

Abstract
Mycoplasma contamination in cell culture is considered as serious problem in the manufacturing of biological products. Our goal in this research is to find the best standard and rapid method with high sensitivity, specificity, accuracy and predictive values of positive and negative results for detection of mycoplasma contamination in cell cultures of the National Cell Bank of Iran. In this study, 40 cell lines suspected to mycoplasma contamination were evaluated by three different methods: microbial culture, enzymatic mycoalert® and molecular. Enzymatic evaluation was performed using the mycoalert® kit while in the molecular technique, a universal primer pair was designed based on the common and fixed 16SrRNA ribosomal sequences used. Mycoplasma contaminations in cell cultures with molecular, enzymatic and microbial culture methods were determined as 57.5, 52.5 and 40 %, respectively. These results confirmed the higher rate of sensitivity, specificity and accuracy for the molecular method in comparison with enzymatic and microbial methods. Polymerase chain reaction (PCR) assay based on fixed and common sequences in the 16SrRNA, is a useful valuable and reliable technique with high sensitivity, specificity and accuracy for detection of mycoplasma contamination in cell cultures and other biological products. The enzymatic mycoalert® method can be considered as a substitution for conventional microbial culture and DNA staining fluorochrome methods due to its higher sensitivity, specificity and speed of detection (<20 min).
Keywords: Mycoalert®, Cell culture, Mycoplasma contamination, Human-animal cell lines
Introduction
In 1956 for the first time, Robinson et al. (1956) observed that their cell cultures were contaminated by mycoplasma. In 1960s, mycoplasma were introduced as the most important and prevalent contaminatons in cell cultures (Razin et al. 1998; Yavlovich et al. 2006; Zhang et al. 2006). Mycoplasma are considered as the smallest living organism within the submicron size (200–800 nm). These polymorphic microorganisms that are categorized in the class of mollicutes and order of tenericutes are also characterized by the lack of a cell wall, self multiplication within 1–9 h and genomic size of 600–2,200 kilobases. Of around 200 species available in the class of mollicutes, some are saprophytes with a commensal life in human, animals, insects, plants and some others like Mycoplasma pneumonia, Mycoplasma genitalium, Mycoplasma hominis and Ureaplasma urealyticum are pathogenic species infecting genitourinary and nasopharyngeal tracts (Harasawa et al. 2005; Waites et al. 2005).
A mycoplasma contamination may affect physiology, growth, morphology as well as biochemical, immunological and genetic characteristics of the cultured cells. Therefore, interpretation of the biological tests on these cells will be affected by incorrect results. In order to increase the quality and safety of the biological products prepared in animal cell cultures, US Food and Drug Administration (FDA) strongly recommends the application of mycoplasma-free cells approved by proper detection tests (Cheng et al. 2007; Dabrazhynetskaya et al. 2011; Huang et al. 2008; Uphoff and Drexler 2011). It has been reported (in different studies) that between 5 and 87 % of the cell lines have been contaminated by mycoplasmas. From more than 200 mycoplasma species, 20 have been isolated from cell cultures (Uphoff and Drexler 2013). Eight species including M. arginini, M. fermentans, M. hominis, M. orale, M. hyorhinis, M. salivarium, M. pirum and A. laidlawii are responsible for more than 95 % of cell culture contaminations (Harlin and Gajewski 2008; Markoullis et al. 2009; Timenetsky et al. 2006). The main sources of mycoplasma contamination in cell cultures are laboratory staff, serum, culture media, materials, reagents, instruments and previously contaminated cells. Meanwhile, mollicutes may be present in cell cultures without any superficial detectable effects like turbidity or pH changes (Zhao et al. 2008; Nikfarjam and Farzaneh 2012; McGarrity et al. 1985). Several techniques have been developed for mycoplasma detection in cell cultures including direct microbial culture or indirect non culture methods. These techniques have differences in accuracy, reliability, sensitivity, specificity, precision and also cost-efficiency (Kong et al. 2007; Peredeltchouk et al. 2011). Culture-based methods are considered as time consuming (days to several weeks) and low sensitive methods with relatively high false negative results, which needs expert interpretation. In addition, some of mycoplasma species such as M. hyorhinis are hardly grown in culture and not detectable with culture-based methods. Nonetheless, microbial culture is still considered as the gold standard for mycolplasma detection. Non-culture-based methods include screening of Adenosine phosphorylase (Adop), cell markers, biochemical and immunological techniques, DNA fluorochrome staining, electron microscopy, DNA-RNA hybridization, one-step PCR, nested PCR, PCR-ELISA, genus and species specific PCR with universal and multiplex primers and real time PCR (Hopert et al. 1993; Lawrence et al. 2010; Störmer et al. 2009; Zhi et al. 2010). These methods are generally more cost-effective and easier to perform, however, they have their own disadvantages from the view of sensitivity, specificity and accuracy. In this research, microbial culture (as a gold standard), enzymatic (mycoalert® kit) and PCR methods were utilized for the detection of mycoplasma contamination in the cell collections of the National Cell Bank of Iran. The sensitivity, specificity, accuracy and operation time (speed) of these methods were compared in order to identify the optimal method of choice.
Materials and methods
Cell cultures
Different animal and human cell lines available in the National Cell Bank of Iran were randomly selected and evaluated by microbial culture, enzymatic (mycoalert®, Lonza, Basel, Switzerland) and molecular detection methods (Table 1). These cell lines were incubated at 37 °C in 88 % humidified and 5 % CO2 atmosphere. The culture medium for each cell line was prepared according to the recommended instructions and supplemented by 10–20 % fetal bovine serum (FBS) and growth factors. The following reagents were used: Fetal Bovine Serum (FBS, Gibco®-Invitrogen, Cat No: 10270-106), Roswell Park Memorial Institute medium (RPMI, Gibco®, Cat No: 51800-035), Dulbecco’s Modified Eagle Medium High Glucose (DMEM, Gibco®, Cat No: 52100-021), F12 nutrient mixture (Hams’F12, Gibco®-Invitrogen, Cat No: 21700-075), Non Essential Amino Acid (NEAA, Gibco® MEM, Cat No: 11140076), Penicillin/Streptomycin (Gibco®, Cat No: 15140-130), Horse serum (Gibco®, Cat No: 16050-130 (origin: New Zealand)), Trypsin-EDTA (Gibco®, Cat No: 25300-054), 100 mM Sodium pyruvate (Gibco®, Cat No: 11360070), were supplied by Gibco/Invitrogen Company (Carlsbad, CA, USA). Oxalate, Pyruvate, and Insulin (Sigma-Aldrich®, OPI, Cat No: O 5003), Bovine Insulin (Sigma-Aldrich®, Cat No: I 6634), Human Insulin (Sigma-Aldrich®, Cat No: I 9278), Epidermal Growth Factor (Sigma-Aldrich®, EGF, Cat No: E 9644), Fibroblast Growth Factor-Basic from bovine pituitary (Sigma-Aldrich®, bFGF, Cat No: F 5392), Human Endothelial Cell Growth Factor (Sigma-Aldrich®, Cat No: E 9640), Heat-Inactivated FBS (Sigma-Aldrich®, Cat No: F4135), 2-mercaptoethanol (0.05 mM 2ME, Sigma-Aldrich®, Cat No: M3148) and Macrophage-Colony Stimulating Factor (M-CSF, Sigma-Aldrich®, Cat No: M9170) were purchased from Sigma (St. Louis, MO, USA). All the reagents including FBS, culture medium, phosphate buffer saline (PBS) (Sigma-Aldrich®, Cat No: P-4417) and trypsin/EDTA (Gibco®, Cat No: 25300-054) were initially evaluated for mycoplasma contamination by PCR, microbial culture and DAPI-staining methods. Fluorescence DNA staining with DAPI (4′, 6-Diamidine-2-phenyl indole dihydro chloride, Roche, Cat No: 10236276001, Mannheim, Germany) was performed based on the rapid uptake of the dye by cells and also binding selectively to minor grooves of cell and mycoplasmal DNA. The fluorescent DAPI dye (Roche) was dissolved in water to make a 1 mg/ml stock. The working solution was freshly prepared by diluting the DAPI stock into 1 μg/ml with methanol. Cells cultured on cover slip slides (Thermo Scientific, USA) were rinsed once with the working solution, incubated with the working solution at 37 °C for 15 minutes and rinsed with methanol. Slides were mounted with glycerol and examined under a fluorescence microscope with 340/380 nm excitation filter. When the cells are contaminated with mycoplasmas, discrete fluorescent foci are readily detected over the cytoplasm and sometimes in intercellular spaces (Jung et al. 2003). In order to detect the mycoplasma contamination after harvesting, each cell line was cultured in an antibiotic-free medium for at least 4 days without exchanging the medium (Freshney et al. 2006). All cells were examined under the quality control of microbial cultures to establish whether cells were not contaminated with other microorganisms. Accordingly, Nutrient Broth (Sigma-Aldrich®, Cat No: 70122 FLUKA), Sabouraud Dextrose Broth (BD-Difco, Cat No: 238230, Franklin Lakes, NJ, USA), Thioglycolate Broth (BD-BBL™, Cat No: 297642), Brain Heart Infusion Broth (BD-BBL™, Cat No: 221812), Trypticase Soy Broth (BD-BBL™, Cat No:221715), Yeast Malt Broth (Sigma-Aldrich®, Cat No: Y3752), Blood Agar (Sigma-Aldrich®, Cat No: 70133 FLUKA), Nutrient Agar (BD-Difco™, Cat No: 212000), MacConkey Agar (Sigma-Aldrich®, Cat No: 70143 FLUKA), Sabouraud Dextrose Agar (BD-RODAC™, Cat No: 295872), and Brain Heart Infusion agar (BD-BBL™, Cat No: 211065) were used for investigation of microbial contaminants and quality control (Hay and Ikonomi 2005).
Table 1.
List of selected cell lines for checking of mycoplasma contamination in the present study
| No | NCBI code | Cell lines | Cell type | Culture medium |
|---|---|---|---|---|
| 1 | NCBI C450 | 5637 | Human bladder carcinoma | RPMI 1640 + 10 % FBS |
| 2 | NCBI C135 | MCF7 | Human breast adenocarcinoma | |
| 3 | NCBI C516 | MOLT-17 | Human T cell leukemia | |
| 4 | NCBI C161 | L929 | Mouse connective tissue fibroblast | |
| 5 | NCBI C549 | C1300 Clone NA | Mouse neuroblastoma | |
| 6 | NCBI C437 | HT 1080 | Human fibrosarcoma | |
| 7 | NCBI C131 | AGS | Human caucasian gastric adenocarcinoma | |
| 8 | NCBI C124 | RAJI TK+ | Human Burkitt,s lymphoma (Thymidine kinase positive) | |
| 9 | NCBI C138 | RAJI TK− | Human Burkitt,s lymphoma (Thymidine kinase deficient) | |
| 10 | NCBI C181 | KE37 | Human T cell acute lymphoblastic leukemia | |
| 11 | NCBI C578 | MDA-MB231 | Human breast adenocarcinoma | |
| 12 | NCBI C428 | DU145 | Human prostatic carcinoma | |
| 13 | NCBI C433 | MDA-MB361 | Human breast adenocarcinoma | |
| 14 | NCBI C209 | SKOV3 | Human ovary adenocarcinoma | |
| 15 | NCBI C160 | Luckes | Human Burkitt,s lymphoma | |
| 16 | NCBI C103 | BL 28 | Human Burkitt,s lymphoma | |
| 17 | NCBI C146 | SW742 | Human colorectal adenocarcinoma | |
| 18 | NCBI C459 | MIA Paca-2 | Human pancreatic carcinoma | |
| 19 | NCBI C110 | B95.8 | Marmoset-EBV transformed lymphocytes | |
| 20 | NCBI C207 | SKBR3 | Human breast adenocarcinoma | |
| 21 | NCBI C515 | NB4 | Human acute promyelocytic leukemia | |
| 22 | NCBI C212 | Nalm6 | Pre B cell leukemia | |
| 23 | NCBI C453 | Saos2 | Human osteogenic sarcoma | |
| 24 | NCBI C612 | C6/36 | Aedesalbopictus (mosquito, Asian tiger) | DMEM + 10 % FBS |
| 25 | NCBI C149 | MOLT4 | Human acute T lymphoblastic leukemia | |
| 26 | NCBI C483 | J774A.1 | Mouse monocyte/macrophage | |
| 27 | NCBI C565 | QU-DB | Human large cell lung carcinoma | |
| 28 | NCBI C540 | B16/F10 | Mouse melonema | |
| 29 | NCBI C118 | 1321N1 | Human brain astrocytoma | |
| 30 | NCBI C577 | BE(2)-C | Human neuroblastoma | |
| 31 | NCBI C456 | Mel3 | Rhesus mammary gland carcinoma | |
| 32 | NCBI C143 | COS7 | Monkey kidney SV40 transformed | |
| 33 | NCBI C114 | EL4 | Mouse T Cell lymphoma | |
| 34 | NCBI C137 | A549 | Human-lung-carcinoma | Ham’s F12 (DMEM + 2 mM Glutamine) + 10% FBS |
| 35 | NCBI C111 | CHO | Chinese hamster ovary | |
| 36 | NCBI C141 | G-8 | Mouse Swiss Webster myoblast | DMEM + 10% Horse Serum + 10% FBS |
| 37 | NCBI C555 | MG63 | Human osteosarcoma | DMEM + 0.1 Mm NEAA*+1.0 mM SP** + 10 % heat-inactivated FBS |
| 38 | NCBI C598 | HSKMC | Human fetal skeletal muscle cells | Muscle Cell Growth Medium |
| 39 | NCBI C153 | PC12 | Rat Adrenal fibroblast pheochromocytoma | RPMI 1640 + 5 % FBS + 10 % horse serum |
| 40 | NCBI C482 | M-NFS-60 | Mouse Myeloid Leukemia | RPMI 1640 + 0.05 mM 2ME*** + 2000 U/ml M-CSF**** + 10% FBS |
NEAA* Non-essential amino acids, SP** Sodium pyruvate, 2ME*** 2-Mercapto ethanol, CSF**** Macrophage-Colony Stimulating Factor
Positive and negative controls for PCR technique
The DNA of different mollicutes and bacterial strains were supplied from American Type Culture Collection (ATCC, Manassas, VA, USA) and National Collection of Type Cultures (NCTC, Salisbury, UK). Ureaplasma urealiticum (NCTC 10177T), M. genitalium (NCTC 10195), M. salivarium (NCTC 10113), M. pneumonia (NCTC 10119), A. laidlawii (ATCC 23206), M. orale (ATCC 23714), M. hyorhinis (ATCC 17981), M. fermentans (ATCC 19989), M. hominis (ATCC 23114), M. arginini (ATCC 23828) and M. pirum (NCTC 11702) were used as positive controls for genus specific PCR and genomic DNA from gram positive and negative bacteria like Staphylococcus aureus (ATCC 25923), Proteus mirabilis (ATCC 49565), Bacillus subtilis (ATCC 6633) and Pseudomonas aeruginosa (ATCC 49189) were utilized as negative controls to verify the non-cross reactivity of mycoplasma universal primers.
Microbial culture for detection of mycoplasma contamination in cell lines
For microbial culture, 1 ml of a cell culture medium plus contaminated cells were added to PPLO broth (supplemented with glucose and arginine) and incubated for 72 h. Afterwards, PPLO medium was vigorously mixed to obtain monotonous turbidity. This medium was centrifuged for 10 min and 100 μl of the precipitate transferred to a solid PPLO agar culture plate. The plate was carefully sealed to prevent contamination and evaporation and incubated at 37 °C for 4 weeks. The formation of egg form or non- typical colonies of mycoplasm was investigated by light microscopy every 3 days (Dabrazhynetskaya et al. 2011; Volokhov et al. 2011; Peredeltchouk et al. 2011).
Mycoalert® mycoplasma detection kit for mycoplasma contamination in cell lines
The enzymatic Mycoalert® is a selective biochemical test (bioluminescent reaction) that reveals the activity amount of mycoplasma enzymes. As described earlier, detection of mycoplasma is performed by measuring the acetate kinase or carbamate kinase Activity (Pitt et al. 2012). These specific enzymes provide a rapid screening method for sensitive detection of mycoplasma contamination in cell cultures. The enzymes are released during viable mycoplasma lysis and able to transform adenosine diphosphate (ADP) into adenosine triphosphate (ATP) by reaction with carbamoyl phosphate or acetyl phosphate (Mariotti et al. 2008; Volokhov et al. 2008). The level of ATPs in each sample was measured before (Reading A) and after (Reading B) the addition of Mycoalert® substrate. This ratio (B/A) was considered as an indicator for the presence of mycoplasma contamination. The ratio (Reading B/Reading A) >1, confirms the existence of contamination. If these enzymes are not present, the second reading shows no increase over the first, while reaction of mycoplasmal enzymes with their specific substrate, leads to elevated ATP levels. This increase in level of ATP was determined by luminometer (Berthold, FB12, Bad Wildbach, Germany) at the optimum temperature for luciferase activation (20 °C) according to the following formula (equation).
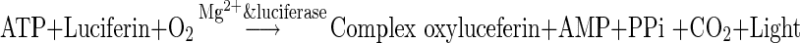 |
The procedure was performed according to the manufacture’s instruction and the intensity of the emitted light is correlated linearly with released ATP concentration and is measured using a luminometer.
Mycoplasma detection with the molecular method (PCR)
The specific PCR with universal primers was initially started by DNA extraction from suspicious samples based on a previously published method with a modification (Tang et al. 2000). Briefly, Cells (1 × 105) in final logarithmic growth phase were harvested and centrifuged at 12,000g for 1 min. The cell pellet was suspended in STE buffer (NaCl 10 mM, TrisHCl 20 mM and EDTA 1 mM, pH = 8.0) and incubated with sodium dodecyl sulfate (SDS, 1 % w/v) and proteinase K (40 μg/ml, Promega, Madison, WI, USA) for 2 h at 37 °C. Afterwards, a solution of phenol–chloroform-isoamyl alcohol (1:24:25) was added to an equal volume of the suspended cells to precipitate the proteins. The total nucleic acids were precipitated by the addition of 1 volume of sodium acetate (3 M, pH = 5.2) and 25 volume 96 % ethanol. The cell pellet was washed with ethanol (70 % v/v) and dried for 30 min at room temperature. Finally, the precipitated DNA was suspended in RNA-DNA free sterile deionized water and kept at −20 °C (Molla Kazemiha et al. 2009, 2011).
Oligonucleotides and specific PCR for mycoplasma detection in cell lines
In order to detect the mycoplasma contamination of cell lines with the PCR method, a universal primer pair in addition to 11 species-specific primer pairs were designed based on the 16SrRNA of mollicutes according to the previously published reports (Molla Kazemiha et al. 2009, 2011) (Table 2). PCR master mix was prepared by 2.5 μl PCR 10x Buffer, 1 μl dNTPs (50 μM), 3 μl forward and reverse primers (15 pmol), 1 μl Taq DNA polymerase enzyme (1 U), 1 μl magnesium chloride (1.5 mmol), 12.5 μl distilled water and 1 μl mycoplasma genomic DNA (0.1 μg/μl) or cell DNA (1 μg/μl). Afterwards, the mixture was heated at 94 °C for 3 min and then 32 cycles of amplification including 94 °C for 60 s, 60 °C for 30 s, 72 °C for 1 min were applied to the mixture. The temperature for universal primers and species-specific primers were set at 55 and 60 °C, respectively. Finally, the PCR products were separated by agarose gel (1 % w/v) electrophoresis and visualized by UV transilluminometer.
Table 2.
The sequences of oligonucleotide primers used for detection of mycoplasmas
| Mycoplasma species | Primer sequence | Amplicon size | GC % | Tm |
|---|---|---|---|---|
| Universal primer | S: GTG GGG AGG AAA YAG GAT TAG A AS: GGC ATG ATG ATT TGA CGT CRT |
425 bp | 45–50 | 53–54.8 |
| 45–48 | 50.5–52.4 | |||
| M. arginini | S: TGA TCA TTA GTC GGT GGA GAG TTC AS: TAT CTC TAG AGT CCT CGA CAT GAC TC |
326 bp | 46 | 55.7 |
| 46 | 58 | |||
| M. orale | S: TGA TCA TTA GTC GGT GGA AAA CTA | 325 bp | 38 | 52.3 |
| AS: TAT CTC TAG AGT CCT CGA CAT GAC TC | 46 | 58 | ||
| M. hyorhinis | S: CGA TGA TCA TTA GTT GGT GGA ATA AAT AS: AGG CAG TAT CTC TAG AGT CCT TAA CTT A |
334 bp | 33 | 53.7 |
| 39 | 57 | |||
| M. fermentans | S: TGA TCA TTA GCT GAT GGG GAA CT AS: TCT CTT AGA GTC CTC AAC TAA ATG |
324 bp | 43 | 53.5 |
| 38 | 52.3 | |||
| M. genitalium | S: ATA GAT ACT AGC TGT CGG AGC GAT AS: CCA ATT TAC ATT AGC AGT CTC GTT AA |
335 bp | 46 | 55.7 |
| 35 | 53.2 | |||
| A. laidlawii | S: GAT GAG AAC TAA GTG TTG GCC ATA A AS: CGC TAG AGT CCC CAA CTT AAT GA |
300 bp | 40 | 54.4 |
| 48 | 55.3 | |||
| M. hominis | S: ATC ATT AGT CGG TGG AGA ATC A AS: GCA GTA TCT CTA CTA GAG TCC TCA ACT TAAT |
301 bp | 41 | 55.1 |
| 39 | 59.1 | |||
| M. pirum | S: TGG ATG TTA GAT GTC GGG GTA AA AS: GTT GGC AGT ATC GCT AGA CAA A |
324 bp | 43 | 53.5 |
| 41 | 56.7 | |||
| M. pneumoniae | S: GAT ACT AGC TGT CGG GGC GAT AS: AAT TTG CAT TAG TAG CAG TCT CGC TAG |
329 bp | 57 | 56.3 |
| 41 | 56.7 | |||
| M. salivarium | S: GAT CAT TAG TCG GCA GAG AAC TCG AS: TAT CTC TAG AGT CCT CGA CAT GAC TC |
324 bp | 50 | 57.4 |
| 46 | 58 | |||
| U. urealyticum | S: CAT CAT TAA ATG TCG GCT CGA A AS: CGG TAG CAG TAT CGC TAG AAA AGC |
323 bp | 41 | 51.1 |
| 50 | 57.4 |
Tm = melting temprature
Statistical evaluation
The data analysis was done using the SPSS statistical software version 20. P values <0.05 were considered statistically significant. Descriptive statistics (i.e. frequencies and percentages) were used to summarize the quantitative variables. Data were analyzed by t test for comparing two tests and Freidman test for comparing three tests. Sensitivity, specificity, accuracy and predictive value of positive and negative results were calculated using Microbial Culture as the Gold Standard (Hopert et al. 1993; Uphoff and Drexler 2002; Galen and Gambino 1975). Sensitivity: true positives/(true positives + false negatives); Specificity: true negatives/(true negatives + false positives); Predictive value of a positive result: true positives/(true positives + false positives) and Predictive value of a negative result: true negatives/(true negatives + false negatives), respectively; Accuracy: (true positives + true negatives)/total number of cases.
Results
In this research, 40 different human and animal cell lines were randomly selected and assessed for mycoplasma contamination using microbial culture, enzymatic and PCR methods (Table 1). Vero and NSO cell lines were evaluated by these methods and considered as positive and negative controls, respectively (Fig. 1). Three different strains including M. hyorhinis, M. arginini and M. fermentans were detected in the positive control (Vero cell line) by species-specific PCR primers (Fig. 2). Microbial culture, enzymatic mycoalert® and PCR tests, respectively, showed 40 % (16 cases), 52.5 % (21 cases) and 57.5 % (23 cases) contamination among the 40 studied cell lines. Indeed, microbial culture and enzymatic tests indicated 7 and 2 false negative without any false positive (Table 3).
Fig. 1.

PCR gel electrophoresis results for detection of mycoplasma contamination in Vero (positive control), NSO (negative control) and other cell lines. Lane 1 DNA free water (negative control), lane 2 NSO (negative control), lane 3 CHO (negative), lane 4 Vero (positive control), lane 5 L929 (positive), lane 6 DU145 (positive), lane 7 MOLT17 (positive), lane 8 Saos2 (positive), lane 9 B16F10 (positive), lane 10 Vero (positive control), lane 11 NSO (negative control), lane 12 5,637 (negative), lane 13 J774.A (negative), lane 14 DNA size marker (100 bp DNA Ladder, Roche VIII)
Fig. 2.

PCR gel electrophoresis results for detection of mycoplasma contamination with three different mycoplasma species in Vero cell line as positive control with mycoplasma species-specific primers. Lane 1 DNA size marker (100 bp DNA Ladder, Roche VIII), lane 2 M. hominis-specific primer (negative), lane 3 M. pirum-specific primer (negative), lane 4 M. pneumoniae-specific primer (negative), lane 5 A. laidlawii-specific primer (negative), lane 6 M. salivarium-specific primer (negative), lane 7 M. hyorhinis-specific primer (positive with Amplicone size 334 bp), lane 8 M. arginini-specific primer (positive with Amplicone size 326 bp), lane 9 M. fermentans-specific primer (positive with Amplicone size 324 bp), lane 10 M. orale-specific primer (negative), lane 11 M. genitalium-specific primer (negative), lane 12 U. urealyticum-specific primer (negative), lane 13 DNA-free water (negative control)
Table 3.
The evaluation results of 40 cell lines for the detection of mycoplasma contamination by three different methods (microbial culture, molecular PCR and mycoalert® enzymatic assay)
| Number | NCBI code | Cell lines | Test results | ||
|---|---|---|---|---|---|
| Microbial culture | Mycoalert® | PCR | |||
| 1. | NCBI C612 | C6/36 | Negative | Negative | Negative |
| 2. | NCBI C149 | MOLT4 | Negative | Negative | Negative |
| 3. | NCBI C450 | 5,637 | Negative | Negative | Negative |
| 4. | NCBI C483 | J774A.1 | Negative | Negative | Negative |
| 5. | NCBI C135 | MCF7 | Positive | Positive | Positive |
| 6. | NCBI C516 | MOLT17 | Negative (false-negative) | Positive | Positive |
| 7. | NCBI C161 | L929 | Positive | Positive | Positive |
| 8. | NCBI C565 | QUDB | Positive | Positive | Positive |
| 9. | NCBI C540 | B16F10 | Negative (false-negative) | Positive | Positive |
| 10. | NCBI C549 | C1300 Clone NA | Positive | Positive | Positive |
| 11. | NCBI C437 | HT-1080 | Negative | Negative | Negative |
| 12. | NCBI C118 | 1321N1 | Negative | Negative | Negative |
| 13. | NCBI C131 | AGS | Positive | Positive | Positive |
| 14. | NCBI C124 | RAJI TK+ | Negative (false-negative) | Positive (weak positive) | Positive |
| 15. | NCBI C138 | RAJI TK− | Negative | Negative | Negative |
| 16. | NCBI C181 | KE-37 | Positive | Positive | Positive |
| 17. | NCBI C577 | BE(2)-C | Positive | Positive | Positive |
| 18. | NCBI C578 | MDA-MB231 | Negative (false-negative) | Positive | Positive |
| 19. | NCBI C153 | PC12 | Positive | Negative (false-negative) | Positive |
| 20. | NCBI C428 | DU145 | Positive | Positive | Positive |
| 21. | NCBI C433 | MDA-MB361 | Negative | Negative | Negative |
| 22. | NCBI C209 | SKOV3 | Negative | Negative | Negative |
| 23. | NCBI C160 | Luckes | Negative | Negative | Negative |
| 24. | NCBI C103 | BL28 | Negative | Negative | Negative |
| 25. | NCBI C146 | SW742 | Negative | Negative | Negative |
| 26. | NCBI C459 | MIA Paca-2 | Negative (false-negative) | Negative (false-negative) | Positive |
| 27. | NCBI C482 | M-NFS-60 | Negative (false-negative) | Positive | Positive |
| 28. | NCBI C456 | MEL-III | Negative (false-negative) | Positive | Positive |
| 29. | NCBI C141 | G-8 | Positive | Positive | Positive |
| 30. | NCBI C207 | SKBR3 | Positive | Positive | Positive |
| 31. | NCBI C137 | A549 | Negative | Negative | Negative |
| 32. | NCBI C143 | COS7 | Positive | Positive | Positive |
| 33. | NCBI C110 | B95.8 | Negative | Negative | Negative |
| 34. | NCBI C114 | EL4 | Positive | Positive | Positive |
| 35. | NCBI C111 | CHO | Negative | Negative | Negative |
| 36. | NCBI C515 | NB4 | Positive | Positive | Positive |
| 37. | NCBI C598 | HSKMC | Negative | Negative | Negative |
| 38. | NCBI C555 | MG63 | Positive | Positive | Positive |
| 39. | NCBI C212 | Nalm6 | Negative | Negative | Negative |
| 40. | NCBI C453 | Saos2 | Positive | Positive | Positive |
Sensitivity of mycoalert® test has been announced <50 cfu/ml by manufacturer. For example, sensitivity of this kit for A. laidlawii, M. hyorhinis and M. orale was reported 10–20 cfu/ml. In this experiment, serial dilutions (1/2–1/4,096) were prepared from the positive control of the kit, Vero cell line and also A. laidlawii strain. The limit of detection (LOD) for all those three samples was obtained until 1/256 dilution (Table 4). Besides in molecular PCR, at annealing temperature of 55 °C, the universal pair primers could amplify the DNA of all mollicute strains so that a 425 bp product was obtained (Fig. 3). However, there was no cross-reaction indicated for these primers with rat DNA, human DNA, mouse DNA as well as prokaryotes and bacteria DNA such as S. aureus, P. mirabilis, B. subtilis and P. aeruginosa (data not shown). In order to examine the sensitivity of the universal primer, different serial dilutions of extracted DNA from A. laidlawii were prepared. For the detection of amplified product, the detection limit was sensitive 10-7 (10 fg). This proves the high sensitivity, specificity and accuracy of the PCR method in comparison with other direct and indirect mycoplasma detection methods (Fig. 4). Of note, to ensure that cell lines were not contaminated by other gram positive and negative bacteria samples taken from the cell line cultures were transferred to various microbial culture media. No bacterial growth was detected for these cultures (data not shown), which confirmed the specificity of mycoalert® results.
Table 4.
LOD (limit of detection) evaluation of positive control in mycoalert® mycoplasma detection kit. Different serial dilutions in the range of 1/2 to 1/4,096 were prepared
| Control positive kit | Reading A | Reading B | B/A | Test results |
|---|---|---|---|---|
| Undiluted positive control kit | 1/364 | 206/435 | 151/345 | Positive >1 |
| Dilution 1/2 | 1/334 | 103/456 | 77/553 | Positive >1 |
| Dilution 1/4 | 1/668 | 51/683 | 30/985 | Positive >1 |
| Dilution 1/8 | 2/209 | 47/161 | 21/349 | Positive >1 |
| Dilution 1/16 | 2/075 | 24/618 | 11/864 | Positive >1 |
| Dilution 1/32 | 2/842 | 12/893 | 4/536 | Positive >1 |
| Dilution 1/64 | 2/359 | 5/556 | 2/355 | Positive >1 |
| Dilution 1/128 | 3/579 | 4/190 | 1/170 | Positive >1 |
| Dilution 1/256 | 2/392 | 3/479 | 1/454 | Positive >1 |
| Dilution 1/512 | 2/751 | 2/190 | 0/796 | Negative <1 |
| Dilution 1/1,024 | 3/168 | 1/185 | 0/374 | Negative <1 |
| Dilution 1/2,048 | 2/203 | 0/985 | 0/447 | Negative <1 |
| Dilution 1/4,096 | 1/996 | 0/571 | 0/286 | Negative <1 |
| Control negative kit | 1/080 | 0/681 | 0/630 | Negative <1 |
Fig. 3.

PCR gel electrophoresis of different mycoplasma DNA strains with specific universal primers of mycoplasma genus. Lane 1 DNA Size marker (100 bp DNA Ladder, Roche VIII), lane 2 M. orale, lane 3 M. hyorhinis, lane 4 M. arginini, lane 5 M. genitalium, lane 6 M. hominis, lane 7 A. laidlawii, lane 8 M. salivarium, lane 9 M. pirum, lane 10 M. pneumoniae, lane 11 U. urealyticum, lane 12 M. fermentans, lane 13 DNA-free water (negative control)
Fig. 4.

PCR gel electrophoresis results, sensitivity analysis of optimized PCR assay performed with universal primers were designed and different concentrations of DNA/μL obtained by dilution from a culture of A. laidlawii (limit of detection). Lane 1 DNA-free water (negative control), lane 2 100 ng (positive), lane 3 10 ng (positive), lane 4 1 ng (positive), lane 5 100 pg (positive), lane 6 10 pg (positive), lane 7 1 pg (positive), lane 8 100 fg (positive), lane 9 10 fg (positive), lane 10 1 fg (negative), lane 11 100 atg (negative), lane 12 DNA-free water (negative control), lane 13 DNA Size marker (100 bp DNA Ladder, Roche VIII)
Statistical results
The specificity for all three methods was calculated 100 % while the sensitivity of PCR, enzymatic mycoalert® and microbial culture methods was determined 100, 91.30 and 69.56 %, respectively. In addition, the highest and lowest accuracy belonged to PCR (100 %) and microbial culture (85.10 %) methods, respectively. The predictive value of positive results were assessed as 100 % for these methods, while the predictive value of negative results was estimated 100 % for PCR, 90.47 % for enzymatic mycoalert® and 77.41 % for microbial culture techniques (Fig. 5). Table 5 compares the statistical validity of the methods two-by-two using standard t-test analysis. The differences between enzymatic mycoalert® and microbial culture methods and also enzymatic and PCR methods were not statistically significant. On the other hand, the comparison of PCR and microbial culture methods showed the statistically significant differences with P value <0.05. According to the ranking method using the Friedman test, variable with the lowest average ranking becomes the most important. The average ranking for PCR, enzymatic mycoalert® and microbial culture were calculated as 1.89, 1.96 and 2.15, respectively. Moreover, the P value in the Friedman test was also significant (P value = 0.008). Therefore, the similarities in the ranking of these three methods were discarded. Correspondingly, these three methods were ranked as molecular PCR, enzymatic mycoalert® and microbial culture.
Fig. 5.

Comparison of the statistical parameters related to each method. The specificity, sensitivity and accuracy for all three methods were calculated
Table 5.
Pairwise multiple comparison test procedures (the test statistics t test and P value)
| Type of testing | Statistics of t test | P value |
|---|---|---|
| Microbial culture | ||
| Mycoalert® | 1.955 | 0.058 |
| PCR | 2.876 | 0.006 |
| Mycoalert® | ||
| PCR | 1.433 | 0.16 |
Discussion
Cell cultures are extensively used in biological research in order to evaluate the biopharmaceutical products. Mycoplasma is an inevitable and common infectious agent in cultures which can change the biological and biochemical response of the cells and also the reliability of the results (Smith and Mowles 1996). Therefore, cells must be screened for mycoplasma contamination before any experiment. The prevalence of mycoplasma contamination has been reported between 5 and 35 % throughout the world (Harlin and Gajewski 2008; McGarrity and Kotani 1985). Nowadays, several direct and indirect techniques have been developed by researchers. In direct methods, microbial colony growth on agar medium can be evaluated directly without any false positive results. On the other hand, enzymatic and metabolic activities or genomic productions of mycoplasma are indirectly checked to determine the presence of contamination. The characteristics of an ideal method are sensitivity, specificity, accuracy, quickness, cost effectiveness and ease of interpretation. In order to ensure the validity of microbial test, it is recommended to verify the experiment with at least another method. Some of the mycoplasma strains cannot grow in artificial culture mediums (M. hyorhinis, M. genitalium, M. orale, M. amphoriforme and M. vulturii). This is considered as an important disadvantage of microbial culture method which results in false negative results, as we also detected 7 false negative cases by microbial culture. The slow growth rate of mycoplasma (duplication time 1–9 h) is another problem that spends 2–6 weeks for mycoplasma colony appearance (Del Giudice et al. 1980).
Because of incomplete respiration pathway in mycoplasma and the absence of tricarboxylic cycle and cytochrome, the ATP production that depends on oxidative phosphorylation (except malate dehydrogenase avtivity) is stopped. Therefore, the fermentative arginine and carbohydrate pathways are considered as ATP production survival systems in mollicultes (except in ureaplasmas for which ATP production depends on urea hydrolysis) (Markoullis et al. 2009).
Mycoalert® kit as a biochemical test, detects the mycoplasma enzymes such as acetate kinases and carbamate kinases. Theses enzymes are able to catalyze and change the ADP to ATP in the presence of phosphate acetyl and phosphate carbamoyl substrates. This produces a detectable luminescence signal related to the luciferase activity. The sensitivity, accuracy, specificity and high speed (<20 min) are the main advantages of enzymatic test. In addition, in opposite to PCR method, detection of enzymatically-active viable mycoplasma germs is highly valuable, particularly in antibiotic therapies. In this study, false negative results for the enzymatic method were detected in only two cell lines including PC12 (NCBI C153) and MIA paca-2 (NCBI C459). In all other 38 cases, the PCR results for contamination were concordant with Mycoalert® results. The main disadvantage of this method is related to the false negative results due to the function of enzymes. The quality, quantity and stability of acetate kinases and carbamate kinases must be in appropriate level for detection by luminometer. In addition, this method is unable to detect different genus and species of ureaplasmas such as U. urealyticum and also specific mycoplasma species (Volokhov et al. 2011; Mariotti et al. 2008).
PCR techniques with the protocols based on 16SrRNA and 16S-23SrRNA of different mycoplasma species include various strategies such as one- and two-step PCR (nested PCR), genus and species specific PCR, gel electrophoresis and real time PCR. The results obtained in this study reveal that the fix and common ribosomal sequences (16SrRNA) can be considered as an appropriate target for detection of different mycoplasma strains in cell cultures. This strategy is not only capable of detecting eight typical mycoplasma strains including M. hyorhinis, M. arginini, M. orale, M. fermentans, M. salivarium, M. hominis, M. pirum and A. laidlawii (that are responsible for 98 % of culture contaminations), but also may identify other mollicute species belonging to mycoplasma genus, Acholeplasma, Ureaplasma and Spiroplasma (which are responsible for 2 % of culture contaminations). The main advantages of the PCR method in comparison with enzymatic and microbial techniques are higher sensitivity, specificity, accuracy and predictive value of positive and negative results.
The preference of microbial and enzymatic tests over the PCR method is that these methods act on living organisms instead of DNA of live or dead organisms. Viable mycoplasma may be successfully removed during treatment periods while its DNA still remains. The presence of false positive results is considered as an important disadvantage of PCR-based methods especially in nested PCR (Cheong et al. 2011). Although the hybridization technique is basically established on PCR method principles, discrimination between specific and non specific signals due to the possibility of the presence of gram positive bacteria is very difficult. Thus, the true positive results of hybridization are acceptable when no gram positive bacteria are detected via microbiological culture. The sensitivity of DNA-rRNA hybridization is approximately supposed in the range of 103–104 organisms while this range for PCR is considered as 1–10 organisms. Therefore, higher sensitivity and specificity are obtained by the PCR method in comparison with the other techniques (Uphoff and Drexler 2002).
In a previous study by Mc Garrity et al. mycoplasma contamination was evaluated in 30 cell lines using microbial and PCR methods, which respectively detected 10 and 14 positive ones. Some of the M. hyorhinis strains were not able to grow in microbial cultures (McGarrity et al. 1985; Del Giudice et al. 1980). In contaminated cultures, more than 1,000 mycoplasma cfu are necessary to be detectable by microbial culture while in PCR and PCR-ELISA methods <10 mycoplasma cfu can be detected. This explains the advantageous of the PCR over the classical methods especially in the case of low level contaminations (right before invasion of infection or after eradication of agents) (Young et al. 2010).
In spite of precaution and preventive observations, the risk of PCR products contamination in molecular diagnosis is probable. However, utilizing appropriate controls different steps is helpful in decreasing false positive and negative results. Low quantities of mycoplasma in sample, non homogenous distribution of mycoplasma in the cell culture and spurious PCR products in extraction and purification procedures are the main causes of false negative results in the PCR technique. Consequently, PCR detection can be chosen as the best method with one pair or two pair primers (nested PCR). The higher sensitivity and specificity is achieved by nested PCR while the risk of false positive results still remains. It should be noted that titer of mycoplasma organisms in PCR method must be sufficient. However, this method (nested PCR) is more effective in some cases such as antibiotic therapy or detection of mycoplasma in biological products like FBS, trypsin, etc.
Reverse transcriptase PCR (RT-PCR) is considered as one of the suggestive methods for increasing the sensitivity of PCR (Peredeltchouk et al. 2011). This technique identifies the presence of free ribosomal RNA in contrast to the DNA coded with 16S rRNA gene (Puppe et al. 2013). In brief, we suggest that for recognition of routine mycoplasma contamination in cell cultures single-step PCR and not nested-PCR should be used (Uphoff and Drexler 2013).
In a study by Uphoff et al., sensitivity and specificity of different methods including direct and indirect DNA DAPI staining, DNA-RNA hybridization, ELISA and enzymatic 6-MPDR were assessed on adherent and suspension cells. In comparison with microbial culture, sensitivity and specificity were obtained as 100 and 100 % for indirect DNA DAPI staining, 100 and 98 % for DNA-RNA hybridization, 87 and 94 % for direct DNA DAPI staining, 72 and 100 % for ELISA, and finally 75 and 90 % for enzymatic 6-MPDR (Uphoff et al. 1992). In another study by Hobert and Drexler on 42 cancer cell lines, the sensitivity and specificity of nested PCR was compared with DNA-RNA hybridization, DNA DAPI staining, immunofluorescence staining by monoclonal antibody (IFA) and ELISA. It was concluded that nested PCR exhibited positive and negative results less than other methods. The designed primers were able to detect mycoplasma contamination until 10−4 serial dilutions (Hopert et al. 1993). In another research, for the validation of a microbial (reference) method, nested PCR and enzymatic Mycoalert® detection kits as alternative techniques were used (Cheong et al. 2011). The LOD of nested PCR was similar to the sensitivity of microbial culture and higher than for Mycoalert®. Uphoff and Drexler also reported that in comparison with microbial culture and DNA-RNA hybridization, molecular PCR technique could be considered as a sensitive, economic, accurate, precise, cheap, simple and rapid method for detection of mycoplasma contamination in cell cultures (Uphoff and Drexler 2002). On the other hand, according to the Young et al. suggestion, at least two or three tests must be synchronously performed for contamination recognition (Young et al. 2010). However, few researches declared that mycoalert® method can be considered as selective technique in comparison with PCR-ELISA and microbial culture, due to the higher sensitivity, specificity and accuracy (Mariotti et al. 2008). It has been stated by Garner and coworkers that interpretation, reproducibility and acceptability of the results with a more convenient procedure for ELISA and PCR-ELISA can be achieved, compared to DNA staining and PCR for detecting mycoplasma contamination in cell cultures (Garner et al. 2000). Another study by Hong and colleagues for the detection of mycoplasma contamination in clinical specimens by PCR and Dot blot hybridization was performed. In this research, DNA amplification primers (in PCR method) were designed to identify and reproduce DNA of known mycoplasma species with LOD of 100 pg for pure mycoplasma DNA. The quantity of LOD for DOT blot hybridization technique with digoxigenin labeled probe was reported about 10,000 pg DNA whereas, this value for PCR-Dot blot hybridization (PCR/DBH) method decreased to 10 pg DNA (Hong et al. 2011).
Conclusion
In this study three different methods including Mycoalert® detection kit, microbial culture and molecular PCR method were compared. Sensitivity, specificity, accuracy and predictive value of positive and negative results were obtained 91.30, 100, 95.23, 100 and 90.47 % for the Mycoalert® enzymatic method and 69.56, 100, 85.10, 100 and 77.41 % for the microbial culture method, respectively, and 100 % for all parameters for PCR. The enzymatic Mycoalert® method with respect to its sensitivity, specificity and very high speed of detection of mycoplasma contamination (<20 min) after PCR can be used instead of conventional microbial culture and DNA staining fluorochrome methods. Therefore, the PCR assay based on fix and common sequences in ribosomal 16SrRNA is confirmed as a valuable, reliable, highly sensitive, specific and accurate method for mycoplasma detection in cell cultures and biological products.
Acknowledgments
The authors would like to express their appreciation to Dr. Ehsan Mostafavi (head of Department of Epidemiology, Pasteur Institute of Iran) and also would like to thank National Cell Bank of Iran Pasteur Institute for their financial assistance.
Contributor Information
Mohammad Ali Shokrgozar, Phone: +98-21-66492595, Email: mashokrgozar@pasteur.ac.ir.
Reza Mahdian, Phone: +98-21-66480780, Email: rezamahdian@yahoo.com.
References
- Cheng H-S, Shen C-W, Wang S-R. Effect of storage conditions on detection of mycoplasma in biopharmaceutical products. In Vitro Cell Dev Biol Anim. 2007;43:113–119. doi: 10.1007/s11626-007-9015-7. [DOI] [PubMed] [Google Scholar]
- Cheong KA, Agrawal SR, Lee AY. Validation of nested PCR and a selective biochemical method as alternatives for mycoplasma detection. J Basic Microbiol. 2011;51:215–219. doi: 10.1002/jobm.201000066. [DOI] [PubMed] [Google Scholar]
- Dabrazhynetskaya A, Volokhov D, David S, Ikonomi P, Brewer A, Chang A, Chizhikov V. Preparation of reference strains for validation and comparison of mycoplasma testing methods. J Appl Microbiol. 2011;111:904–914. doi: 10.1111/j.1365-2672.2011.05108.x. [DOI] [PubMed] [Google Scholar]
- Del Giudice R, Gardella R, Hopps H. Cultivation of formerly noncultivable strains ofMycoplasma hyorhinis. Curr Microbiol. 1980;4:75–80. doi: 10.1007/BF02602896. [DOI] [Google Scholar]
- Freshney RI, Vunjak-Novakovic G, Freshney R (2006) Basic principles of cell culture. Cult Cells Tissue Eng 7:11–14
- Galen RS, Gambino SR. Beyond normality: the predictive value and efficiency of medical diagnoses. New York: Wiley; 1975. [Google Scholar]
- Garner C, Hubbold L, Chakraborti P. Mycoplasma detection in cell cultures: a comparison of four methods. Br J Biomed Sci. 2000;57:295. [PubMed] [Google Scholar]
- Harasawa R, Mizusawa H, Fujii M, Yamamoto J, Mukai H, Uemori T, Asada K, Kato I. Rapid detection and differentiation of the major mycoplasma contaminants in cell cultures using real-time PCR with SYBR Green I and melting curve analysis. Microbiol Immunol. 2005;49:859–863. doi: 10.1111/j.1348-0421.2005.tb03675.x. [DOI] [PubMed] [Google Scholar]
- Harlin H, Gajewski TF. Diagnosis and treatment of mycoplasma-contaminated cell cultures. Curr Protocol Cytom. 2008;43:A. 3C. 1–A. 3C. 7. doi: 10.1002/0471142956.cya03cs43. [DOI] [PubMed] [Google Scholar]
- Hay RJ, Ikonomi P (2005) Detection of microbial and viral contaminants in cell lines. In: Cell biology, vol 1, p 49.
- Hong S, Lee H-A, Park S-H, Kim O. Sensitive and specific detection of mycoplasma species by consensus polymerase chain reaction and dot blot hybridization. Lab Anim Res. 2011;27:141–145. doi: 10.5625/lar.2011.27.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopert A, Uphoff CC, Wirth M, Hauser H, Drexler HG. Specifity and sensitivity of polymerase chain reaction (PCR) in comparison with other methods for the detection of mycoplasma contamination in cell lines. J Immunol Methods. 1993;164:91–100. doi: 10.1016/0022-1759(93)90279-G. [DOI] [PubMed] [Google Scholar]
- Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, Habet SA, Baweja RK, Burckart GJ. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48:662–670. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- Jung H, Wang SY, Yang IW, Hsueh DW, Yang WJ, Wang TH, Wang HS (2003) Detection and treatment of mycoplasma contamination in cultured cells. Chang Gung Med J 26:250–258 [PubMed]
- Kong H, Volokhov DV, George J, Ikonomi P, Chandler D, Anderson C, Chizhikov V. Application of cell culture enrichment for improving the sensitivity of mycoplasma detection methods based on nucleic acid amplification technology (NAT) Appl Microbiol Biotechnol. 2007;77:223–232. doi: 10.1007/s00253-007-1135-1. [DOI] [PubMed] [Google Scholar]
- Lawrence B, Bashiri H, Dehghani H. Cross comparison of rapid mycoplasma detection platforms. Biologicals. 2010;38:218–223. doi: 10.1016/j.biologicals.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Mariotti E, Mirabelli P, Di Noto R, Fortunato G, Salvatore F. Rapid detection of mycoplasma in continuous cell lines using a selective biochemical test. Leuk Res. 2008;32:323–326. doi: 10.1016/j.leukres.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Markoullis K, Bulian D, Hölzlwimmer G, Quintanilla-Martinez L, Heiliger K-J, Zitzelsberger H, Scherb H, Mysliwietz J, Uphoff CC, Drexler HG. Mycoplasma contamination of murine embryonic stem cells affects cell parameters, germline transmission and chimeric progeny. Transgenic Res. 2009;18:71–87. doi: 10.1007/s11248-008-9218-z. [DOI] [PubMed] [Google Scholar]
- McGarrity GJ, Kotani H (1985) Cell culture mycoplasmas. In: Razin S, Barile MF (eds) The Mycoplasmas, vol 4, Academic Press Inc., New York, pp 353–390
- McGarrity G, Sarama J, Vanaman V. Cell culture techniques. ASM News. 1985;51:170–183. [Google Scholar]
- Molla Kazemiha V, Shokrgozar MA, Arabestani MR, Moghadam MS, Azari S, Maleki S, Amanzadeh A, Tehrani MJ, Shokri F. PCR-based detection and eradication of mycoplasmal infections from various mammalian cell lines: a local experience. Cytotechnology. 2009;61:117–124. doi: 10.1007/s10616-010-9252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla Kazemiha V, Azari S, Amanzadeh A, Bonakdar S, Moghadam MS, Anbouhi MH, Maleki S, Ahmadi N, Mousavi T, Shokrgozar MA. Efficiency of Plasmocin™ on various mammalian cell lines infected by mollicutes in comparison with commonly used antibiotics in cell culture: a local experience. Cytotechnology. 2011;63:609–620. doi: 10.1007/s10616-011-9378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikfarjam L, Farzaneh P (2012) Prevention and detection of mycoplasma contamination in cell culture. Cell J 13:203–212 [PMC free article] [PubMed]
- Peredeltchouk M, Wilson David S, Bhattacharya B, Volokhov D, Chizhikov V. Detection of mycoplasma contamination in cell substrates using reverse transcription PCR assays. J Appl Microbiol. 2011;110:54–60. doi: 10.1111/j.1365-2672.2010.04853.x. [DOI] [PubMed] [Google Scholar]
- Pitt A, Crouch SPM, Slater KJ, Cox A (2012) Assay for detecting mycoplasma by measuring acetate kinase or carbamate kinase activity. European Patent No. EP 2264181
- Puppe W, Weigl J, Gröndahl B, Knuf M, Rockahr S, von Bismarck P, Aron G, Niesters H, Osterhaus A, Schmitt H-J. Validation of a multiplex reverse transcriptase PCR ELISA for the detection of 19 respiratory tract pathogens. Infection. 2013;41:77–91. doi: 10.1007/s15010-012-0298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LB, Wichelhausen RH, Roizman B (1956) Contamination of human cell cultures by pleuro pneumonia like organisms. Science 124:1147–1148 [DOI] [PubMed]
- Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev. 1998;62:1094–1156. doi: 10.1128/mmbr.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, Mowles J (1996) Prevention and control of mycoplasma infection of cell cultures. In: Tully JG, Razin S (eds) Molecular and diagnostic procedures in mycoplasmology, Academic Press, San Diego pp 445–451
- Störmer M, Vollmer T, Henrich B, Kleesiek K, Dreier J. Broad-range real-time PCR assay for the rapid identification of cell-line contaminants and clinically important mollicute species. Int J Med Microbiol. 2009;299:291–300. doi: 10.1016/j.ijmm.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Tang J, Hu M, Lee S, Roblin R. A polymerase chain reaction based method for detecting MycoplasmaAcholeplasma contaminants in cell culture. J Microbiol Methods. 2000;39:121–126. doi: 10.1016/S0167-7012(99)00107-4. [DOI] [PubMed] [Google Scholar]
- Timenetsky J, Santos L, Buzinhani M, Mettifogo E. Detection of multiple mycoplasma infection in cell cultures by PCR. Braz J Med Biol Res. 2006;39:907–914. doi: 10.1590/S0100-879X2006000700009. [DOI] [PubMed] [Google Scholar]
- Uphoff CC, Drexler HG (2002) Comparative PCR analysis for detection of mycoplasma infections in continuous cell lines. In Vitro Cell Dev Biol Anim 38:79–85 [DOI] [PubMed]
- Uphoff CC, Drexler HG (2011) Detecting Mycoplasma contamination in cell cultures by polymerase chain reaction. In: Cree IA (ed) Cancer cell culture. Humana Press, Chicago, pp 93–103 [DOI] [PubMed]
- Uphoff CC, Drexler HG (2013) Detection of mycoplasma contaminations. In: Helgason CD, Miller CL (eds) Basic cell culture protocols. Humana Press, Chicago, pp 1–13
- Uphoff CC, Gignac SM, Drexler HG. Mycoplasma contamination in human leukemia cell lines. I. Comparison of various detection methods. J Immunol Methods. 1992;149:43–53. doi: 10.1016/S0022-1759(12)80047-0. [DOI] [PubMed] [Google Scholar]
- Volokhov DV, Kong H, George J, Anderson C, Chizhikov VE. Biological enrichment of Mycoplasma agents by cocultivation with permissive cell cultures. Appl Environ Microbiol. 2008;74:5383–5391. doi: 10.1128/AEM.00720-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volokhov DV, Graham LJ, Brorson KA, Chizhikov VE. Mycoplasma testing of cell substrates and biologics: review of alternative non-microbiological techniques. Mol Cell Probes. 2011;25:69–77. doi: 10.1016/j.mcp.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Waites KB, Katz B, Schelonka RL. Mycoplasmas and ureaplasmas as neonatal pathogens. Clin Microbiol Rev. 2005;18:757–789. doi: 10.1128/CMR.18.4.757-789.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavlovich A, Kohen R, Ginsburg I, Rottem S. The reducing antioxidant capacity of Mycoplasma fermentans. FEMS Microbiol Lett. 2006;259:195–200. doi: 10.1111/j.1574-6968.2006.00271.x. [DOI] [PubMed] [Google Scholar]
- Young L, Sung J, Stacey G, Masters JR. Detection of Mycoplasma in cell cultures. Nat Protoc. 2010;5:929–934. doi: 10.1038/nprot.2010.43. [DOI] [PubMed] [Google Scholar]
- Zhang S, Tsai S, Lo S-C. Alteration of gene expression profiles during mycoplasma-induced malignant cell transformation. BMC Cancer. 2006;6:116. doi: 10.1186/1471-2407-6-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Dreses-Werringloer U, Davies P, Marambaud P. Amyloid-beta peptide degradation in cell cultures by mycoplasma contaminants. BMC Res Notes. 2008;1:38. doi: 10.1186/1756-0500-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi Y, Mayhew A, Seng N, Takle GB. Validation of a PCR method for the detection of mycoplasmas according to European Pharmacopoeia section 2.6. 7. Biologicals. 2010;38:232–237. doi: 10.1016/j.biologicals.2009.11.003. [DOI] [PubMed] [Google Scholar]