

Abstract
Objective
Macrophages are critical contributors in abdominal aortic aneurysm (AAA) disease. We examined the ability of MKEY, a peptide inhibitor of CXCL4-CCL5 interaction, to influence AAA progression in murine models.
Methods and Results
AAAs were created in 10-week-old male C57BL/6 mice by transient infrarenal aortic porcine pancreatic elastase (PPE) infusion. Mice were treated with MKEY via intravenous injection either 1) before PPE infusion, or 2) after aneurysm initiation. Immunostaining demonstrated CCL5 and CCR5 expression on aneurysmal aortae and mural monocytes/macrophages, respectively. MKEY treatment partially inhibited transmural AAA migration of adaptively transferred leukocytes in recipient mice. While all vehicle-pretreated mice developed AAA, aneurysms formed in only 60% (3/5) and 14% (1/7) of mice pretreated with MKEY at 10 and 20 mg /kg, respectively. MKEY pretreatment reduced aortic diameter enlargement, preserved medial elastin fibers and smooth muscle cells, and attenuated mural macrophage infiltration, angiogenesis, and aortic MMP2 & 9 expression following PPE infusion. MKEY initiated after PPE infusion also stabilized and/or reduced enlargement of existing AAAs. Finally, MKEY treatment was effective in limiting AAA formation following angiotensin II infusion in apolipoprotein E deficient mice.
Conclusion
MKEY suppresses AAA formation and progression in two complementary experimental models. Peptide inhibition of CXCL4-CCL5 interactions may represent a viable translational strategy to limit progression of human AAA disease.
Keywords: abdominal aortic aneurysm, chemokines, CCL5, mice
INTRODUCTION
Abdominal aortic aneurysm (AAA) is a potentially life-threatening degenerative vascular condition. Histologically, AAA disease is characterized by transmural aortic macrophage infiltration, medial neovascularization, elastin degradation, and smooth muscle cell attenuation1. Accumulating evidence suggests that medial and adventitial macrophage activity promotes aneurysmal degeneration via production of matrix-degrading metalloproteinases, pro-inflammatory cytokines, reactive oxygen species, and lipid-derived mediators2–10. To date, no pharmacological treatment strategy has proven effective in limiting aneurysm enlargement or rupture. Surgical repair of late stage disease, with its attendant morbidity, remains the only effective method of reducing aneurysm-related morality11. Given the crucial role of macrophages in AAA pathogenesis, pharmacological strategies to reduce transmural monocyte migration and macrophage localization may provide an effective alternative for medical disease management.
Transmural leukocyte migration is tightly controlled by receptor interactions with adhesion molecules and chemokines in vascular structures. These interactions mediate leukocyte tethering and rolling, activation-dependent arrest or firm adhesion, and eventual transendothelial diapedesis12. In human AAA disease, serum and tissue levels of monocyte-attracting chemokines, such as CCL2/monocyte chemotactic protein-1 and CCL5/RANTES, are significantly elevated13–16, and CXCL4, a platelet-derived chemokine, is over-expressed in intraluminal aneurysm thrombus13, 14. Chemokine receptor polymorphisms, such as CCR2 heterozygote V64l and CCR5 delta 32 deletion, are also associated with increased AAA disease risk17, 18. In experimental models, increased CCL2 and CCL5 mRNA and/or protein expression is present in aneurysmal aortic tissue19–23, and either leukocyte CCR2-gene deletion, or siRNA-mediated expression knockdown attenuates AAA development24–29. While the significance of CCL2/CCR2 interaction in AAA pathogenesis is well described, the role of CCL5/CCR5 remains controversial and uncertain25.
Formation of a C-type CXCL4/CCL5 heterodimer substantially augments CCL5-mediated monocyte adhesion, arrest and transmigration in vitro. MKEY, a mouse CCL5-based synthetic cyclic peptide, prevents CXCL4-CCL5 heterodimer formation by competition with CXCL4 for CCL5 binding sites and/or releases CXCL4 from existing heterodimers. In in vitro assays, MKEY inhibits CCL5-mediated monocyte chemotaxis and arrest on activated endothelial cells. In apolipoprotein E-deficient (ApoE−/−) mice, MKEY inhibits monocyte recruitment to atherosclerotic lesions30. The critical importance of macrophage localization and activation in AAA pathogenesis led us to hypothesize that MKEY, on the basis of its known inhibitory effects in atherosclerosis-prone mice, might alter the course of aneurysm pathogenesis as well. We thus designed a series of experiments to evaluate the efficacy of MKEY in limiting initiation and progression of experimental AAA disease.
METHODS
Mice
Male C57BL/6 mice and ApoE−/− mice on C57BL/6J genetic background at 10 weeks of age were purchased from The Jackson Laboratory, Bar Harbor, Maine. Experimental procedures and care for laboratory animals were conducted in compliance with Stanford Laboratory Animal Care Guidelines and approved by the Administrative Panel on Laboratory Animal Care (APLAC - labanimals.stanford.edu).
Experimental aneurysm creation
In most experiments, AAAs were created via intra-aortic porcine pancreatic elastase (PPE) infusion as previously described31. Briefly, under inhaled anesthesia with operative magnification, the infrarenal abdominal aorta was exposed and controlled proximally and distally with 6-0 silk suture. Heat-tapered PE-10 tubing was inserted into the controlled segment just proximal to the aortic bifurcation. PPE was infused for five minutes into the controlled segment (30 μL of 1.5 U/ml type I PPE in saline, cat# 098K7008, Sigma-Aldrich, St. Louis, MO). After PPE infusion, the residual infusate was aspirated, the tubing was withdrawn, and the aortotomy closed using 10-0 nylon suture. In additional experiments, AAAs were created in ApoE−/− mice via a 28 day-subcutaneous infusion of angiotensin II (Ang II, 1000 ng/kg/min) via implanted osmotic minipumps (Azet model 2004, Durect Corporation, Cupertino)32, 33. After recovery from surgery and anesthesia, mice were housed in separated cages with free access to chow and water.
Serial aortic diameter determination via ultrasound imaging
Aneurysm formation and progression was monitored by serial aortic diameter measurements using transabdominal ultrasound at 40 MHz (Vevo 770; Visualsonics, Toronto, Canada), as previously described by ourselves and others31, 34, beginning immediately prior to PPE infusion (day 0), and at 3, 7 and 14 days post-operation. For the Ang II/ApoE−/− experiments, measurements were obtained at day 0 (prior to initiating Ang II infusion) and at 3, 7, 14, 21 and 28 days after pump implantation. The presence of an aneurysm was defined as > 50% increase in infused aortic segment diameter in the PPE model, and either a > 50% diameter increase or the onset of suprarenal aortic dissection in the Ang II/ApoE−/− mice. All diameter measurements were performed by a single investigator blinded to study group assignment, with less than 2% variation of repeated measurements.
Analysis of CCR5 expression on single leukocyte suspensions
In selected PPE-infused mice, aortae were harvested at 14 days, digested using elastase and collagenase, and passed through 40 μm filters to obtain single cell populations. In additional experiments, leukocytes were also isolated from whole blood for analysis. Single leukocyte suspensions were stained with mAbs against CD45, CD11b and CCR5, and analyzed using flow cytometry (BD FACSCalibur, BD Biosciences, San Diego, CA). Data are presented as the percentage of CCR5+ cells in CD45+CD11b+ cells (monocytes/macrophages) or CD45+CD11b−leukocytes.
In vivo leukocyte migration assays
Donor leukocytes were isolated from spleens and bone marrow of mice 2 wks after PPE infusion, and labelled with fluorescent dye CFSE as previously described35. Additional aneurysmal recipient mice were injected intravenously with either vehicle alone, or MKEY (Formula CT-2009ca, Carolus Therapeutics, Inc, San Diego) at 20 mg/kg in vehicle. Thirty minutes after infusion, 5×107 CFSE-labelled donor leukocytes were intravenously injected into both the vehicle and MKEY groups. To ensure sufficient MKEY levels for blocking CXCL4-CCL5 interaction in vivo, an additional 20 mg/kg was given intravenously at the time of donor cell transfer. Recipient mice were sacrificed 2 hours following cell transfer. CFSE+ donor leukocytes in the spleen, peripheral lymph nodes and blood of each recipient mouse were determined by flow cytometric analysis. At least 5×104 total leukocytes were analysed for each sample. Donor cells in lymphoid tissues and blood of recipient mice were calculated as the percentage of total leukocytes. Donor cells in aneurysmal lesions were identified via fluorescence microscopy, and calculated as the number of donor cells/aortic cross section (ACS). At least 10 aortic sections, 50 μm apart, were evaluated per mouse aorta. Donor cell migration in MKEY-treated group is expressed as the percentage of that in vehicle-treated mice, in which migration was set at 100.
MKEY influence on AAA progression
Both the PPE and AngII/ApoE−/− models were used to examine the effect of MKEY on AAA formation and progression. In the PPE/C57BL/6J model, mice were injected intravenously with 10 mg or 20 mg/kg MKEY daily starting 3 days prior to PPE infusion for 17 days, or starting 5 days after PPE infusion for 9 days. In control mice, treatment with vehicle alone was provided in equal volume and at identical time points following PPE infusion. In the Ang II/ApoE−/− model, mice were treated daily with 10 mg/kg MKEY for 3 days prior to Ang II pump implantation, and for 27 days thereafter. Infrarenal and suprarenal aortic diameters were recorded for 14 days following PPE infusion and for 28 days following Ang II pump implantation, respectively, using transabdominal ultrasound.
Tissue analysis and immunostaining
At sacrifice, aortae were harvested, fixed with 4% paraformaldehyde in phosphate-buffered saline, embedded in paraffin and sectioned (4 μm in thickness). Selected aortic tissues were embedded in OCT medium for frozen sectioning. Elastic-Masson (EM) and immunohistochemical staining were performed as previously described31. The primary antibodies for immunohistochemistry were a rabbit anti-mouse SMC α-actin polyclonal antibody (Laboratory Vision, Fremont, CA), a rat anti-mouse MAC-2 monoclonal antibody (Clone M3/38, Cedarlane Laboratories, Burlington, Ontario, Canada), a goat anti-mouse CCL5 polyclonal antibody or normal goat IgG (R&D systems, Minneapolis), a rabbit anti-mouse CD31 polyclonal antibody (Laboratory Vision), a rabbit anti-mouse MMP2 polyclonal antibody and a rabbit anti-mouse MMP9 polyclonal antibody (Chemicon International Inc, Temecula, CA). Other reagents, including biotinylated anti-goat, rat or rabbit secondary antibodies, streptavidin-peroxidase conjugates, and perioxidase substrate kits (DAB and ACE) were purchased from the Vector Laboratories, Burlingame, CA. Destruction of medial elastin and SMCs was graded as I (mild) to IV (severe)36. Data on mural macrophage infiltration and angiogenesis are provided as the number of MAC2+ cells and CD31+ blood vessels per ACS, respectively.
Statistical Analysis
Data are represented as mean ± standard deviation (SD). Depending on the type of data analysed, nonparametric Mann-Whitney test or two-way ANOVA followed by Newman-Keuls post-test were used to determine significance between groups. The difference in AAA incidence or mortality between groups was examined via Kaplan-Meier analysis. P<0.05 was considered to be significant.
RESULTS
CCL5 and its receptor CCR5 are strongly expressed in aneurysmal aortae
An anti-CCL5 antibody was used to stain frozen aortic sections from PPE- or saline (control)-infused C57BL/6J mice. There was no CCL5 staining in the control aortae (Fig. 1A, upper right panel). In contrast, strong medial and adventitial anti-CCL5 antibody staining was present in aneurysmal aortae (Fig. 1A, upper left panel). Most CCL5 staining was located within the areas of leukocyte infiltration. Normal goat IgG (negative control for CCL5 antibody) did not stain aneurysmal or control aortae (Fig. 1A, lower panels). Immunostaining for CD45, CD11b and CCR5 was performed on isolated tissue leukocytes from aneurysm specimens. As seen in Fig. 1B, 32% of CD45+CD11b+ cells expressed CCR5, versus 4% of CD45+CD11b−, suggesting that most CCR5-expressing cells were monocytes/macrophages. In contrast, only 5% of circulating CD11b+ leukocytes (Fig. 1C & 1D) and CD11b− leukocytes (not shown) expressed CCR5. Thus CD11b+CCR5+ monocytes/macrophages appear to be preferentially localized within aneurysm tissue in PPE-infused mice.
Figure 1. Expression of CCL5 and its leukocyte receptor CCR5 in aneurysmal aortae.

(A): Acetone-fixed frozen sections from mice 2 wk after PPE (left panels) or saline (right panels) infusion were stained with a goat anti-mouse CCL5 polyclonal antibody (upper panels) or an equal amount of normal goat IgG (negative control, lower panels). This staining pattern was reproduced at least 3 mice. Original magnification: X400.
(B): Single leukocyte suspensions from enzyme-digested aneurysmal aortae were stained with the mAbs against CD45, CD11b and CCR5 (or its negative control antibody), and analyzed using flow cytometry. The percentages of CCR5+ cells in CD45+CD11b+ (monocytes/macrophages) and CD45+CD11b− cells (other leukocytes) are shown in the left and right panels, respectively. Each flow cytometric histograph is the overlay image for the staining of anti-CCR5 mAb (unshaded) and its negative control antibody (shaded). This experiment was repeated 3 times, and the cells pooled from 3 mouse aortae were used for each staining.
(C, D): Whole blood leukocytes from mice 2 wk after PPE infusion were stained with CD11b and CCR5 (or its negative control antibody), and analyzed using flow cytometry. Both small and large leukocytes expressed CD11b (C). A representative flow cytometric plot shows that 9.4% of CD11b+ leukocytes expressed CCR5 and most of them were small leukocytes as indicated by side scatter size (D). This experiment was repeated in 5 mice.
CCL5 expression influences trans-mural aortic leukocyte migration in aneurysmal aortae
In vivo short-term leukocyte migration assays were performed to determine the significance of CCL5 expression on aortic leukocyte (including monocyte) recruitment in PPE-induced AAAs. Pre-treatment with MKEY reduced aortic accumulation of injected, CFSE-labeled, mixed spleen and bone marrow cells by 25%, without apparent affect on PLN and bone marrow migration (Fig. 2). These results indicate that CCL5 expression influences, at least to some degree, trans-aortic leukocyte migration in this model.
Figure 2. Leukocyte migration in experimental AAA.
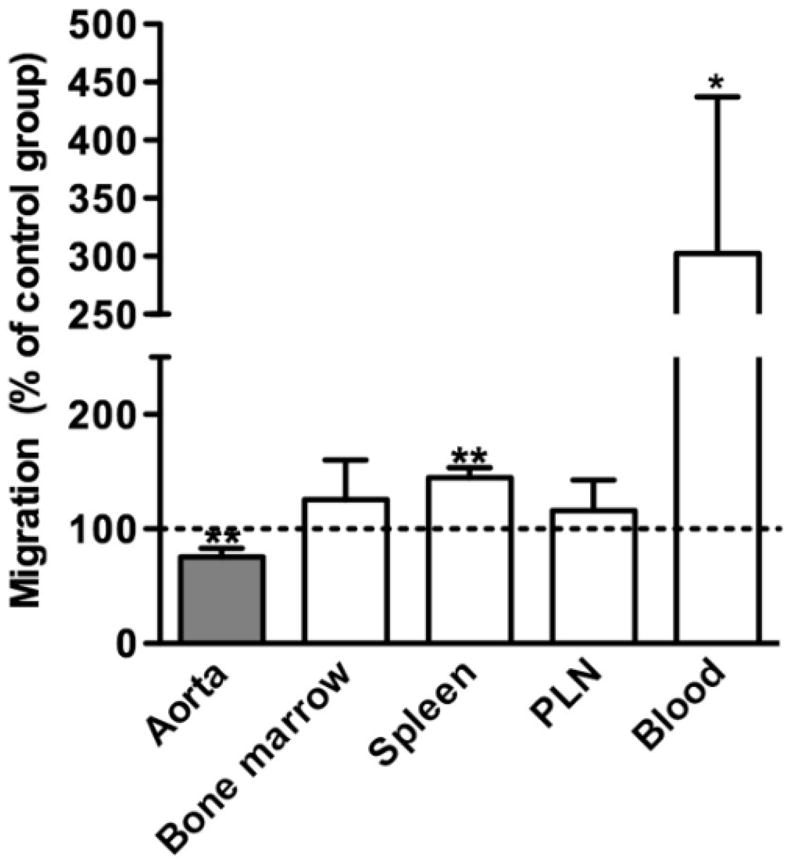
A mixed population of spleen and bone marrow cells from aneurysmal mice were labeled with CFSE, and intravenously transferred into aneurysmal recipient mice pretreated with MKEY 30 min before cell transfer. Recipient mice were sacrificed 2 hours after cell transfer. Donor cells in the spleen, peripheral lymph nodes (PLN), bone marrow and aneurysmal aortae of recipient mice were evaluated using either flow cytometric or tissue immunofluorescence analysis. Migration of donor cells in MKEY-treated group was expressed as the percentage of that in vehicle-treated group, in which migration was set up at 100. Nonparametric Mann-Whitney test, *P<0.05 and **P<0.01 compared to vehicle-treated group. n=4 mice in each group.
MKEY treatment suppresses the development and progression of AAAs
Based on the apparent functional consequences of CCL5 expression in aneurysmal aortae, we hypothesized that CCL5 inhibition would influence the formation and progression of AAAs following PPE infusion. To test this, mice were intravenously injected with vehicle or MKEY starting 3 days prior to aortic PPE infusion, and continuing daily for the next 17 days. Following PPE infusion, aortic diameters enlarged in a progressive fashion from day 3 onward (Fig. 3A, upper panels). In MKEY-treated mice, at either dose (Fig. 3A middle panels for 10 mg/kg, Fig. 3A lower panels for 20 mg/kg), PPE-infusion produced significantly smaller aneurysms. At 20 mg/kg, aneurysm formation was nearly completely obliterated. At both 7 and 14 days, mean aortic diameters in both MKEY-treated groups were significantly smaller than their respective controls (Fig. 3B). Mean aortic diameter in the 20 mg/kg MKEY-treated group was also significantly smaller than that in the 10 mg/kg MKEY-treated group (Fig. 3B). In terms of aneurysm incidence following PPE infusion, AAAs (defined by a >50% or more increase in aortic diameter) developed in all vehicle-treated mice (8/8) within 7 days (Fig. 3C). In contrast, AAA developed in three mice (60%, 3/5) and one mouse (14%, 1/7) treated with 10 and 20 mg/kg MKEY, respectively, within 14 days (Fig. 3C). Although AAA incidence in both MKEY-treated groups was lower than that noted in the vehicle-treated group, a significant difference was only seen between vehicle- and 20 mg/kg MKEY-treated groups. These results indicate that MKEY therapy suppresses experimental AAA formation and progression in a dose-dependent fashion.
Figure 3. Influence of MKEY treatment on AAA formation and progression.

Male C57BL/6 mice were treated with vehicle (n=8), 10 mg/kg MKEY (n=5) or 20 mg/kg MKEY (n=7) starting on day 3 prior to PE infusion for 17 days. AAAs were imaged by measuring infrarenal aortic diameter for each mouse using noninvasive transabdominal ultrasonography. An AAA was defined as a more than 50% increase in the aortic diameter over baseline level.
(A): Representative ultrasound images of aortae from PPE-infused mice treated with vehicle or MKEY.
(B): Mean and SD of aortic diameters. ANOVA followed by Newman-Keuls post-test, *P<0.05 and **P<0.01 between two groups.
(C): AAA incidence in PPE-infused mice treated with vehicle or MKEY. Kaplan-Meier analysis, **P<0.01 compared to vehicle-treated mice.
MKEY treatment preserves aortic mural integrity
To identify the mechanisms responsible for MKEY-mediated AAA suppression, Elastic-Masson staining and SMC immunostaining was performed on aortic sections. As illustrated in Fig. 4A, PPE infusion severely reduced medial elastin density and SMC cellularity in vehicle-treated mice. Both elastin fragmentation and SMC depletion were significantly attenuated in MKEY-treated mice (Figs. 4B & 4C). Though the mean scores were lower in the 20 mg/kg MKEY group than those in the 10 mg/kg MKEY group, the difference between two groups did not reach statistical significance.
Figure 4. Influence of MKEY treatment on AAA pathology.

Aortic sections from mice 2 wk after PPE infusion were stained with Elastin Masson stain for elastin fibers or immunostained with an antibody against SMC α-actin for SMCs, MAC2 for macrophages or CD31 for blood vessels. There were 8 mice in the vehicle group, 5 mice in 10 mg/kg MKEY treatment groups and 7 mice in 20 mg/kg MKEY treatment group.
(A): Representative aortic histology images for elastin, SMCs (SMC alpha actin), macrophages (MAC2) and blood vessels (CD31) from PPE-infused mice treated with vehicle or MKEY. Lum: lumen; Med: media; Adv: adventitia.
(B, C): Medial elastin fragmentation (C) and SMC destruction (D) were scored as mild (I) to severe (IV) using a histology grading system. Data are mean and SD of the scores in individual groups.
(D, E): MAC2+ macrophages and CD31+ blood vessels in media and adventitia were counted on each ACS, and data are given as mean and SD for macrophages or blood vessels per ACS. In all experiments, Nonparametric Mann-Whitney test, *P<0.05 and **P<0.01 between two groups.
In addition to mural SMC and elastin preservation, only a small numbers of aortic MAC2+ cells and CD31+ vessels were observed in MKEY-treated mice. By semi-quantitative histological analysis, monocyte/macrophage infiltration and mural neovascularization were significantly reduced in MKEY-treated mice compared to control (Figs. 4A–4D). Consistent with reduced aortic accumulation of monocytes/macrophages, immunostaining for MMP2 and MMP9 was attenuated in the aortae from MKEY-treated mice as compared to vehicle-treated mice (Supplemental file). Thus, reduction of monocyte/macrophage infiltration, resultant MMP2 & 9 expression, and mural neoangiogenesis may contribute to MKEY-mediated AAA suppression.
MKEY treatment stabilizes existing AAAs
To gain insight into the translational value of MKEY-mediated suppression of existing aneurysms, AAA mice were treated with 20 mg/kg MKEY beginning 5 days after PPE infusion, for 9 additional days. As shown in Figs. 5A & 5B, aortic diameters in PPE-infused, vehicle-treated mice continued to enlarge during this timeframe. In contrast, further enlargement was nearly completely suppressed in PPE-infused mice by the third day of MKEY treatment. Consistent with the observed effect on aneurysm diameter, qualitatively there appeared to be reduced elastin degradation and SMC depletion in delayed treatment mice, although the impact on SMC density did not reach statistical significance (Figs. 5C & 5D). Mural macrophage and neovessel density were significantly reduced in delayed-treatment as compared to those in vehicle-treated mice (Figs. 5E & 5F). These results indicate that MKEY treatment stabilizes existing AAAs by limiting mural monocyte/macrophage infiltration, further elastin degradation and angiogenesis.
Figure 5. MKEY treatment in existing aneurysms.
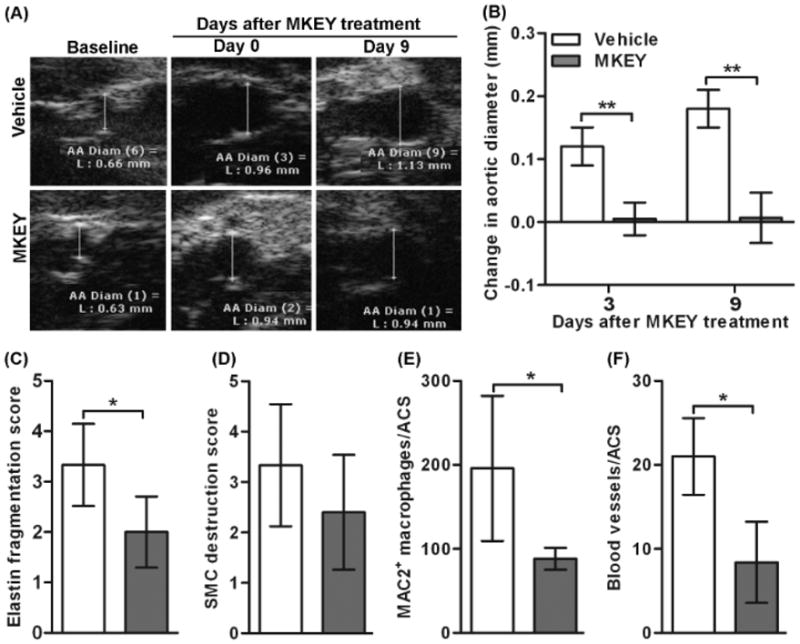
Mice were treated with vehicle (n=5) or 20 mg/kg MKEY (n=6) starting on day 5 after PPE infusion for 9 days. Changes in aortic diameters were measured on days 3 and 9 after MKEY treatment. Mice were sacrificed 2 wk after PPE infusion, and aortic sections were prepared for histopathologic analysis.
(A): Representative ultrasound aortic images from PPE-infused mice treated with vehicle or MKEY.
(B, C): Medial elastin fragmentation (B) and SMC destruction (C) were scored as mild (I) to severe (IV) using a histology grading system. Data are mean and SD of the scores in individual groups.
(D, E): MAC2+ macrophages and CD31+ blood vessels in media and adventitia were counted on each ACS, and data are given as mean and SD for macrophages or blood vessels per ACS. In all experiments, Nonparametric Mann-Whitney test, *P<0.05 between two groups.
MKEY treatment suppresses Ang II infusion-induced AAAs in ApoE−/− mice
Finally, the translational value of MKEY-mediated aneurysm suppression was further examined in the AngII/ApoE−/− AAA model. This represents a mechanistically distinct model, complementary to the PPE model in C57BL/6J mice. MKEY treatment starting prior to Ang II infusion significantly lowered AAA incidence (20%) as compared vehicle treatment (60%) (Fig. 6A). AAA-associated mortality in MKEY-treated ApoE−/− mice (20%) was also lower than that in vehicle-treated mice (40%), although there was no statistical difference between two treatment groups (Fig. 6B). Consistent with its influence on AAA incidence and mortality, MKEY treatment inhibited aortic enlargement during the first 14 days after Ang II infusion as compared to vehicle treatment. There was a significance difference in aortic diameters between two treatment groups on day 3 after Ang II infusion. These results extend our findings in the PPE model, suggesting that CXCL4-CCL5 heterodimer-mediated aortic monocyte migration may be a common mechanism for aneurysm formation in multiple AAA models.
Figure 6. Influence of MKEY treatment on Ang II-induced AAAs in ApoE−/− mice.

MKEY at the dose of 10 mg/kg/day was given to 10 weeks old male ApoE−/− mice stating from 3 days prior to Ang II infusion and continued for 28 days thereafter. Suprarenal aortae in individual mice were imaged for the onsets of AAA using ultrasonography. An AAA was defined by a more than 50% increase in the aortic diameter over baseline level or the onset of aortic dissection. (A): The percentage of AAA free mice. (B) Mortality due to AAA rupture. (C): Changes in suprarenal aortic diameters after Ang II infusion. There were 10 mice in each treatment group. ANOVA followed by Newman-Keuls post-test, *P<0.05 compared to vehicle treatment group at same time point.
DISCUSSION
These experiments demonstrate that CCL5, and its receptor CCR5, are expressed in experimental aneurysm tissue and mural CD45+CD11b+ monocytes/macrophages, respectively. MKEY, a peptide inhibitor to CXCL4-CCL5 heterodimer formation, significantly inhibits migration of adoptively transferred donor leukocytes into recipient aneurysmal aortae. Moreover, MKEY treatment initiated after aneurysm formation stabilizes aortic mural architecture and limits further aneurysm progression. The relevance of these findings to aneurysm disease was generally validated by the observation that MKEY suppressed aneurysms initiated by Ang II infusion in male ApoE−/− mice. Together, these experiments highlight the significance of CCL5 in experimental aneurysm pathogenesis and underscore its potential role in human AAA disease.
Chemokines CCL2 and CCL5 recruit inflammatory monocytes into target tissues, promoting macrophage-driven inflammatory conditions such as vascular disease28, 37–42. Prior work suggests significant potential roles for CCL2 and CCR2 in experimental aneurysm pathogenesis 24–29. In the current experiments, using similar models22 we demonstrated significant CCL5 production in infiltrating leukocytes, mostly monocytes/macrophages. Its production may occur from other constituitive or infiltrative aortic cell types as well14, 22. While both clearly are overexpressed in experimental aneurysm tissue, whether CCL2 and CCL5 exert redundant or coordinated roles in AAA pathogenesis remains to be determined.
A prior study reported no apparent impact of targeted CCR5 deletion on aneurysm initiation or progression in mice25. In that study, aneurysms were created via abluminal aortic application of calcium chloride. This apparent discrepancy is likely attributable to pathological features that distinguish the PPE and Ang II/ApoE−/− models from the calcium chloride -application model43, 44. In the former two models, aneurysm formation is accompanied by abundant transmural monocyte/macrophage infiltration and marked medial elastin and SMC depletion. Aneurysms in the calcium chloride model are distinguished primarily by elastin fragmentation, however, with relative preservation of total elastin content and medial SMC density. In their pathological features, aneurysms created by either luminal PPE infusion or systemic Ang II administration have substantially more fidelity to the human condition than those generate by calcium chloride. Additionally, reciprocal up-regulation of CCR2 activity and/or expression may occur following CCL5 deletion, sustaining the ability to form aneurysms following calcium chloride-induced injury. Regardless of the alternative potential explanations considered, however, in these experiments, in either WT or ApoE−/− mice, MKEY was highly effective in suppressing aneurysm formation.
Monocytes/macrophages are significant contributors to AAA pathogenesis in both human and experimental disease3–8, 10, 33, 45. In this study, MKEY therapy significantly reduced aortic monocyte/macrophage infiltration while suppressing AAA formation and progression. Previously, intravenous MKEY has been shown to inhibit arrest/firm adhesion of adaptively transferred donor monocytes onto inflamed arteries, binding to both leukocytes and endothelial cells30. In these experiments, MKEY partially but significantly inhibited donor leukocyte migration into aneurysmal aortae of recipient mice. Because of the heterogeneity of the donor cells used in the in-vivo short-term leukocyte migration assays, the efficacy of monocyte inhibition specifically could not be determined. Flow cytometric analysis, however, confirmed that the majority of circulating CCR5+ cells in these mice were small CD11b+ myeloid cells, likely monocytes rather than neutrophils or lymphocytes. Thus, it appears that attenuation of inflammation following PPE infusion in C57BL/6J mice resulted at least in part from MKEY-mediated inhibition of aortic monocyte migration. Furthermore, consistent with previous studies, this results demonstrate that chemokines such as CCL5 or CCL2 are produced by infiltrating leukocytes including monocytes/macrophages14, 22. Inhibition of monocyte migration by MKEY may reduce the density of chemokine-producing cells and thus the regional chemokine gradient available for attracting additional leukocytes.
MKEY therapy also substantially attenuated aortic adventitial neovessel formation in these experiments. Mural neoangiogenesis is a salient histologic feature of both experimental and human AAA disease32, 46, 47. Subsets of monocytes/macrophages produce the pro-angiogenic cytokine VEGF-A48, 49, and CCR5 deletion results in sustained inhibition of experimental corneal neovascularization50. Additionally, CCR5 deficiency in a skin wound healing model is associated with both reduced local VEGF-A levels and endothelial progenitor accumulation51, both prevalent in experimental AAA52. Thus, the anti-aneurysmal influences of MKEY therapy may also related to influences on aortic mural endothelial progenitor cell accumulation. MKEY may also exert anti-angiogenic effects in a VEGF-A and/or endothelial progenitor-independent manner via influences on monocyte migration or phenotypic differentiation of resident macrophages53.
MKEY treatment downregulated PPE-induced aortic MMP 2 & 9 expression, proteases essential to extracellular matrix degradation in experimental and clinical aneurysm disease2, 7. Macrophages are recognized sources of MMP production during aneurysmal degeneration. The specific mechanism by which MKEY minimizes MMP expression, however, remains uncertain. No existing evidence indicates that MKEY directly inhibits MMP production in macrophages or other cells30. Thus, it is more likely that reduced MMP expression in MKEY-treated aneurysmal aortae results from attenuated monocyte/macrophage migration. Regardless of the mechanism, however, reduced MMP expression in response to MKEY treatment will likely limit elastin degradation, monocyte/macrophage migration and consequent aneurysmal degradation 32, 44, 54.
The ability of delayed MKEY treatment to stabilize or attenuate established experimental AAAs has substantial clinical implications. Most if not all inhibitor studies reported to date, regardless of the agent being tested, initiated therapy prior to aneurysm creation, a situation at odds with the clinical reality of AAA diagnosis and pre-surgical disease management. The ability to impair or arrest existing aortic mural inflammation, rather than preventing initiation, is a critical requirement for successful medical AAA inhibition strategies. As compared to CCL2/CCR2 axis antagonists, MKEY and similar CCL5/CCR5 inhibitors offer distinct advantages. First, CCL5/CCR5 inhibition will have less impact on host innate immunity compared to CCL2/CCR2 inhibition12, 55, 56,57. In a prior study, MKEY did not suppress T cell proliferation, viral clearance or macrophage survival30. In the present study, MKEY suppressed transmural aortic leukocyte migration without affecting migration of leukocytes/lymphocytes migration into lymph nodes and the spleen, secondary lymphoid organs critical for adaptive immunity. Secondly, due to prevalent prior studies of CCL5 in atherosclerosis and macrophage-tropic HIV-1, more clinical trials on CCL5/CCR5 antagonists have been completed or are ongoing as compared to CCL2/CCR2 inhibitors (www.clinicaltrial.gov)57. Maraviroc, a CCR5 antagonist, is approved for clinical use in HIV-1 pateints. Thus, CCL5/CCR5 inhibition therapies are primed for translational application in AAA disease.
MKEY, as a mouse CCL5-based antagonist for CXCL4-CCL5 heterodimer formation, does not cross-react with its human homologue. CKEY2, a human pepptide ortholog of MKEY, disrupts human CXCL4-CCL5 heterodimer and has previously been shown to suppress human monocyte chemotaxisis to CCL5, as well as CCL5-triggered monocyte arrest on endothelial cells in vitro30. The experimental method employed in these experiments did not allow for assessment of the influence of CKEY therapy on experimental or clinical aneurysm progression. In a prior human study, however, CXCL4 and CCL5 were noted to be co-localized in aneurysmal aortae, released from aortic tissue and intraluminal thrombus, and elevated in plasma obtained from AAA patients14. Futher studies are required to examine whether CKEY2 alters the formation and progression of AAAs in the murine model in which entire mouse hematopietic lineages are replaced with human hematopoic cell lineases. Alternatively, once all safety data for CKEY2 is acquired, a clinical trial may be indicated to evaluate its therapeutic efficacy in human AAA disease.
In conclusion, these experiments demonstrate for the first time the ability of the peptide CXCL4-CCL5 heterodimer inhibitor MKEY to both suppress experimental AAA initiation and stabilize existing aneurysms, through mechanisms likely related to impaired mural monocytes/macrophage infiltration and angiogenesis. These findings add to prior findings suggesting that CXCL4-CCL5 inhibition may hold substantial translational value for both atherosclerotic and aneurysmal arterial diseases.
Supplementary Material
Acknowledgments
This study was supported in part by grants from the National Heart, Lung and Blood Institute (5R21HL109750-02) and Carolus Therapeutics, Inc., San Diego, CA, USA.
References
- 1.Nordon IM, Hinchliffe RJ, Holt PJ, Loftus IM, Thompson MM. Review of current theories for abdominal aortic aneurysm pathogenesis. Vascular. 2009;17:253–263. doi: 10.2310/6670.2009.00046. [DOI] [PubMed] [Google Scholar]
- 2.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahluwalia N, Lin AY, Tager AM, Pruitt IE, Anderson TJ, Kristo F, Shen D, Cruz AR, Aikawa M, Luster AD, Gerszten RE. Inhibited aortic aneurysm formation in BLT1-deficient mice. J Immunol. 2007;179:691–697. doi: 10.4049/jimmunol.179.1.691. [DOI] [PubMed] [Google Scholar]
- 4.Xiong W, MacTaggart J, Knispel R, Worth J, Persidsky Y, Baxter BT. Blocking TNF-alpha attenuates aneurysm formation in a murine model. J Immunol. 2009;183:2741–2746. doi: 10.4049/jimmunol.0803164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S, Fishbein MC, Blaschke F, Kintscher U, Graf K, Law RE, Hsueh WA. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112:1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao L, Moos MP, Grabner R, Pedrono F, Fan J, Kaiser B, John N, Schmidt S, Spanbroek R, Lotzer K, Huang L, Cui J, Rader DJ, Evans JF, Habenicht AJ, Funk CD. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat Med. 2004;10:966–973. doi: 10.1038/nm1099. [DOI] [PubMed] [Google Scholar]
- 7.Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 9.Thomas M, Gavrila D, McCormick ML, Miller FJ, Jr, Daugherty A, Cassis LA, Dellsperger KC, Weintraub NL. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation. 2006;114:404–413. doi: 10.1161/CIRCULATIONAHA.105.607168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, Berk BC. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15:649–656. doi: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. doi: 10.1161/CIRCULATIONAHA.107.735274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. 2004;4:432–444. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- 13.Choke E, Cockerill GW, Laing K, Dawson J, Wilson WR, Loftus IM, Thompson MM. Whole genome-expression profiling reveals a role for immune and inflammatory response in abdominal aortic aneurysm rupture. Eur J Vasc Endovasc Surg. 2009;37:305–310. doi: 10.1016/j.ejvs.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 14.Houard X, Touat Z, Ollivier V, Louedec L, Philippe M, Sebbag U, Meilhac O, Rossignol P, Michel JB. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc Res. 2009;82:532–541. doi: 10.1093/cvr/cvp048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koch AE, Kunkel SL, Pearce WH, Shah MR, Parikh D, Evanoff HL, Haines GK, Burdick MD, Strieter RM. Enhanced production of the chemotactic cytokines interleukin-8 and monocyte chemoattractant protein-1 in human abdominal aortic aneurysms. Am J Pathol. 1993;142:1423–1431. [PMC free article] [PubMed] [Google Scholar]
- 16.Golledge J, Clancy P, Moran C, Biros E, Rush C, Walker P, Norman P. The novel association of the chemokine CCL22 with abdominal aortic aneurysm. Am J Pathol. 2010;176:2098–2106. doi: 10.2353/ajpath.2010.090416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katrancioglu N, Manduz S, Karahan O, Yilmaz MB, Sezgin I, Bagci G, Berkan O. The role of the CCR2 gene polymorphism in abdominal aortic aneurysms. Angiology. 2011;62:140–143. doi: 10.1177/0003319710385335. [DOI] [PubMed] [Google Scholar]
- 18.Sandford B, Bown M, London N, Sayers R. The role of the CCR5 Delta32 polymorphism in abdominal aortic aneurysms. Int J Immunogenet. 2009;36:199–205. doi: 10.1111/j.1744-313X.2009.00845.x. [DOI] [PubMed] [Google Scholar]
- 19.Rush C, Nyara M, Moxon JV, Trollope A, Cullen B, Golledge J. Whole genome expression analysis within the angiotensin II-apolipoprotein E deficient mouse model of abdominal aortic aneurysm. BMC Genomics. 2009;10:298. doi: 10.1186/1471-2164-10-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Vickle-Chavez SJ, Tung WS, Absi TS, Ennis TL, Mao D, Cobb JP, Thompson RW. Temporal changes in mouse aortic wall gene expression during the development of elastase-induced abdominal aortic aneurysms. J Vasc Surg. 2006;43:1010–1020. doi: 10.1016/j.jvs.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, Dyer JS, Maegdefessel L, Dalman RL, Tsao PS. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiol Genomics. 2011;43:993–1003. doi: 10.1152/physiolgenomics.00044.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colonnello JS, Hance KA, Shames ML, Wyble CW, Ziporin SJ, Leidenfrost JE, Ennis TL, Upchurch GR, Jr, Thompson RW. Transient exposure to elastase induces mouse aortic wall smooth muscle cell production of MCP-1 and RANTES during development of experimental aortic aneurysm. J Vasc Surg. 2003;38:138–146. doi: 10.1016/s0741-5214(03)00125-3. [DOI] [PubMed] [Google Scholar]
- 23.Middleton RK, Lloyd GM, Bown MJ, Cooper NJ, London NJ, Sayers RD. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: a protein array study. J Vasc Surg. 2007;45:574–580. doi: 10.1016/j.jvs.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 24.de Waard V, Bot I, de Jager SC, Talib S, Egashira K, de Vries MR, Quax PH, Biessen EA, van Berkel TJ. Systemic MCP1/CCR2 blockade and leukocyte specific MCP1/CCR2 inhibition affect aortic aneurysm formation differently. Atherosclerosis. 2010;211:84–89. doi: 10.1016/j.atherosclerosis.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 25.MacTaggart JN, Xiong W, Knispel R, Baxter BT. Deletion of CCR2 but not CCR5 or CXCR3 inhibits aortic aneurysm formation. Surgery. 2007;142:284–288. doi: 10.1016/j.surg.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 26.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 2010;118:681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishibashi M, Egashira K, Zhao Q, Hiasa K, Ohtani K, Ihara Y, Charo IF, Kura S, Tsuzuki T, Takeshita A, Sunagawa K. Bone marrow-derived monocyte chemoattractant protein-1 receptor CCR2 is critical in angiotensin II-induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2004;24:e174–178. doi: 10.1161/01.ATV.0000143384.69170.2d. [DOI] [PubMed] [Google Scholar]
- 29.Moehle CW, Bhamidipati CM, Alexander MR, Mehta GS, Irvine JN, Salmon M, Upchurch GR, Jr, Kron IL, Owens GK, Ailawadi G. Bone marrow-derived MCP1 required for experimental aortic aneurysm formation and smooth muscle phenotypic modulation. J Thorac Cardiovasc Surg. 2011;142:1567–1574. doi: 10.1016/j.jtcvs.2011.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- 31.Schultz G, Tedesco MM, Sho E, Nishimura T, Sharif S, Du X, Myles T, Morser J, Dalman RL, Leung LL. Enhanced abdominal aortic aneurysm formation in thrombin-activatable procarboxypeptidase B-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30:1363–1370. doi: 10.1161/ATVBAHA.109.202259. [DOI] [PubMed] [Google Scholar]
- 32.Tedesco MM, Terashima M, Blankenberg FG, Levashova Z, Spin JM, Backer MV, Backer JM, Sho M, Sho E, McConnell MV, Dalman RL. Analysis of in situ and ex vivo vascular endothelial growth factor receptor expression during experimental aortic aneurysm progression. Arterioscler Thromb Vasc Biol. 2009;29:1452–1457. doi: 10.1161/ATVBAHA.109.187757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barisione C, Charnigo R, Howatt DA, Moorleghen JJ, Rateri DL, Daugherty A. Rapid dilation of the abdominal aorta during infusion of angiotensin II detected by noninvasive high-frequency ultrasonography. J Vasc Surg. 2006;44:372–376. doi: 10.1016/j.jvs.2006.04.047. [DOI] [PubMed] [Google Scholar]
- 35.Xu B, Wagner N, Pham LN, Magno V, Shan Z, Butcher EC, Michie SA. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by L-selectin/PNAd, alpha4beta1 integrin/VCAM-1, and LFA-1 adhesion pathways. J Exp Med. 2003;197:1255–1267. doi: 10.1084/jem.20010685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamblin M, Chang L, Zhang H, Yang K, Zhang J, Chen YE. Vascular smooth muscle cell peroxisome proliferator-activated receptor-gamma deletion promotes abdominal aortic aneurysms. J Vasc Surg. 2010;52:984–993. doi: 10.1016/j.jvs.2010.05.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braunersreuther V, Steffens S, Arnaud C, Pelli G, Burger F, Proudfoot A, Mach F. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol. 2008;28:1090–1096. doi: 10.1161/ATVBAHA.108.165423. [DOI] [PubMed] [Google Scholar]
- 38.Braunersreuther V, Zernecke A, Arnaud C, Liehn EA, Steffens S, Shagdarsuren E, Bidzhekov K, Burger F, Pelli G, Luckow B, Mach F, Weber C. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2007;27:373–379. doi: 10.1161/01.ATV.0000253886.44609.ae. [DOI] [PubMed] [Google Scholar]
- 39.Olzinski AR, Turner GH, Bernard RE, Karr H, Cornejo CA, Aravindhan K, Hoang B, Ringenberg MA, Qin P, Goodman KB, Willette RN, Macphee CH, Jucker BM, Sehon CA, Gough PJ. Pharmacological inhibition of C-C chemokine receptor 2 decreases macrophage infiltration in the aortic root of the human C-C chemokine receptor 2/apolipoprotein E−/− mouse: magnetic resonance imaging assessment. Arterioscler Thromb Vasc Biol. 2010;30:253–259. doi: 10.1161/ATVBAHA.109.198812. [DOI] [PubMed] [Google Scholar]
- 40.Potteaux S, Combadiere C, Esposito B, Lecureuil C, Ait-Oufella H, Merval R, Ardouin P, Tedgui A, Mallat Z. Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:1858–1863. doi: 10.1161/01.ATV.0000231527.22762.71. [DOI] [PubMed] [Google Scholar]
- 41.Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, Ley K, Weber C. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002;106:1523–1529. doi: 10.1161/01.cir.0000028590.02477.6f. [DOI] [PubMed] [Google Scholar]
- 42.von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, Weber C. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103:1772–1777. doi: 10.1161/01.cir.103.13.1772. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Sun J, Lindholt JS, Sukhova GK, Sinnamon M, Stevens RL, Adachi R, Libby P, Thompson RW, Shi GP. Mast cell tryptase deficiency attenuates mouse abdominal aortic aneurysm formation. Circ Res. 2011;108:1316–1327. doi: 10.1161/CIRCRESAHA.111.243758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaneko H, Anzai T, Takahashi T, Kohno T, Shimoda M, Sasaki A, Shimizu H, Nagai T, Maekawa Y, Yoshimura K, Aoki H, Yoshikawa T, Okada Y, Yozu R, Ogawa S, Fukuda K. Role of vascular endothelial growth factor-A in development of abdominal aortic aneurysm. Cardiovasc Res. 2011;91:358–367. doi: 10.1093/cvr/cvr080. [DOI] [PubMed] [Google Scholar]
- 45.Daugherty A, Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor −/− mice. Ann N Y Acad Sci. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- 46.Choke E, Cockerill GW, Dawson J, Howe F, Wilson WR, Loftus IM, Thompson MM. Vascular endothelial growth factor enhances angiotensin II-induced aneurysm formation in apolipoprotein E-deficient mice. J Vasc Surg. 2010;52:159–166 e151. doi: 10.1016/j.jvs.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 47.Choke E, Thompson MM, Dawson J, Wilson WR, Sayed S, Loftus IM, Cockerill GW. Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler Thromb Vasc Biol. 2006;26:2077–2082. doi: 10.1161/01.ATV.0000234944.22509.f9. [DOI] [PubMed] [Google Scholar]
- 48.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. Macrophages and angiogenesis. J Leukoc Biol. 1994;55:410–422. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- 50.Ambati BK, Anand A, Joussen AM, Kuziel WA, Adamis AP, Ambati J. Sustained inhibition of corneal neovascularization by genetic ablation of CCR5. Invest Ophthalmol Vis Sci. 2003;44:590–593. doi: 10.1167/iovs.02-0685. [DOI] [PubMed] [Google Scholar]
- 51.Ishida Y, Kimura A, Kuninaka Y, Inui M, Matsushima K, Mukaida N, Kondo T. Pivotal role of the CCL5/CCR5 interaction for recruitment of endothelial progenitor cells in mouse wound healing. J Clin Invest. 2012;122:711–721. doi: 10.1172/JCI43027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sho E, Sho M, Nanjo H, Kawamura K, Masuda H, Dalman RL. Hemodynamic regulation of CD34+ cell localization and differentiation in experimental aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:1916–1921. doi: 10.1161/01.ATV.0000142805.20398.74. [DOI] [PubMed] [Google Scholar]
- 53.Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, Takamura T, Yamamoto H, Miyamoto KI, Ginsberg HN, Mukaida N, Kaneko S, Ota T. CCR5 Plays a Critical Role in Obesity-Induced Adipose Tissue Inflammation and Insulin Resistance by Regulating Both Macrophage Recruitment and M1/M2 Status. Diabetes. 2012 doi: 10.2337/db11-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest. 2008;118:3012–3024. doi: 10.1172/JCI32750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, Leiner I, Li MO, Frenette PS, Pamer EG. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 57.Jones KL, Maguire JJ, Davenport AP. Chemokine receptor CCR5: from AIDS to atherosclerosis. Br J Pharmacol. 2011;162:1453–1469. doi: 10.1111/j.1476-5381.2010.01147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.