L-to-I amino acid substitution at the N terminus of BM2 (Fig. 1B). Despite these changes, proteins reactive with the BM2 peptide antiserum were detected in transfected 293T human embryonic kidney cells (Fig. 2 and 3). However, the intracellular localization of the proteins expressed from these constructs differed from that of those produced by the wild-type, pPolBLeeBM2stop#1, and pPolBLeeBM2stop#2 plasmids (Fig. 2).
Western blot analysis demonstrated that the molecular size of the presumed BM2 protein expressed from pPolBLeeBM2stop#3 was slightly smaller than that of the wild-type BM2 protein, possibly due to the three-amino-acid deletion at the N terminus of BM2. The expression level of the protein from this construct was also lower than that of the wild-type construct. By contrast, the molecular size of the presumed BM2 protein from pPolBLeeBM2stop#4 was identical to that of the wild-type BM2 protein, but the former was expressed at a substantially lower level, detectable only when increased amounts of cell lysate (more than twice the wild-type amount) were loaded onto gels (Fig. 3 and data not shown). Even though we detected proteins reactive with the anti-BM2 peptide serum, we were unable to generate viruses possessing these mutant M genes.
To completely eliminate the expression of proteins from the BM2 coding region, we next constructed pPolBLeeBM2del, which encodes a mutant M segment with a deletion of the entire BM2 coding region (Fig. 4). This mutant M segment did not express proteins detectable with the anti-BM2 peptide serum when cells were transfected with pPolBLeeBM2del and with protein expression plasmids for polymerase proteins and NP (Fig. 2 and 3). A mutant virus possessing this M segment was not generated. We considered the possibility that mutations in the above-described construct might have affected the expression of M1 protein, but the results in Fig. 3 indicate negligible differences in the production of M1 by mutant and wild-type constructs.
Generation of a mutant virus expressing BM2 protein from the NA but not the M segment.
To confirm the importance of BM2 protein in the viral life cycle, we attempted to generate a mutant virus whose BM2 protein was provided from the NA segment. We recently generated an influenza A virus whose NA coding sequence was replaced with a reporter gene by exogenously supplementing the viral sialidase activity with a bacterial sialidase (4). Using this strategy, we attempted to generate a mutant virus that expresses BM2 protein from the NA segment. We first constructed plasmid pPolBNAMCS, containing a gene cassette (200 bases of the 3′ end of NA viral RNA, a multiple cloning site, and 344 bases of the 5′ end of NA viral RNA) (Fig. 4) that was flanked by the human RNA polymerase I promoter and the mouse RNA polymerase I terminator. In this construct, the initiation codons of the NA and NB proteins were changed to GCG to eliminate the expression of any proteins. A cyto-GFP or BM2 gene with a Kozak sequence (GCCGCCACC) upstream of the initiation codon of BM2 protein was introduced into the multiple cloning site of pPolBNAMCS, yielding pPolBNAGFP and pPolBNABM2, respectively (Fig. 4).
To determine if the NA segment of influenza B virus can stably express a foreign protein, enabling the use of this system to express BM2 protein, we transfected 293T cells with pPolBNAGFP together with seven other viral RNA expression plasmids and eight plasmids for the expression of three polymerase proteins plus HA, NP, NA, M1, and BM2, and NS1 and NS2. At 48 h posttransfection, supernatant was recovered and incubated with MDCK cells. Virus grew in MDCK cells to a TCID50 of 105. As shown in Fig. 5, GFP was expressed in BNAGFP-infected MDCK cells even after five passages in these cells in the presence of exogenous Vibrio cholerae sialidase (1 mU/ml; Roche), used to provide the deleted viral sialidase activity, indicating that the mutant NA segment was incorporated into virions and stably maintained. We next attempted to generate a mutant virus whose BM2 protein was expressed from the NA but not the M segment by transfecting 293T cells with pPolBNABM2 (expressing a mutant NA segment encoding BM2 protein) and pPolBM2del (expressing a mutant M segment expressing M1 but not BM2) as well as six other viral RNA expression plasmids and seven protein expression plasmids (excluding the M1 and BM2 protein expression plasmids). The transfectant virus was successfully produced and was able to undergo multiple cycles of replication in MDCK cells in the presence of exogenous sialidase, achieving a TCID50 of 105. BM2 protein was stably maintained even after five passages in MDCK cells (Fig. 5).
FIG. 5.

Detection of proteins encoded by the NA segment in virus-infected MDCK cells. (Left panel) BNAGFP virus after five passages in MDCK cells. (Right panel) BNABM2 virus after five passages in MDCK cells.
Virus growth in a BM2-expressing cell line.
To further confirm the necessity of BM2 protein for virus replication, we established a cell line expressing BM2 protein (BM2CK#1-3) (Fig. 6A) and attempted to amplify the mutant viruses, which we were unable to do with normal MDCK cells. Supernatants of 293T cells which had been transfected with all protein expression plasmids including the BM2 expression plasmid and eight PolI plasmids were incubated with BM2CK#1-3 cells. The BLeeRG, BLeeBM2stop#1, BLeeBM2stop#2, BLeeBM2stop#4, and BLeeBM2del viruses grew in BM2CK#1-3 cells, while the BLeeBM2stop#3 and BLeeBM2stop#1(4Met−) viruses did not (data not shown). Using BLeeRG, BLeeBM2stop#4, and BLeeBM2del viruses amplified in BM2CK#1-3 cells, we next tested their replicative properties in MDCK and BM2CK#1-3 cells (Fig. 6B and 7). The BLeeBM2del virus, which lacks the entire BM2 coding region, did not grow in MDCK cells at all, although this mutant grew as well as the wild-type BLeeRG virus did in BM2CK#1-3 cells, indicating that the BM2 protein is necessary for virus growth. Interestingly, BLeeBM2stop#4 virus grew in MDCK cells up to 103 TCID50, while the mutant viruses BLeeBM2stop#3 and BLeeBM2stop#1(4Met−) did not grow in either cell line, suggesting that the BM2 protein expressed from pPolBLeeBM2stop#4 might be functional to some extent. The results also suggested that the BM2 protein expressed from BLeeBM2stop#3 and BLeeBM2stop#1(4Met−) might not be functional or might function in a dominant-negative manner, interfering with the functions of native BM2 protein expressed in BM2CK cells, prohibiting virus generation.
FIG. 6.
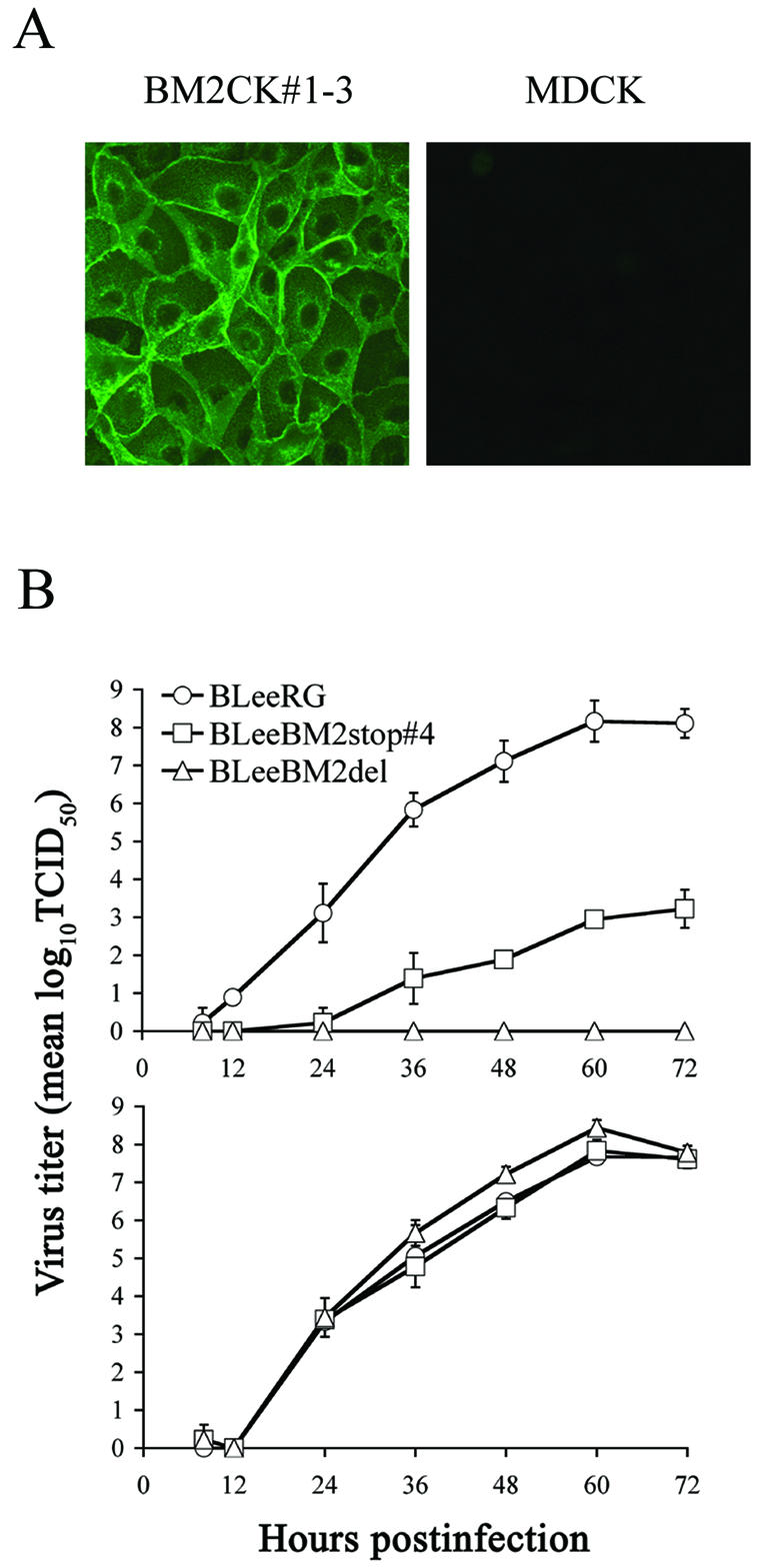
Generation of cells constitutively expressing the BM2 protein and growth curves for BLeeRG and mutant viruses in MDCK cells and BM2-expressing cells. (A) BM2CK#1-3 (left panel) and MDCK cells (right panel) were fixed with 3% formaldehyde solution and permeated with 0.1% Triton X-100. The BM2 protein was detected with rabbit anti-BM2 peptide serum, used as a primary antibody, and FITC-conjugated anti-rabbit IgG as a secondary antibody. (B) MDCK and BM2CK#1-3 cells were infected with virus (103 TCID50) which had been amplified in BM2CK#1-3 cells. At the indicated times after infection, the titers of virus in the supernatant from infected MDCK (upper panel) and BM2CK#1-3 (lower panel) cells were determined with BM2CK#1-3 cells. The values are means (± standard deviation) of three determinations.
FIG. 7.

BLeeRG- and mutant virus-infected MDCK and BM2-expressing cells. MDCK and BM2CK#1-3 cells were infected with 104 TCID50 of wild-type BLeeRG, BLeeBM2stop#4, or BLeeBM2del virus (which had been grown in BM2CK#1-3 cells) and overlaid with 1.0% agarose. The cells were fixed at different times and treated with 0.1% Triton X-100 in 3% formaldehyde solution. The viral proteins were detected by immunostaining with anti-influenza B virus nucleoprotein monoclonal antibody as the primary antibody and biotinylated secondary antibody with the Vectastain ABC kit. The left, middle, and right dishes in each row show cells infected with the wild-type BLeeRG, BLeeBM2stop#4, and BLeeBM2del viruses, respectively.
DISCUSSION
Mould et al. (14) recently demonstrated that the BM2 protein has ion channel activity analogous to that of the M2 protein of influenza A virus. In our study, which unequivocally demonstrates an essential role for the BM2 protein in influenza B virus replication, a virus possessing a mutant BM2 protein with a three-amino-acid truncation at the amino terminus (BLeeBM2stop#3) was not viable. Since the mutant BM2 protein did not appear to be efficiently transported to the cell surface and thus was unlikely to be incorporated into virions, this finding is consistent with the reported function of BM2 protein as an ion channel. That is, the protein must be incorporated into virions before it can function during uncoating.
Although the stop-start translation system (overlapping codons composed of a stop codon for one gene followed by the initiation codon for another) is common in prokaryotes but not in eukaryotes (for a review, see reference 21). This strategy was suggested to be operational for the expression of BM2 protein. Our results demonstrate that mutations in the pentanucleotide and a single-nucleotide deletion downstream of the pentanucleotide did not abolish the production of a protein reactive with antiserum against the C terminus of BM2 protein, suggesting that the ATG in the pentanucleotide may not be essential for the initiation of translation of the BM2 protein in the system tested. Since mutation of the pentanucleotide destroyed the ATG initiation codon, translation might start from a non-ATG codon around the mutated stop-start pentanucleotide lacking this codon. Alternatively, BM2 protein might be expressed from a spliced mRNA. However, like Horvath et al. (9), we were unable to detect a spliced mRNA in plasmid-transfected cells (data not shown). Interestingly, the replacement of the coding region of BM2 protein with that of GFP resulted in expression of the latter protein, indicating that the sequence downstream of the stop-start motif does not affect the stop-start translation system. Direct protein sequencing will be required to fully understand the BM2 expression strategy.
Since BM2 is essential for influenza B virus replication and is highly conserved among these viruses, attenuating mutations can be introduced into this protein for live vaccine development, possibly by combining them with NB protein knockout mutations (7). Two classes of anti-influenza drugs with different inhibitory mechanisms are currently available (for a review, see reference 2), the M2 ion channel inhibitors amantadine and rimantadine and the NA inhibitors zanamivir and oseltamivir. Since M2 ion channel inhibitors do not inhibit influenza B virus replication, only NA inhibitors are effective against influenza B virus infection. Although there has been only one report of an influenza B virus resistant to the NA inhibitors in a clinical trial (6), they obviously do emerge. Also, the results with a BM2-expressing cell line indicated that nonfunctional BM2 proteins may interfere with native BM2, prohibiting viral replication. Thus, we consider BM2 a prudent target for the development of alternative drugs to inhibit influenza B virus infection.
Yamamoto-Goshima et al. (27) suggested that assembly or budding of influenza B virus requires the presence of NA at the plasma membrane, unlike that of influenza A virus. Our results demonstrate that influenza B virus does not require NA protein for its replication so long as sialidase activity is provided exogenously. Thus, by replacing the HA coding sequence with one encoding a protein with receptor binding and fusion capability, e.g., vesicular stomatitis virus G protein (25), the NA segment could be used as a carrier for a foreign gene. This suggestion expands on the potential of influenza A virus as a platform for foreign gene delivery and expression in diverse applications (1, 5, 13, 15, 17, 19, 24, 28). Indeed, an influenza B virus that we generated maintains the GFP gene stably, suggesting its candidacy as a gene delivery vector. Furthermore, we established a cell line expressing BM2 protein. This cell line may be useful for the amplification of large amounts of a virus whose BM2 coding region has been deleted or replaced with a foreign gene for the generation of a live attenuated vaccine or a gene delivery vector. Since influenza B virus infection is limited to humans (26), the risk of its spreading to other species seems negligible, enhancing its potential as a human-specific virus vector.
Acknowledgments
We thank Krisna Wells and Martha McGregor for excellent technical assistance and John Gilbert for editing the manuscript. Automated sequencing was performed at the University of Wisconsin—Madison Biotechnology Center.
This work was supported by grants from the National Institutes of Health, NIAID, by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology and the Ministry of Health, Labor and Welfare, Tokyo, Japan, and by grants from Core Research for Evolutional Science and Technology from the Japan Science and Technology Corporation, Tokyo, Japan.
REFERENCES
- 1.Castrucci, M. R., S. Hou, P. C. Doherty, and Y. Kawaoka. 1994. Protection against lethal lymphocytic choriomeningitis virus (LCMV) infection by immunization of mice with an influenza virus containing an LCMV epitope recognized by cytotoxic T lymphocytes. J. Virol. 68:3486-3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crumpacker, C. 2001. Antiviral therapy, p. 393-433. In D. M. Knipe et al. (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 3.DuBridge, R. B., P. Tang, H. C. Hsia, P. M. Leong, J. H. Miller, and M. P. Calos. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7:379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujii, Y., H. Goto, T. Watanabe, T. Yoshida, and Y. Kawaoka. 2003. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 100:2002-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Sastre, A., T. Muster, W. S. Barclay, N. Percy, and P. Palese. 1994. Use of a mammalian internal ribosomal entry site element for expression of a foreign protein by a transfectant influenza virus. J. Virol. 68:6254-6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gubareva, L. V., M. N. Matrosovich, M. K. Brenner, R. C. Bethell, and R. G. Webster. 1998. Evidence for zanamivir resistance in an immunocompromised child infected with influenza B virus. J. Infect. Dis. 178:1257-1262. [DOI] [PubMed] [Google Scholar]
- 7.Hatta, M., and Y. Kawaoka. 2003. The NB protein of influenza B virus is not necessary for virus replication in vitro. J. Virol. 77:6050-6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann, E., K. Mahmood, C. F. Yang, R. G. Webster, H. B. Greenberg, and G. Kemble. 2002. Rescue of influenza B virus from eight plasmids. Proc. Natl. Acad. Sci. USA 99:11411-11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horvath, C. M., M. A. Williams, and R. A. Lamb. 1990. Eukaryotic coupled translation of tandem cistrons: identification of the influenza B virus BM2 polypeptide. EMBO J. 9:2639-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson, D., A. Cadman, T. Zurcher, and W. S. Barclay. 2002. A reverse genetics approach for recovery of recombinant influenza B viruses entirely from cDNA. J. Virol. 76:11744-11747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobasa, D., M. E. Rodgers, K. Wells, and Y. Kawaoka. 1997. Neuraminidase hemadsorption activity, conserved in avian influenza A viruses, does not influence viral replication in ducks. J. Virol. 71:6706-6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamb, R. A., and R. M. Krug. 2001. Orthomyxoviridae: the viruses and their replication, p. 1487-1503. In D. M. Knipe et al. (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 13.Luytjes, W., M. Krystal, M. Enami, J. D. Pavin, and P. Palese. 1989. Amplification, expression, and packaging of foreign gene by influenza virus. Cell 59:1107-1113. [DOI] [PubMed] [Google Scholar]
- 14.Mould, J. A., R. G. Paterson, M. Takeda, Y. Ohigashi, P. Venkataraman, R. A. Lamb, and L. H. Pinto. 2003. Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. Dev. Cell 5:175-184. [DOI] [PubMed] [Google Scholar]
- 15.Muster, T., B. Ferko, A. Klima, et al. 1995. Mucosal model of immunization against human immunodeficiency virus type 1 with a chimeric influenza virus. J. Virol. 69:6678-6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neumann, G., M. R. Castrucci, and Y. Kawaoka. 1997. Nuclear import and export of influenza virus nucleoprotein. J. Virol. 71:9690-9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann, G., and G. Hobom. 1995. Mutational analysis of influenza virus promoter elements in vivo. J. Gen. Virol. 76:1709-1717. [DOI] [PubMed] [Google Scholar]
- 18.Neumann, G., T. Watanabe, H. Ito, et al. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 96:9345-9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neumann, G., A. Zobel, and G. Hobom. 1994. RNA polymerase I-mediated expression of influenza viral RNA molecules. Virology 202:477-479. [DOI] [PubMed] [Google Scholar]
- 20.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193-199. [DOI] [PubMed] [Google Scholar]
- 21.Normark, S., S. Bergstrom, T. Edlund, T. Grundstrom, B. Jaurin, F. P. Lindberg, and O. Olsson. 1983. Overlapping genes. Annu. Rev. Genet. 17:499-525. [DOI] [PubMed] [Google Scholar]
- 22.Odagiri, T., J. Hong, and Y. Ohara. 1999. The BM2 protein of influenza B virus is synthesized in the late phase of infection and incorporated into virions as a subviral component. J. Gen. Virol. 80:2573-2581. [DOI] [PubMed] [Google Scholar]
- 23.Paterson, R. G., M. Takeda, Y. Ohigashi, L. H. Pinto, and R. A. Lamb. 2003. Influenza B virus BM2 protein is an oligomeric integral membrane protein expressed at the cell surface. Virology 306:7-17. [DOI] [PubMed] [Google Scholar]
- 24.Percy, N., W. S. Barclay, A. Garcia-Sastre, and P. Palese. 1994. Expression of a foreign protein by influenza A virus. J. Virol. 68:4486-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe, T., S. Watanabe, T. Noda, Y. Fujii, and Y. Kawaoka. 2003. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 77:10575-10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright, P. F., and R. G. Webster. 2001. Orthomyxoviruses, p. 1538. In D. M. Knipe et al. (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 27.Yamamoto-Goshima, F., K. Maeno, T. Morishita, M. Ueda, Y. Fujita, K. Nakajima, and S. Yoshii. 1994. Role of neuraminidase in the morphogenesis of influenza B virus. J. Virol. 68:1250-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou, Y., M. Konig, G. Hobom, and E. Neumeier. 1998. Membrane-anchored incorporation of a foreign protein in recombinant influenza virions. Virology 246:83-94. [DOI] [PubMed] [Google Scholar]