

Abstract
A remarkable pathological difference between grey matter lesions (GML) and white matter lesions (WML) in Multiple Sclerosis (MS) patients is the paucity of infiltrating leukocytes in GML. To better understand these pathological differences, we hypothesize that the chemokine monocyte chemotactic protein-1 (MCP-1 or CCL2), of importance for leukocyte migration, and its receptor CCR2 are more abundantly expressed in WML than in GML of MS patients. To this end, we analyzed CCL2 and CCR2 expression in the hippocampus, comprising WML and GML, of post-mortem MS patients, and of control subjects.
CCL2 and CCR2 mRNA were significantly increased in demyelinated MS hippocampus. Semi-quantification of CCL2 and CCR2 immunoreactivity showed that CCL2 is present in astrocytes only in active WML. CCR2 is upregulated in monocytes/macrophages or amoeboid microglia in active WML, and in ramified microglia in active GML, although to a lesser extent. As a follow-up, we observed a significantly increased CCL2 production by WM-, but not GM-derived astrocytes upon stimulation with bz-ATP in vitro. Finally, upon CCL2 stimulation, GM-derived microglia significantly increased their proliferation rate.
We conclude that within hippocampal lesions, CCL2 expression is mainly restricted to WML, whereas the receptor CCR2 is upregulated in both WML and GML. The relative absence of CCL2 in GML may explain the lack of infiltrating immune cells in this type of lesions. We propose that the divergent expression of CCL2 and CCR2 in WML and GML explains or contributes to the differences in WML and GML formation in MS.
Keywords: Multiple sclerosis, Hippocampus, Monocyte chemotactic protein-1, Microglia, Astrocyte, Proliferation
Introduction
Multiple sclerosis (MS) is a chronic neuroinflammatory and degenerative disease affecting mostly young adults in the prime of their life. Clinical features are extremely varied and include cognitive deficits, e.g. memory impairment [1]. Pathologically, MS is characterized by areas of focal demyelination which are spread throughout the entire central nervous system [1]. These demyelinated lesions can be found in several white matter (WM) as well as grey matter (GM) areas, ranging from subcortical and subventricular WM, and tracts within the spinal cord [2-4] and of the brainstem [2] to GM structures such as the thalamus [5,6], hypothalamus [7], hippocampus [8,9], cerebellum [5,10] and cortex [5,6,11].
White matter lesions (WML) are characterized by the infiltration of large numbers of leukocytes [12-14], which are considered to play a crucial role in the formation of WML [15-17]. Although the presence of these cells has been described in GM lesions (GML) biopsy material of MS patients [18,19], a range of studies using post-mortem MS material demonstrated the relative absence of infiltrated immune cells in GML [11,20-22]. This difference in immune cell infiltration between post-mortem WML and GML is best illustrated by leukocortical lesions which encompass both WM and GM (type I lesions), with more T-cells, monocytes/macrophages and/or activated microglia present in the WM than GM part of the lesions [20].
Various hypotheses have been put forward to explain the apparent pathological differences between WML and GML, including a putative role for a divergent expression of chemo-attractant molecules important for the influx of immune cells into the brain. One such molecule is the chemokine monocyte-chemotactic protein-1 (MCP-1, also known as CCL2), which is reported to contribute to the pathogenesis of MS [23,24], through its involvement in the migration of leukocytes into the CNS [25-27]. In addition, CCL2 has been shown to induce migration [28,29], proliferation [30] and activation [30,31] of microglia in vitro. CCL2 mediates its effects by binding to and activating the chemokine receptor CCR2. CCL2-CCR2 interaction is essential to evoke the clinical and histopathological characteristics of experimental autoimmune encephalomyelitis (EAE), an animal model of MS, by regulating the infiltration of immune cells [26] and activation of microglial cells [32]. During EAE, a higher number of infiltrated immune cells correlated with increased disease severity [33] and blockade of CCL2-CCR2 signaling ameliorated progression of EAE together with a decrease in the number of infiltrating immune cells [34]. In post-mortem human MS tissue, CCL2 expressing astrocytes have been described to be present in WML [35-37] which may then contribute to the attraction of immune cells into WM leading to WML formation.
Based on the described histopathological differences between WML and GML, we hypothesize that CCL2 and its receptor CCR2 are more abundantly expressed in WML than in GML of MS patients. To this end, we studied post-mortem human hippocampus, a brain region known to be affected during MS, and to contain WML, GML, and mixed WML and GML [8,9]. We analyzed CCL2 and CCR2 expression in the hippocampus of MS patients and control subjects using semi-quantitative qPCR analysis and immunohistochemistry. An in vitro approach was used to study bz-ATP-induced CCL2 production by astrocytes derived from WM versus GM rat brain. Finally, we determined in vitro whether the CCR2 present in GM-derived microglia is functionally active, i.e. involved in CCL2 induced proliferation of microglial cells.
Materials and methods
Human subjects
For semi-quantitative qPCR analysis, post-mortem hippocampal tissue was collected as part of the “tissue procurement program” approved by the Cleveland clinic Institutional Review Board. Hippocampal fresh frozen tissue of 10 MS patients (age range: 40-73 years) and 5 control subjects (age range: 52-77 years) was included in this study and pathologically characterized previously [38]. Clinicopathological data of the MS patients and non-neurological controls used for qPCR are provided in Table 1.
Table 1.
Clinicopathological data MS patients and control subjects (qPCR study)
| Case | Gender | Age | PMD (h) | DD | MS Type | Demyelination | COD |
|---|---|---|---|---|---|---|---|
| MS | |||||||
| 1 | F | 58 | 7.3 | 13 | RR | no | Abdominal Adenocarcinoma |
| 2 | F | 54 | 5.9 | 37 | SP | no | Unknown Infection |
| 3 | M | 63 | 4.9 | 36 | PP | no | Sepsis |
| 4 | F | 54 | 7.1 | 5 | SP | no | Drug Overdose |
| 5 | F | 51 | 7.0 | 15 | PP | no | Attempted Suicide (asphyxiation) |
| 6 | F | 73 | 7.0 | 46 | PP | yes | Dehydration/Gastroenteritis |
| 7 | F | 46 | 5.6 | 14 | SP | yes | Unknown |
| 8 | M | 52 | 6.7 | 30 | SP | yes | Pulmonary Infections |
| 9 | F | 40 | 5.4 | 13 | SP | yes | Pneumonia |
| 10 | F | 66 | 13.0 | 35 | SP | yes | Unknown |
| Control | |||||||
| 1 | M | 65 | 8.5 | Cardiac arrest | |||
| 2 | F | 53 | 36.0 | Cardiac thrombosis | |||
| 3 | F | 52 | 12.0 | Polymicrobial sepsis | |||
| 4 | M | 53 | 12.0 | Myocardial infarct | |||
| 5 | M | 77 | 12.0 | Mesenteric bleeding |
PMD = Postmortem delay; DD = Disease duration; COD = Cause of death; SP = Secondary progressive; PP = Primary progressive.
For immunohistochemistry, post-mortem human hippocampal tissue was obtained from the Netherlands Brain Bank (NBB, Amsterdam, The Netherlands) or from the Department of Pathology (VU University Medical Center in Amsterdam, The Netherlands). Formalin-fixed, paraffin-embedded hippocampal tissue sections were included from 18 clinically diagnosed and neuropathologically verified MS patients (age range: 43-77 years) and 9 control subjects (age range: 50-92 years) without neurological or psychiatric disease. Clinicopathological data of the MS patients and non-neurological controls used for immunohistochemistry are provided in Table 2.
Table 2.
Clinicopathological data MS patients and control subjects (immunohistochemistochemical study)
| Case | Gender | Age | PMD (h) | DD | MS Type | Demyelination (lesion type) | COD |
|---|---|---|---|---|---|---|---|
| MS | |||||||
| 11 | M | 43 | 8:30 | 17 | SP | yes (active) | Pneumonia |
| 12 | F | 66 | 6:00 | 23 | SP | yes (active) | Unknown |
| 13 | F | 60 | 10:40 | 7 | PP | yes (active) | Euthanasia |
| 14 | M | 54 | 8:15 | 12 | PP | yes (active) | Euthanasia |
| 15 | F | 50 | 7:35 | 17 | SP | yes (active) | Euthanasia |
| 16 | F | 62 | 6:45 | 25 | Unknown | yes (inactive) | Unknown |
| 17 | M | 63 | 7:05 | 28 | SP | yes (inactive) | Cardiac arrest after rupture of abdominal aorta |
| 18 | M | 47 | 7:15 | 7 | SP | yes (inactive) | Urosepsis with organ failure |
| 19 | M | 49 | 8:00 | 26 | PP | yes (inactive) | Pneumonia by MS |
| 20 | M | 66 | 7:30 | 26 | PP | yes (inactive) | Ileus |
| 21 | M | 61 | 9:15 | 31 | SP | yes (inactive) | Euthanasia |
| 22 | F | 57 | 8:40 | 27 | Unknown | yes (inactive) | Respiratory insufficiency by (uro)sepsis |
| 23 | F | 77 | 5:43 | 6 | Unknown | no | Respiratory insufficiency with aspiration pneumonia |
| 24 | M | 71 | 7:00 | 26 | PP | no | Pneumonia by aspiration |
| 25 | F | 76 | 9:45 | 20 | PP | no | Unknown |
| 26 | M | 50 | 9:30 | 24 | PP | no | Unknown |
| 27 | M | 51 | 11:00 | 20 | Unknown | no | Unknown |
| 28 | M | 56 | 9:50 | 14 | PP | no | Cachexia and exhaustion by end stage MS |
| Control | |||||||
| 6 | M | 84 | 7:05 | Exacerbation of COPD | |||
| 7 | M | 56 | 9:15 | Myocardial infarction | |||
| 8 | F | 62 | 7:55 | Euthanasia | |||
| 9 | F | 92 | 7:00 | Acute death, probably pulmonary embolism | |||
| 10 | F | 50 | 4:10 | Metastasized large cell bronchocarcinoma | |||
| 11 | F | 62 | 7:20 | Metastases | |||
| 12 | M | 82 | 5:10 | Pneumonia/cardiovascular accidents | |||
| 13 | M | 78 | Unknown | Cardiac arrest after rupture of abdominal aorta | |||
| 14 | F | 84 | 6:55 | Myelodysplasia |
PMD = Postmortem delay; DD = Disease duration; COD = Cause of death; SP = Secondary progressive; PP = Primary progressive.
qPCR analysis on human hippocampus material
Fresh hippocampal tissue was homogenized in Qiazol and total RNA was isolated as described by the manufacturer using the RNeasy Microarray tissue kit (Qiagen Inc, Valencia, USA). RNA concentration was determined spectrophotometrically at 260 nm using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, USA) and purity of the RNA samples was determined by measuring the absorbance ratio at 260/230 nm (samples were excluded when ratio was outside the 2.00-2.20 range) and 260/280 nm (samples were excluded when ratio was outside the 1.90.-2.10 range). When RNA quality was approved, 1 μg of RNA was reverse transcribed into cDNA and the PCR reaction was carried out using RT2 First Strand kit (Qiagen Inc, Valencia, USA) according to the manufacturer’s instructions. For the qPCR reaction, qPCR assays (CCL2:PPH00192E; CCR2:PPH00612F) were purchased (Qiagen Inc, Valencia, USA). All reactions were normalized to the reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the qPCR Assay (PPH00150E, Qiagen Inc.), which have been found to produce reproducible results using RT-PCR analysis previously performed using control and MS hippocampus tissues [38,39].
Amplification of cDNA was performed in MicroAmp Optical 96-well Reaction Plates (Applied Biosystems Inc, NY, USA) and the analysis was done using an ABI Biosystems 7300 RT-PCR System (Applied Biosystems Inc, NY, USA). The 25 μl reaction mixture was composed of RT2 SYBR Green Rox Master Mix (Qiagen Inc.), 1 μl cDNA and 1 μl of the forward and reverse primers each. As a negative control, no template (no cDNA) reactions were performed to exclude DNA contamination, and to confirm the absence of primer-dimer artifacts from amplification plots. The reaction conditions were an initial 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C each. All samples were run in triplicate and mRNA levels were normalized to the levels of the reference gene GAPDH using previously published methods [38,39].
Immunohistochemical detection of MBP, MHC-II, CCL2 and CCR2
After rapid autopsy (mean postmortem delay: 7.5 h) hippocampal tissue samples were fixed in 10% formalin for 30 days and embedded in paraffin. Of 32 paraffin-embedded tissue blocks, 5-μm sections were cut and mounted on positively charged glass slides (Menzel-Glaser SuperFrost plus, Braunschweig, Germany), and dried overnight at 37°C. Upon use, sections were heated in an incubator for 30 min at 56°C, before they were deparaffinized in xylene, and rehydrated through a series of 100%, 96%, and 70% ethanol and distilled water. For subsequent antigen retrieval, sections were rinsed in 0.01 M citrate buffer (pH 6.0) or in 10 mM Tris buffer (pH 9.0) containing 1 mM EDTA (Tris-EDTA) and subsequently heated in a steamer for 30 min at 90-99°C in the same buffers. After antigen retrieval, the sections were allowed to regain room temperature (RT), rinsed in Tris-buffered saline (TBS), and incubated for 20 min in TBS containing 0.3% H2O2 and 0.1% sodiumazide. Non-specific binding sites were blocked with 5% non-fat dried milk (Campina) in TBS containing 0.5% Triton (TBS-T; blocking solution) for 30 min at RT. Subsequently, sections were incubated overnight at 4°C with the following primary antibodies: myelin basic protein (MBP), MHC-II (LN3), CCL2 or CCR2 (see Table 3 for details on primary antibodies), diluted in blocking solution. The next day, sections were washed in TBS and incubated for 2 h at RT in corresponding biotinylated IgG’s (1:400, Jackson Immunoresearch, Westgrove, PA, USA; see Table 3 for details on secondary antibodies), followed by HRP-labeled avidin-biotin complex (ABC complex 1:400; Vector Laboratories, Burlingame, CA, USA) for 1 h at RT. To detect CCL2 immunoreactivity, an amplification step was included at this stage, and thus sections were subsequently incubated in biotinylated tyramide (1:800, kindly provided by Dr. I. Huitinga, Netherlands Institute for Neuroscience, Amsterdam) and 0.01% peroxide in TBS-T for 30 min, followed by another incubation with ABC complex (1:800) in TBS-T for 1 h. Detection of all antigens was visualized using 3,3-diaminobenzidine (DAB, Sigma, St. Louis, MO, USA) as a chromogen and counterstaining was performed with hematoxylin. After dehydration in graded ethanol solutions, the sections were cleared in xylene and coverslipped in Entellan (Merck, Darmstadt, Germany). Negative controls were performed by omitting the primary antibody resulting in no immunohistochemical signal (data not shown).
Table 3.
Primary and secondary antibodies used for single labeling
| Antigen | Antigen retrieval | Species | Final dilution | Source primary antibody | Secondary antibody | Source secondary antibody |
|---|---|---|---|---|---|---|
| Human MBP | Tris-EDTA | Mouse | 1:100 | Boehringer, 1118099 | Biotinylated donkey-anti-mouse IgG | Jackson, 715-065-151 |
| Human MHC-II | Citrate | Mouse | 1:100 | Gift, Clone LN3 | Biotinylated donkey-anti-mouse IgG | Jackson, 715-065-151 |
| Human CCL2 | Citrate | Mouse | 1:200 | R&D Systems, MAB2791 | Biotinylated goat-anti-mouse IgG | Jackson, 115-065-146 |
| Human CCR2 | Citrate | Rabbit | 1:1000 | Abcam, ab32144 | Biotinylated goat-anti-rabbit IgG | Jackson, 111-065-144 |
Immunofluorescent double labeling
Based on the morphological appearance of CCL2 and CCR2 positive cells, double-labeling of astrocytes and CCL2 or of microglia and CCR2 was performed. To that end, antibodies for astrocytes, i.e. glial fibrillary acidic protein (GFAP), and for CCL2 or antibodies for microglia, i.e. ionized calcium-binding adapter molecule 1 (Iba-1) and for CCR2 were used. In addition, a double labeling of CCR2 and the cell proliferation marker BM28 [40] was performed.
Sections were co-incubated (Iba-1/CCR2 and Iba-1/BM28) with the appropriate primary antibodies. To prevent steric hindrance of the CCL2 antibody and GFAP antibody, sections were sequentially incubated (GFAP/CCL2) with the appropriate primary antibodies (see Table 3 for details on primary antibodies used).
Sections were deparaffinized and antigen retrieval was performed with citrate or Tris-EDTA buffer as described above. Non-specific binding sites were blocked with 3% bovine serum almunin (BSA) (Sigma) in TBS-T for 30 min at RT. All antibodies were diluted in 3% BSA in TBS-T. After a 24 h incubation at 4°C, the sections were washed and subsequently incubated for 90 min at RT with appropriate Alexa Fluor 488 or Alexa Fluor 594 labeled IgG’s (1:400; Molecular Probes) or with streptavidin-labeled Alexa Fluor 488 (1:400, Molecular Probes) when the secondary antibodies were biotinylated (see Table 4 for detailed information on secondary antibodies and conjugates). After washing, the sections were coverslipped with Vectashield (Vector Laboratories, Burlingame, CA, USA). Sections were examined using a confocal laser scanning microscope (Leica TSC-SP2-AOBS; Leica Microsystem, Wetzlar, Germany).
Table 4.
Primary and secondary antibodies used for double labeling
| Antigen | Antigen retrieval | Species | Final dilution | Source primary antibody | Secondary antibody | Source secondary antibody | Conjugate | Source conjugate |
|---|---|---|---|---|---|---|---|---|
| Iba-1 | Tris-EDTA | Goat | 1:300 | Abcam, ab5076 | Alexa Fluor 488 coupled donkey anti goat IgG | Molecular Probes, A11055 | ||
| GFAP | Citrate | Rabbit | 1:4000 | DAKO, Z0334 | Alexa Fluor 594 coupled donkey anti rabbit IgG | Molecular Probes, A21207 | ||
| CCL2 | Citrate | Mouse | 1:200 | R&D Systems, MAB2791 | Biotinylated goat anti mouse IgG | Jackson, 115-065-146 | Alexa Fluor 488 coupled streptavidin | Molecular Probes, S11223 |
| CCR2 | Tris-EDTA | Rabbit | 1:1000 | Abcam, ab32144 | Alexa Fluor 594 coupled donkey anti rabbit IgG | Molecular Probes, A21207 | ||
| BM28 | Tris-EDTA | Mouse | 1:600 | BD Transduction Laboratories, 610700 | Biotinylated donkey anti mouse IgG | Jackson, 715-065-151 | Alexa Fluor 488 coupled streptavidin | Molecular Probes, S11223 |
Definition of lesion center and border
Lesions were identified by the loss of myelin basic protein (MBP) immunoreactivity. The border of a lesion is represented by the edge of demyelinated and myelinated WM or GM. The center of a lesion is the demyelinating or demyelinated area between the borders of a WML or a GML (Figure 1A, F). A distinction was made between demyelinated WM and GM regions within the hippocampus, in which the WM regions comprise the stratum radiatum and the alveus and GM regions comprise the cornu ammonis (CA) 1, CA2, CA3 and CA4. The activity status was determined by the presence or absence of MHC-II positive monocytes/macrophages or microglia in the border or center of a lesion. Active/chronic active and inactive WML were defined as described before [41,42], in which active and chronic active lesions show amoeboid monocytes/macrophages in the center (Figure 1B) or at the border (Figure 1C) of the lesions, respectively, while inactive lesions are almost devoid of activated immune cells, but do show ramified microglia (Figure 1G, H) [41-43]. The activity status of GML, which are virtually devoid of immune cells, was determined by the presence or absence of MHC-II positive amoeboid macrophages present within neighboring demyelinated WM which was part of the same lesion (combined WML and GML) and the presence of MHC-II positive microglia in the GML. Although not officially identified as such, we called these active GML (Figure 1D, E). When hardly any MHC-II positive microglia were present, we called these inactive GML (Figure 1I, J). Thus, to be able to carefully localize the CCL2 and CCR2 expressing cells, we discriminated between the center and the borders of WML and GML as well as between active/chronic active and inactive lesions.
Figure 1.

Histopathological features of active and inactive hippocampal MS lesions. MS lesions are recognized by the loss of MBP immunoreactivity in hippocampal WM and/or GM areas (A,F). Active and inactive lesions were distinguished based on the presence or absence of MHC-II positive monocytes/macrophages, respectively. WM within the lesion centre (B) and at the border (C) of the active lesion presents with activated amoeboid MHC-II positive cells. The number of MHC-II positive cells in the lesion centre (D) and at the border of a GML (E) is far lower compared to WML. Moreover, LN3+ cells within the GM have a more ramified morphology. LN3+ cells within an inactive lesion are lower in number compared to active lesions and have a ramified morphology, both within the centre (G) and at the border (H) of WM, as well as in the centre (I) and at the border of (J) GM. Frames in A & F refer to hippocampal areas shown in B-E; G-J. The dashed lines indicate the border between WM (alveus and striatum) and GM (CA1). AV = alveus; Str = Striatum. Scale bar (A, F) = 500 μm. Scale bar (B-E; G-J) = 60 μm.
Semi-quantitative analysis of CCL2- and CCR2-positive cells
Semi-quantification of CCL2- and CCR2-positive cell numbers was performed by unbiased manually counting the number of positive cells with a clearly visible cell nucleus within the region of interest (ROI). In MS lesions, ROIs were positioned at the white matter border, grey matter border, white matter lesion center and grey matter lesion center. The CA4 region was excluded from analysis since this region often showed nonspecific astrocytic staining regardless of the presence of lesions. In control subjects and MS patients without hippocampal lesion, ROIs were placed in hippocampal WM and GM. In each ROI, cells were counted in 2 fields, each measuring 0.1-0.4 mm2. Results were expressed as number of cells (mean and standard error of the mean) per mm2. All pictures were acquired using an Olympus-VANOX-T microscope (Tokyo, Japan) at 10-fold magnification. Cell counting and area determination were performed using Cell^F Olympus Soft Imaging Software (Tokyo, Japan).
Primary culture of astrocytes and microglia
Primary astrocytes and microglia were isolated from 1 day old Wistar rats (Harlan CPB, Zeist, The Netherlands) as described previously [44] and which was approved by the Animal Experiment Committee of the VU University Medical Center, Amsterdam, The Netherlands (approval ID: FGA 11-03). Mixed glial cells were isolated and cultured from pons and cerebral cortices to obtain mainly WM and GM primary astrocytes, respectively. These brain regions were cleared from adhering meninges and blood vessels, and mechanically dissociated in Dulbecco’s modified Eagle’s medium (DMEM)-F10 (Gibco, Life Technologies, Breda, The Netherlands), supplemented with 10% v/v heat-inactivated fetal calf serum (FCS) (Gibco), 2 mM L-glutamine (Sigma-Aldrich), 50 Units/ml penicillin (Sigma-Aldrich) and 50 μg/ml streptomycin (Gibco). Cells were plated in poly-L-lysine (15 μg/ml (2 μg/cm2); Sigma-Aldrich) coated T75 culture flasks (Nunc, Hamstrop, Denmark) and incubated at 37°C in humidified air containing 5% CO2. The medium was changed at day 1, day 6 and day 8 after seeding. After 10 days in culture, microglia and astrocytes were separated by shaking the flasks at 37°C at 260 rpm for 16 h. Cortical microglia were directly plated into poly-L-lysine coated 8-well chamber-slides (Lab-Tek). Astrocytes were further purified by treatment with 5 mM leucine methyl ester (LME, Sigma- Aldrich) in serum free medium, for 24 h at 37°C. Subsequently, cortical and pons-derived astrocytes were plated in poly-L-lysine coated 6-well plates (Thermo Scientific) containing fresh medium with serum. Purity of astrocyte cultures was between 80-90% as determined by immunofluorescent staining using anti-GFAP antibody (1:6000; DAKO; Z0334). Purity of primary microglia cultures was between 90-94% as determined by immunofluorescent staining using anti-Iba1 antibody (1:1000;WAKO Chemicals USA; 019-19741).
Bz-ATP-induced CCL2 expression in white and grey matter derived astrocytes
To determine the level of CCL2 expression by white and grey matter derived astrocytes, 0.5 x 106 cells were plated onto poly-L-lysine coated wells for 24 h at 37°C. Astrocytes were cultured in serum-free medium alone (control) or in the presence of Benzoyl-benzoyl adenosine 5’-triphosphate (bz-ATP) (500 μM) for 2 to 6 h (n = 3 independent experiments). Bz-ATP is a potent analogue of ATP and was chosen as a stimulus because ATP release is a physiological response upon axonal damage [45], such as observed in WML and GML in MS [20,42]. Subsequently, ATP (and bz-ATP) acts on purinergic receptors, e.g. P2X7, present on astrocytes [46] resulting in various responses, including CCL2 production [47]. After incubation with bz-ATP, astrocytes were homogenized in Trizol reagent (Life Technologies, Carlsbad, USA) and total RNA was isolated as described by the manufacturer. RNA concentration was determined spectrophotometrically at 260 nm using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher scientific, Waltham, USA) and purity was determined by measuring the absorbance ratio at 260/280 nm, and was approved when values were between 1.9 and 2.1. When quality criteria were met, 1 μg of RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Invitrogen) according to the manufacturer’s instructions, but using oligo-d(T)16 primers (Applied Biosystems). For the qPCR reaction, primers for CCL2 (GenBank accession number: NM_031530.1) and GAPDH (GenBank accession number: NM_017008.3) were designed and purchased from Eurogentec (Seraing, Belgium).
GAPDH was identified as the most stable reference gene compared to four other reference genes, i.e. hypoxanthine phosphoribosyltransferase 1 (HPRT1), phosphoglycerate kinase 1 (PGK1), peptidylprolyl isomerase A (cyclophilin or PPIA) and tyrosine 3-monooxygenase/tryptophan 5- monooxygenase activation protein, zeta polypeptide (YWHAZ).
Details of the primer sequences are as follows:
CCL2 forward primer: 5′-ACGTGCTGTCTCAGCCAGATG-3′
CCL2 reverse primer: 5′-GACTCATTGGGATCATCTTGCC-3′
GAPDH forward primer: 5′-GAACATCATCCCTGCATCCA-3′
GAPDH reverse primer: 5′-GCCAGTGAGCTTCCCGTTCA-3′
Amplification of cDNA was performed in MicroAmp Optical 96-well Reaction Plates (Applied Biosystems) and the analysis carried out using a StepOnePlus Real-Time-PCR System (Applied Biosystems). The 20 μl reaction mixture was composed of Power SYBR Green PCR Master Mix (Applied Biosystems), 12.5 ng cDNA and 4 pM of the forward and reverse primers each. As a negative control, no template (no cDNA) reactions were performed. The reaction conditions were an initial 2 min at 50°C, followed by 10 min at 95°C and 40 cycles of 15 sec at 95°C and 1 min at 60°C. The mRNA expression levels were quantified relatively to the level of the reference gene glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) using the following calculation: 2-(Cq of target mRNA - Cq of GAPDH) × 100%.
Bromodeoxyuridine labelling assay
In contrast to hippocampal WM, hardly any CCL2 is present in hippocampal GM as observed in the present study (see Results section). We thus questioned whether the CCR2 receptor on GM-derived microglia is activated by CCL2 and results in a functional response. To this end, the effect of CCL2 treatment on GM microglia proliferation was studied. Cortical primary rat microglia were plated on poly-L-lysine coated chamberslides (2 × 104 cells/chamber) and allowed to adhere for 2 h. Then, medium was refreshed and 10 nM of INCB3344 (MedChem Express, Princeton, USA), a specific CCR2 antagonist [48,49] or vehicle (medium) together with 20 μM bromodeoxyuridine (BrdU; Sigma) were added. After 30 min, 50 ng/ml of recombinant rat MCP-1 (CCL2; Peprotech) was added (n = 3 independent experiments). After 48 h, the cells were washed and subsequently fixed with 4% PFA for 20 min, and again washed three times with 1% Triton X-100 (Sigma-Aldrich) in PBS. Then, the fixed cells were incubated with 5 units/100 μl DNAse (Promega, H6101) for 45 min at 37°C. Subsequently, the fixed cells were blocked with 5% normal goat serum in PBS/1% Triton X-100 for 1 h. To identify BrdU incorporation, microglia were incubated with rat anti-BrdU (1:200; Abcam; AB6326) in PBS/1% Triton X-100 for 24 h at RT, washed with PBS/1% Triton X-100 followed by incubation with Alexa Fluor-488 labeled goat anti-rat IgG’s (1:200; Molecular Probes) for 2 h at RT. Cells were washed three times with PBS/1% Triton X-100 and once with PBS, treated for 5 min with 5 μg/ml propidiumiodide (PI) and again washed two times with PBS. Then, the slides were embedded in Vectashield (Vector Laboratories). Immunofluorescent staining was visualized using an Olympus VANOX-T microscope (Tokyo, Japan). In five random 20 × fields per well, the percentage of proliferative cells was calculated as follows: (number of BrdU positive cells/ number of PI positive nuclei) × 100%.
Statistics
Statistical analyses were carried out with SPSS package version 20.0 (Statistical Product and Service Solutions, Chicago, IL, USA). Data of multiple tissue blocks from the same patient were averaged to finally represent each case with 1 value per ROI. Normal distribution of the data was tested using the Shapiro-Wilk procedure. When data were normally distributed, differences between control subjects, MS patients without hippocampal lesions, MS patients with active/chronic active hippocampal lesions and MS patients with inactive hippocampal lesions were compared using one-way ANOVA, followed by Tukey’s post hoc analysis. When data were not normally distributed, a Kruskal-Wallis test was performed followed by a Mann-Whitney U post-hoc analysis.
Differences in CCL2 and CCR2 immunoreactivity within subject WM vs GM were analyzed for controls, MS patients without and with hippocampal demyelination using the Wilcoxon Signed Rank Test. The Wilcoxon Signed Rank Test was also used for analyzing the difference between the number of CCL2 and CCR2 expressing cells within subject lesion center versus lesion border of MS patients with hippocampal demyelination. For evaluating the response of WM and GM astrocytes to bz-ATP, a 2-way ANOVA was performed followed by a Student’s t-test (Bonferroni adjusted). Proliferation of microglia was analyzed using a one-way ANOVA, followed by LSD post hoc analysis. P-values < 0.05 were considered significant.
Results
Lesion characterization
Areas of hippocampal demyelination were identified by loss of MBP immunoreactivity (Figure 1A, F). No demyelination was observed in the sections of non-neurological control subjects (not shown). Of the 18 MS patients, 6 did not show demyelination in the hippocampal tissue block obtained. The other 12 MS patients showed demyelinated areas in hippocampal tissue blocks studied, which were subsequently examined for the presence of MHC-II positive cells to determine the lesion activity. Of these, 5 were classified as having active/chronic active lesions. Together, these 5 patients had 11 lesions, of which 6 mixed WM/GM lesions, 1 purely GM lesion, and 4 purely WM lesions. Seven MS patients were classified as having inactive lesions. In total, this group comprised 22 lesions, of which 12 mixed WM/GM lesions, 6 purely GM lesions, and 4 purely WM lesions. In agreement with previous studies [9,50], only a minimal number of CD3 positive T-cells was observed in active WM lesions, whereas these cells were absent in active GM lesions (data not shown).
CCL2 and CCR2 mRNA in human hippocampus
Levels of CCL2 and CCR2 mRNA within the hippocampus of MS patients and control subjects were determined by qPCR. While CCL2 and CCR2 mRNA in myelinated MS hippocampi (n = 5) was not altered compared to control hippocampi (n = 5), a ~4 fold increase in CCL2 mRNA (ANOVA; F = 18.07; p < 0.001) and a ~3 fold increase in CCR2 mRNA (ANOVA, F = 5.63; p = 0.02) was measured in demyelinated hippocampi (n = 5) compared to myelinated MS hippocampi (p < 0.001; p = 0.03, respectively) and compared to control hippocampi (p < 0.001; p = 0.04, respectively) (Figure 2A, B).
Figure 2.
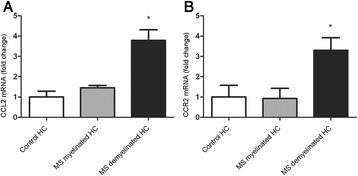
Semi-quantitative RT-PCR analysis of CCL2 and CCR2 mRNA. qPCR of CCL2 (A) and CCR2 (B) mRNA in the hippocampus of control subjects, MS patients without hippocampal lesions and MS patients with hippocampal lesions. Data represent mean ± S.E.M. (n = 5) and are expressed relative to GAPDH.
Semi-quantitative analysis of CCL2 and CCR2 positive cells
For semi-quantitative analysis CCL2 and CCR2 positive cells were counted in several ROIs (see Material and methods). When more tissue blocks of one patient were available, scores for the same ROI in different tissue blocks were averaged. For each group, i.e. control subjects, MS patients without hippocampal lesions, MS patients with active/chronic active hippocampal lesions and MS patients with inactive hippocampal lesions, the mean and standard deviation of CCL2 and CCR2 positive cell numbers per ROI per case are summarized in Table 5.
Table 5.
Mean CCL2 + and CCR2 + cell numbers
| Group | Location | Number of CCL2 + cells (mean ± SD) per mm 2 | Number of CCR2 + cells (mean ± SD) per mm 2 | ||
|---|---|---|---|---|---|
| WM | GM | WM | GM | ||
| Control (n = 9) | Na | 6.6 ± 9.1 | 1.0 ± 2.9 | 5.0 ± 7.5 | 1.9 ± 1.9 |
| NDH (n = 6) | Na | 8.4 ± 11.6 | 0 ± 0 | 9.1 ± 8.5 | 1.1 ± 2.6 |
| Inactive lesions (n = 7) | Center | 8.2 ± 9.7 | 0.7 ± 1.9 | 20.1 ± 25.2 | 1.4 ± 2.4 |
| Border | 16.1 ± 23.8 | 1.8 ± 3.3 | 58.0 ± 84.8 | 2.3 ± 3.9 | |
| Active lesions (n = 5) | Center | 21.1 ± 20.2 | 0.1 ± 0.3 | 189.8 ± 152.6 | 21.8 ± 21.4 |
| Border | 45.3 ± 21.7 | 0 ± 0 | 256.6 ± 90.2 | 63.3 ± 14.3 | |
Na = Not applicable; NDH = Non-demyelinated hippocampi; SD = Standard deviation.
CCL2
In hippocampal WM of control subjects and in WM of myelinated hippocampi of MS patients a few CCL2 positive cells were found upon immunohistochemical analysis (Figure 3A, D). However, at the border of active demyelinated WML an increase in CCL2 positive cells was observed (45.3 ± 21.7 cells/mm2; Figure 3E), which was significantly different from the hippocampal WM of control subjects (6.6 ± 9.1 cells/mm2) and WM of myelinated hippocampi of MS patients (8.4 ± 11.6 cells/mm2) (Kruskall-Wallis, p = 0.031; Mann Whitney U, p = 0.004 and p = 0.017, respectively) (Figure 3 M). CCL2 positive cells were found within the center of active WML (21.1 ± 20.2 cells/mm2) (Figure 3B), albeit to a lesser extent, as well as in the center (Figure 3C) and at the border (Figure 3 F) of inactive WML. In the hippocampal GM areas only scarce CCL2 immunoreactivity was observed, irrespective of the absence or presence of GML (Figure 3G-L).
Figure 3.
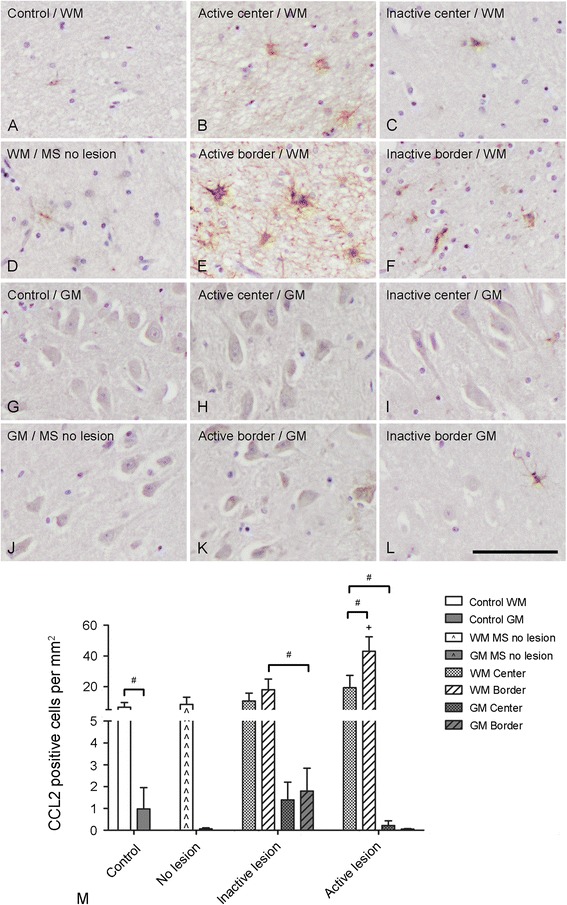
Semi-quantitative analysis of CCL2 positive cell numbers. CCL2 positive cells were only sporadically found within the hippocampal WM of control subjects (A) and MS patients without hippocampal lesions (D). The number of CCL2 positive cells was slightly increased within the WM centre of active (B) and inactive (C) lesions and borders (F). A significant increase in the number of CCL2 positive cells was found within the WM border of active lesions (E). CCL2 positive cells were barely detected within the hippocampal GM of control subjects (G), MS patients without hippocampal lesions (J), the lesion centre (H) and border (K) of active lesions and only sporadically within the lesion centre (I) and border (L) of inactive lesions. Semi-quantification of CCL2 positive cells (M). Scale bar (A-L) = 60 μm; + p < 0.05 versus cell number in WM of non-demyelinated hippocampi of MS patients and in hippocampal WM of control subjects; # p < 0.05.
When we compared CCL2 immunoreactivity in WM and GM of control subjects, MS patients without hippocampal lesions and MS patients with active or inactive lesions, we found significantly more CCL2 positive cells in hippocampal WM (6.6 ± 9.1 cells/mm2) than GM (1.0 ± 2.9 cells/mm2) of control subjects (Wilcoxon Signed Rank test, p = 0.04). Moreover, in the center of active WML more CCL2 positive cells (21.1 ± 20.2 cells/mm2) were found than in the center of active GML (0.1 ± 0.3 cells/mm2) (Wilcoxon Signed Rank test, p = 0.04). CCL2 immunoreactive cells within the WM border of inactive lesions (16.1 ± 23.8 cells/mm2) were significantly outnumbering those found within the hippocampal GM border of inactive lesions (1.8 ± 3.3 cells/mm2) (Wilcoxon Signed Rank test, p = 0.04) (Figure 3M).
Finally, the number of CCL2 positive cells was significantly higher within the WM border compared to the center of active WM hippocampal lesions (Wilcoxon Signed Rank test, p = 0.04) (Figure 3M).
CCR2
CCR2 positive cells were found infrequently within the WM of control subjects and myelinated MS hippocampi (Figure 4A, D). However, within the center (Figure 4B) and at the border (Figure 4E) of active demyelinated WML, CCR2 positive cells were abundantly present. In inactive WML, CCR2 expression was still present but to a lesser extent within the center (Figure 4C) and at the border (Figure 4F) of the lesion. Semi-quantification indicated that inactive hippocampal WML presented with significantly more CCR2 positive cells within the border (58.0 ± 84.8 cells/mm2) compared to hippocampal WM of control subjects (5.0 ± 7.5 cells/mm2) (ANOVA, F = 19.77, p < 0.001; Tukey HSD, p = 0.008). Active hippocampal WML showed significantly more CCR2 immunopositive in the WM border (256.6 ± 90.2 cells/mm2) compared to WM in control hippocampi, and WM in myelinated MS hippocampi (9.1 ± 8.5 cells/mm2) and the WML and border of inactive hippocampal lesions (ANOVA, F = 19.77, p < 0.001; Tukey HSD, p < 0.005 for all comparisons). Similarly, the center of active WML (189.8 ± 152.6 cells/mm2) showed significantly more CCR2 positive cells than WM in control hippocampi, WM in myelinated MS hippocampi and the center of inactive WML (20.1 ± 25.2 cells/mm2) (Kruskal-Wallis, p = 0.003; Mann-Whitney U, p = 0.001, p = 0.004 and p = 0.003, respectively).
Figure 4.
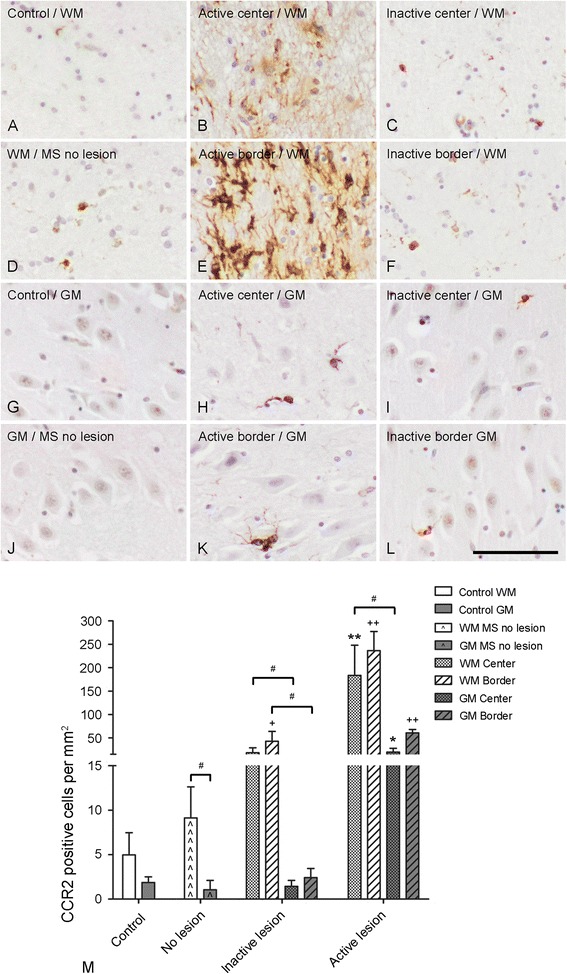
CCR2 immunoreactivity. Low numbers of CCR2 positive cells were observed within the hippocampal WM of control subjects (A), MS patients without hippocampal lesions (D), the lesion centre (C) and border (F) of inactive lesions. A significant increase in the number of CCR2 positive cells was found within the WM centre (B) and border (E) of active lesions. CCR2 positive cells were barely detected within the hippocampal GM of control subjects (G) and MS patients without hippocampal lesions (J). The number of CCR2 positive cells was slightly increased within the WM centre of active (H) and inactive (I) lesions and borders (L). A significant increase in the number of CCR2 positive cells was found within the GM border of active lesions (K). Semi-quantification of CCR2 positive cells (M). Scale bar (A-L) = 60 μm. + p < 0.01 compared to number in the WM of control subjects and the WM border of active lesions, ++ p < 0.01 compared to number in the GM of control subject and the GM border of inactive lesions and +++ p < 0.01 compared to number in the WM border of inactive lesions and WM of non-demyelinated hippocampi of MS patients and control subjects. * p < 0.05 compared to numbers in the GM center of inactive lesions and GM of MS patients without hippocampal lesions and control subjects, and ** p < 0.01 compared to numbers in the WM center of inactive lesions and WM of MS patients without hippocampal lesions and control subjects. ; # p <0.05.
In contrast to the absence of CCR2 positive cells in hippocampal GM of control subjects and in GM of myelinated hippocampal tissue of MS patients (Figure 4G, J), CCR2 immunoreactive cells appeared within the center and at the border of active GML (Figure 4H, K). Less CCR2 positive cells were found in inactive GML (Figure 4I, L). After semi-quantification we measured a significant increase in CCR2 immunoreactivity within the hippocampal GML center (21.8 ± 21.4 cells/mm2) and border (63.3 ± 14.3 cells/mm2) of active lesions compared to hippocampal GM of control subjects (1.9 ± 1.9 cells/mm2), GM of myelinated hippocampi of MS patients (1.1 ± 2.6 cells/mm2) and GML center (1.4 ± 2.4 cells/mm2) and border (2.3 ± 3.9 cells/mm2) of inactive lesions (Kruskal-Wallis, p = 0.01; Mann-Whitney U, p = 0.01, p = 0.009 and p = 0.01, respectively; Kruskal-Wallis, p = 0.007; Mann-Whitney U, p < 0.003; p = 0.01 and p = 0.006, respectively) (Figure 4M).
When we compared CCR2 immunoreactivity in WM and GM of control subjects, MS patients without hippocampal lesions and MS patients with active or inactive lesions we found that the number of CCR2 positive cells was significantly higher in hippocampal WM compared to GM of MS patients without hippocampal lesions (Wilcoxon Signed Rank test, p = 0.03). In addition, CCR2 positive cells were significantly more numerous in the WML center compared to the GML center of active and inactive hippocampal lesions (Wilcoxon Signed Rank test, p = 0.04 and p = 0.05, respectively). Furthermore, the number of CCR2 positive cells was significantly higher within the WML border compared to GML border of hippocampal inactive lesions (Wilcoxon Signed Rank test, p = 0.02) (Figure 4M).
Cellular identification of CCL2 and CCR2 immunoreactivity
Based on the morphological appearance of CCL2 and CCR2 positive cells in hippocampal MS lesions, we performed double labeling studies on lesioned hippocampal material to determine whether CCL2 is expressed by astrocytes and CCR2 by microglia. The experiments revealed extensive co-localization of CCL2 with GFAP in all lesion types (Figure 5A-C), demonstrating that astrocytes indeed expressed CCL2. Additionally, CCR2 co-localized with iba-1 positive ramified microglia in WML and GML (Figure 5D-F), and with infiltrating monocytes/macrophages or amoeboid microglia in WML (Figure 5G-I).
Figure 5.
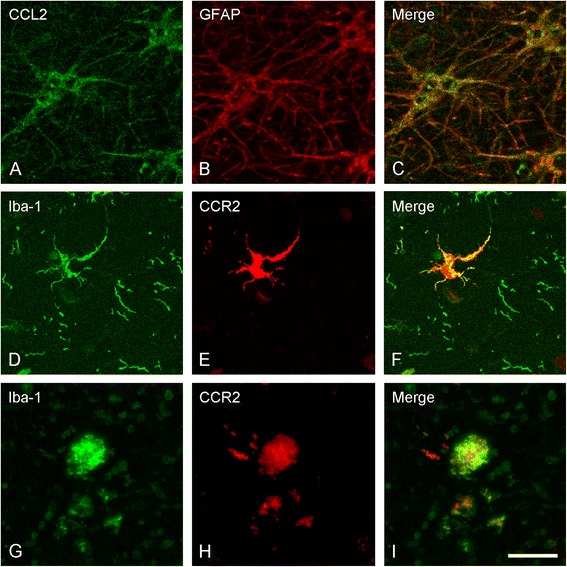
Cellular localization of CCL2 positive and CCR2 positive cells. Representative images of confocal laser scanning microscopy revealed colocalization of CCL2 with GFAP (A-C), whereas CCR2 colocalized with ramified iba-1 positive cells in hippocampal GM (D-F), and amoeboid iba-1 positive cells in hippocampal WM (G-I). Scale bar (A-F) = 10 μm.
Primary culture of astrocytes and microglia
Bz-ATP-induced CCL2 mRNA expression by WM- and GM-derived astrocytes
The observed outnumbering of CCL2 positive cells in active WML compared to active GML was verified by an in vitro study. Upon stimulation with a disease-relevant stimulus, i.e. bz-ATP, primary rat WM astrocytes, showed a significant 6-fold increase in CCL2 mRNA compared to vehicle-treated (medium) astrocytes (p = 0.034). In contrast, GM-derived astrocytes showed a minimal increase in CCL2 mRNA upon bz-ATP stimulation (Figure 6A).
Figure 6.
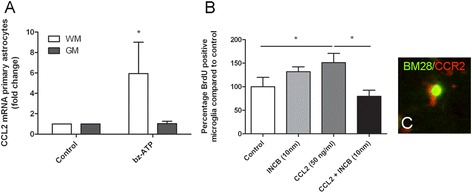
CCL2 mRNA in astrocytes and CCL2 induced microglial proliferation . Primary WM rat astrocytes produce significantly more CCL2 mRNA upon treatment with bz-ATP than GM astrocytes (A). Primary rat microglia significantly increased their proliferation rate upon treatment with CCL2. This effect was abolished when the cells were co-incubated with a CCR2 antagonist (B). Immunofluorescent double labelling study showed colocalization between BM28 and CCR2 in post mortem human hippocampal GML (C). * p < 0,05.
CCR2 mediated proliferation of GM-derived microglia
The relative absence of CCL2 production by GM astrocytes in vivo and in vitro urged the question whether the clearly present CCR2 in GM microglia is bound and activated by CCL2 and results in a functional response. Therefore, primary cortical rat microglia were treated with CCL2 in the absence or presence of the selective CCR2 antagonist INCB3344. Incubation of the microglia with INCB3344 only did not affect microglial proliferation. Upon CCL2 treatment, the proliferation of microglia was significantly increased (LSD, p = 0.04). In the presence of INCB3344, this CCL2-mediated increase in microglial proliferation was eliminated (Figure 6B). In support of a role for CCR2 in cell proliferation in GM of MS patients, we observed BM28 positive/CCR2 positive microglial cells in hippocampal GML (Figure 6C).
Discussion
The present study demonstrates that CCL2 in astrocytes and its receptor CCR2 in monocytes/macrophages and microglia are significantly upregulated at the mRNA and protein level in hippocampal lesions of MS patients. Moreover, we show for the first time that there is a spatial and quantitative discrepancy in the distribution of CCL2 and CCR2 in the hippocampus. CCL2 and CCR2 appear in hippocampal WML whereas in GML only CCR2 is present, and more CCL2 and CCR2 positive cells are present in hippocampal WM compared to hippocampal GM. This discrepancy can contribute to the pathological differences observed in WML versus GML of MS patients and may possibly affect subsequent disease outcome.
Accumulating evidence shows that within the CNS of MS patients besides WML also GML, e.g. hippocampal lesions [8,9], are present which may explain functional deficits seen in MS patients [51,52]. Moreover, GML correlate better with various clinical parameters than WML [53-55]. It is therefore of utmost interest to understand the pathological features of GML versus WML, because that may give direction to the identification of novel therapeutic targets. An important pathological feature of WML is the infiltration of immune cells through the blood-brain barrier (BBB) into the CNS, which is considered to play a crucial role in the pathophysiology of MS [15-17]. In contrast, immunohistochemical studies indicate that the GM is not, or far less, affected by infiltrating immune cells [20]. Therefore, we hypothesized that an important mediator of immune cell infiltration, CCL2, and its receptor CCR2 are present in WML but to a lesser extent in affected GM regions in the hippocampus of MS patients.
Our observation that astrocytes are the source of CCL2 is in agreement with previous studies showing CCL2 expression by cells with an astrocytic morphology [36,56]. In line with the observation that CCL2 is more abundantly present in WM/WML is that WM-derived astrocytes produce significantly more CCL2 upon stimulation with bz-ATP in vitro, whereas GM-derived astrocytes hardly respond. This is not due to a total irresponsiveness of GM-derived astrocytes, since stimulation with pro-inflammatory cytokines interleukin-1β and tumor necrosis factor-α resulted in a strong increase in CCL2 mRNA expression (data not shown). Moreover, the low responsiveness to bz-ATP cannot be attributed to an absence of the P2X7 receptor for ATP on GM astrocytes, since we observed P2X7 receptor immunoreactivity in and on WM- and GM-derived astrocytes (data not shown). Still, it cannot be excluded that the observed difference in bz-ATP-mediated CCL2 production by WM- versus GM-derived astrocytes is due to variation in expression of purinergic receptors other than P2X7, though thus far no such heterogeneity between WM and GM astrocytes has been described [57]. The observed discrepancy in the number of CCL2 positive cells in hippocampal WM/WML versus GM/GML is in line with the observation that CCL2 mRNA levels are significantly higher within the corpus callosum (WM) compared to the cortex (GM) in both control mice as well as a mouse model for MS, e.g. cuprizone treated mice [58]. These differences in CCL2 expression by WM versus GM astrocytes might be explained by regionally dependent differences in astrocytes. Indeed, two different types of astrocytes have been recognized, i.e. fibrous astrocytes that reside mainly in the WM and protoplasmic astrocytes that are preferentially located in the GM [59]. The heterogeneity between fibrous and protoplasmic astrocytes is not restricted to morphology. Molecular differences between fibrous and protoplasmic astrocytes have been described as well (reviewed in [60]) with WM-derived astrocytes expressing more e.g. GFAP and glutamate transporter-1 (GLT-1) [61]. Such differences between astrocytes in WML versus GML as observed in the present study by heterogeneity in CCL2 expression and production can contribute to the variety in pathological and functional outcome of WML and GML.
An alternative explanation for the observed difference between CCL2 positive cell number in hippocampal WM vs hippocampal GM can be the local variation in presence of pro-inflammatory cytokines, e.g interleukin (IL)-1β. These cytokines increase CCL2 production by astrocytes in vitro [62] and could thus, besides ATP, contribute to the CCL2 producing astrocytes found in WML. Indeed, in WML of MS patients, IL-1β positive cells are present [63], but have thus far never been studied in GML. In rats suffering from relapsing EAE, IL-1β appears in certain GM areas, but no IL-1β was observed in the hippocampus of these animals [64], and this absence of IL-1β could contribute to the lack of CCL2 in GML. Since neurons located within the GM are known to suppress the production of pro-inflammatory factors [65-67], this could also indirectly prevent CCL2 production. Thus, astrocyte-specific and environmental conditions can explain the relative lack of CCL2 in GML. Since astrocyte-derived CCL2 is a prominent factor involved in the attraction of leukocytes through the BBB in WML in MS [25,26], the relative absence of CCL2 in GML may subsequently explain the lack of infiltrating immune cells in this type of lesion. Additionally, CCL2 has been described to interact with endothelial cells [68] consequently disrupting the BBB. Thus, the absence of CCL2 in the hippocampal GML could explain a lack of BBB damage in GML [69].
The present immunohistochemical study showed that CCR2 is observed in hippocampal WML as well as GML in myeloid-like cells. Moreover, CCR2 was significantly more expressed in hippocampal WML than in GML. Interestingly, fluorescent double-labelling of CCR2 and Iba1 indicated that in hippocampal WML CCR2 is expressed mainly by infiltrating monocytes/macrophages and amoeboid microglia, while in the GML CCR2 is expressed mainly by ramified microglia, which is in line with studies showing CCR2 expression by microglia and macrophages in vitro and ex vivo, which, however, did not differentiate between their regional origin [70-72]. This suggests that during MS lesion formation, the source of CCR2 positive cells in WML are both resident microglia and infiltrating monocytes/macrophages, while in the GML CCR2 positive cells are mainly resident microglia. Like our data for CCL2, the discrepancy between the higher number of CCR2 positive cells in hippocampal WM versus lower number in hippocampal GM might be explained by differences that reside within WM and GM, e.g. the WM harbors more microglia [73], which are more prone to be pro-inflammatory [74,75].
Another interesting finding is that we observed that CCL2 and CCR2 positive cells are more numerous in the border of the lesion with ongoing demyelinating activity and less in the demyelinated center of the lesion, irrespective of WML or GML. Indeed, in WML it has already been shown that CCL2 and CCR2 are mainly present at the border [35,36], whereas it was unknown for GML. Moreover, the present study showed that CCL2 and CCR2 immunoreactivity in the hippocampus was significantly less when either WML or GML were inactive. This has only been reported for CCL2 in WML [56]. Its receptor CCR2 has been shown to be mostly expressed during active inflammation and demyelination in relapsing EAE [71]. These findings support the notion that CCL2 expression by astrocytes plays an important role during the active phase of disease, and that CCL2-CCR2 interaction in this way likely contributes to infiltration of immune cells occurring during active WML formation in MS.
Although CCL2 was hardly observed in GML, its receptor CCR2 was upregulated in both active WML and GML. We questioned whether the CCR2 receptor expressed by microglia in GM can act as a functional receptor. To this end we studied CCL2-mediated GM microglial proliferation in vitro. Indeed, CCL2 induced a significant increase in the percentage of proliferating GM microglial cells, which was prevented by co-incubation with the specific CCR2 antagonist INCB3344. This indicates that GM-derived microglia express a functional CCR2 receptor, and thus CCL2-CCR2 mediated effects can occur in GML, e.g. the induction of microglial proliferation [30]. This was supported by our observation that CCR2 is expressed by BM28 positive proliferating microglia. Their localization at the border of GML suggests that the proliferating microglial cells are either a consequence of or contribute to GML formation. A recent study indicated that proliferating parenchymal microglia are the main source of microgliosis after ischaemic stroke [76]. However, the role of microglial proliferation during demyelination remains to be established. Since only nanomolar concentrations of CCL2 are needed to activate CCR2 [77], it is possible that, although no evident appearance of CCL2 positive cells in the GM was observed, increased presence of the CCR2 receptor increases the chance of CCL2-CCR2 interaction, and thus minute amounts of CCL2 can result in CCR2 mediated responses e.g. microglia proliferation in GML. Alternatively, it is even more likely that CCL2 diffuses from neighbouring WML into the GML to exert its effect. We can, however, not exclude the possibility that other chemokines, i.e. CCL8 or CCL16, known to be ligands for CCR2, interact with the receptor to induce in vivo effects, e.g. cell migration or proliferation, but no expression of these chemokines in MS GML has been described.
Overall, we conclude that the difference in CCL2 and CCR2 expression in hippocampal WML versus GML of MS patients, at least partly, explains the pathological differences observed in WML and GML. As such CCL2-CCR2 interaction is likely to be involved in leukocyte migration into the CNS contributing to WML formation, whereas the relative absence of CCL2 in GML of MS patients may explain the lack of infiltrating immune cells in this type of lesion. Based on our in vitro experiments, we propose that the CCR2 receptor in GM is a functional receptor and is involved in resident microglial cell proliferation, thereby contributing to microgliosis. Furthermore, the discrepancy in CCL2 and CCR2 expression in hippocampal WML and GML implies that the pathogenesis of WML formation differs from that of GML. As such, our data support several lines of evidence which already pointed towards a possible difference in the pathways underlying demyelination in WM and GM, as there is no, or only low, correlation between the extent of demyelination in WM and GM [6,11]. Although under debate, WML formation is considered to start with perivascular infiltration of CD4+ and CD8+ T-cells [16,78-81], whereas GML formation has been suggested to be initiated by meningeal inflammation [82,83]. Although this may hold true for GM cortical lesions in MS, this does not necessarily explain the occurrence of subcortical GML, e.g. hippocampal lesions. With our observations on the discrepancy in CCL2 and CCR2 expression in WML and GML we have added an additional factor that may explain or contribute to the pathways underlying WML and GML formation in MS.
Acknowledgments
This work was supported by the following grants: RG-4280 awarded to RD and by NIH 38667 and NS35058 awarded to BDT.
Footnotes
Ranjan Dutta and Bart Baselmans contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Marloes Prins, Email: m.prins@vumc.nl.
Ranjan Dutta, Email: DUTTAR@ccf.org.
Bart Baselmans, Email: baselmansb@gmail.com.
John J P Brevé, Email: jjp.breve@vumc.nl.
John G J M Bol, Email: jgjm.bol@vumc.nl.
Sadie A Deckard, Email: deckars@ccf.org.
Paul van der Valk, Email: p.vandervalk@vumc.nl.
Sandra Amor, Email: s.amor@vumc.nl.
Bruce D Trapp, Email: TRAPPB@ccf.org.
Helga E de Vries, Email: he.devries@vumc.nl.
Benjamin Drukarch, Email: b.drukarch@vumc.nl.
Anne-Marie van Dam, Email: amw.vandam@vumc.nl.
References
- 1.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;372:1502–1517. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 2.Charil A, Zijdenbos AP, Taylor J, Boelman C, Worsley KJ, Evans AC, Dagher A. Statistical mapping analysis of lesion location and neurological disability in multiple sclerosis: application to 452 patient data sets. Neuroimage. 2003;19:532–544. doi: 10.1016/S1053-8119(03)00117-4. [DOI] [PubMed] [Google Scholar]
- 3.De Groot V, Beckerman H, Uitdehaag BM, Hintzen RQ, Minneboo A, Heymans MW, Lankhorst GJ, Polman CH, Bouter LM. Physical and cognitive functioning after 3 years can be predicted using information from the diagnostic process in recently diagnosed multiple sclerosis. Arch Phys Med Rehabil. 2009;90:1478–1488. doi: 10.1016/j.apmr.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 4.Zackowski KM, Smith SA, Reich DS, Gordon-Lipkin E, Chodkowski BA, Sambandan DR, Shteyman M, Bastian AJ, van Zijl PC, Calabresi PA. Sensorimotor dysfunction in multiple sclerosis and column-specific magnetization transfer-imaging abnormalities in the spinal cord. Brain. 2009;132:1200–1209. doi: 10.1093/brain/awp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilmore CP, Donaldson I, Bö L, Owens T, Lowe J, Evangelou N. Regional variations in the extent and pattern of grey matter demyelination in multiple sclerosis: a comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord. J Neurol Neurosurg Psychiatry. 2009;80:182–187. doi: 10.1136/jnnp.2008.148767. [DOI] [PubMed] [Google Scholar]
- 6.Vercellino M, Plano F, Votta B, Mutani R, Giordana Maria T, Cavalla P. Grey matter pathology in multiple sclerosis. J Neuropathol Exp Neurol. 2005;64:1101–1107. doi: 10.1097/01.jnen.0000190067.20935.42. [DOI] [PubMed] [Google Scholar]
- 7.Huitinga I, De Groot CJA, Van der Valk P, Kamphorst W, Tilders FJH, Swaab DF. Hypothalamic lesions in multiple sclerosis. J Neuropathol Exp Neurol. 2001;60:1208–1218. doi: 10.1093/jnen/60.12.1208. [DOI] [PubMed] [Google Scholar]
- 8.Geurts JJ, Bo L, Roosendaal SD, Hazes T, Daniels R, Barkhof F, Witter MP, Huitinga I, van der Valk P. Extensive hippocampal demyelination in multiple sclerosis. J Neuropathol Exp Neurol. 2007;66:819–827. doi: 10.1097/nen.0b013e3181461f54. [DOI] [PubMed] [Google Scholar]
- 9.Papadopoulos D, Dukes S, Patel R, Nicholas R, Vora A, Reynolds R. Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol. 2009;19:238–253. doi: 10.1111/j.1750-3639.2008.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kutzelnigg A, Faber-Rod JC, Bauer J, Faber-Rod JC, Bauer J, Lucchinetti CF, Sorensen PS, Laursen H, Stadelmann C, Brück W, Rauschka H, Schmidbauer M, Lassmann H. Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol. 2007;17:38–44. doi: 10.1111/j.1750-3639.2006.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–2712. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 12.Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 13.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 14.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 15.Tran EH, Hoekstra K, Rooijen NV, Dijkstra D, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- 16.Murphy ÁC, Lalor SJ, Lynch MA, Mills KHG. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 17.O’Connor RA, Prendergast CT, Catherine A, Lau CWZ, Leech MD, David C, Anderton SM, Connor RAO, Sabatos CA, Wraith DC. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental Autoimmune Encephalomyelitis. J Immunol. 2008;181:3750–3754. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popescu BFG, Bunyan RF, Parisi JE, Ransohoff RM, Lucchinetti CF. A case of multiple sclerosis presenting with inflammatory cortical demyelination. Neurology. 2011;76:1705–1710. doi: 10.1212/WNL.0b013e31821a44f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lucchinetti CF, Popescu BFG, Bunyan RF, Moll NM, Roemer SF, Lassmann H, Brück W, Parisi JE, Scheithauer BW, Giannini C, Weigand SD, Mandrekar J, Ransohoff RM, Brück W. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188–2197. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 21.Bø L, Vedeler C, Nyland H, Trapp B, Mørk S. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler. 2003;9:323–331. doi: 10.1191/1352458503ms917oa. [DOI] [PubMed] [Google Scholar]
- 22.Brink BP, Veerhuis R, Breij ECW, van der Valk P, Dijkstra CD, Bö L. The pathology of multiple sclerosis is location-dependent: no significant complement activation is detected in purely cortical lesions. J Neuropathol Exp Neurol. 2005;64:147–155. doi: 10.1093/jnen/64.2.147. [DOI] [PubMed] [Google Scholar]
- 23.Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/S1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- 24.Conductier G, Blondeau N, Guyon A, Nahon J-L, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 25.Bose S, Cho J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch Pharm Res. 2013;36:1039–1050. doi: 10.1007/s12272-013-0161-z. [DOI] [PubMed] [Google Scholar]
- 26.Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreno M, Bannerman P, Ma J, Guo F, Miers L, Soulika AM, Pleasure D. Conditional ablation of astroglial CCL2 Suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J Neurosci. 2014;34:8175–8185. doi: 10.1523/JNEUROSCI.1137-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cross AK, Woodroofe MN. Chemokines induce migration and changes in actin polymerization in adult rat brain microglia and a human fetal microglial cell line in vitro. J Neurosci Res. 1999;55:17–23. doi: 10.1002/(SICI)1097-4547(19990101)55:1<17::AID-JNR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 30.Hinojosa AE, Garcia-Bueno B, Leza JC, Madrigal JLM. CCL2/MCP-1 modulation of microglial activation and proliferation. J Neuroinflammation. 2011;8:77. doi: 10.1186/1742-2094-8-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang G, Meng Y, Li W, Yong Y, Fan Z, Ding H, Wei Y, Luo J, Ke Z-J. Neuronal MCP-1 mediates microglia recruitment and neurodegeneration induced by the mild impairment of oxidative metabolism. Brain Pathol. 2011;21:279–297. doi: 10.1111/j.1750-3639.2010.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 34.Brini E, Ruffini F, Bergami A, Brambilla E, Dati G, Greco B, Cirillo R, Proudfoot AE, Comi G, Furlan R, Zaratin P, Martino G. Administration of a monomeric CCL2 variant to EAE mice inhibits inflammatory cell recruitment and protects from demyelination and axonal loss. J Neuroimmunol. 2009;209:33–39. doi: 10.1016/j.jneuroim.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 35.Tanuma N, Sakuma H, Sasaki A, Matsumoto Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006;112:195–204. doi: 10.1007/s00401-006-0083-7. [DOI] [PubMed] [Google Scholar]
- 36.Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84:238–249. doi: 10.1016/S0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- 37.Van Der Voorn P, Tekstra J, Beelen RH, Tensen CP, van der Valk P, De Groot CJ. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol. 1999;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dutta R, Chang A, Doud MK, Mary K, Kidd GJ, Ribaudo MV, Young EA, Fox RJ, Staugaitis SM, Trapp BD. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol. 2011;69:445–454. doi: 10.1002/ana.22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dutta R, Chomyk AM, Chang A, Ribaudo MV, Deckard SA, Doud MK, Edberg DD, Bai B, Li M, Baranzini SE, Fox RJ, Staugaitis SM, Macklin WB, Trapp BD. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann Neurol. 2013;73:637–645. doi: 10.1002/ana.23860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Todorov IT, Werness BA, Wang HQ, Buddharaju LN, Todorova PD, Slocum HK, Brooks JS, Huberman JA. HsMCM2/BM28: a novel proliferation marker for human tumors and normal tissues. Lab Invest. 1998;78:73–78. [PubMed] [Google Scholar]
- 41.Bö L, Mörk S, Kong PA, Nyland H, Pardo CA, Trapp BD. Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions. J Neuroimmunol. 1994;51:135–146. doi: 10.1016/0165-5728(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 42.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 43.van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol. 2000;26:2–10. doi: 10.1046/j.1365-2990.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 44.Ledeboer A, Breve JJP, Wierinckx A, van der Jagt S, Bristow AF, Leysen JE, Tilders FJH, Van Dam A-M. Expression and regulation of interleukin-10 and interleukin-10 receptor in rat astroglial and microglial cells. Eur J Neurosci. 2002;16:1175–1185. doi: 10.1046/j.1460-9568.2002.02200.x. [DOI] [PubMed] [Google Scholar]
- 45.Matute C. Glutamate and ATP signalling in white matter pathology. J Anat. 2011;219:53–64. doi: 10.1111/j.1469-7580.2010.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franke H, Verkhratsky A, Burnstock G, Illes P. Pathophysiology of astroglial purinergic signalling. Purinergic Signal. 2012;8:629–657. doi: 10.1007/s11302-012-9300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panenka W, Jijon H, Herx LM, Armstrong JN, Feighan D, Wei T, Yong VW, Ransohoff RM, MacVicar BA. P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci. 2001;21:7135–7142. doi: 10.1523/JNEUROSCI.21-18-07135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin N, Baribaud F, Wang K, Yang G, Wynn R, Covington MB, Feldman P, Gallagher KB, Leffet LM, Lo YY, Wang A, Xue C-B, Newton RC, Scherle PA. Pharmacological characterization of INCB3344, a small molecule antagonist of human CCR2. Biochem Biophys Res Commun. 2009;387:251–255. doi: 10.1016/j.bbrc.2009.06.135. [DOI] [PubMed] [Google Scholar]
- 49.Brodmerkel CM, Huber R, Covington M, Diamond S, Hall L, Collins R, Leffet L, Gallagher K, Feldman P, Collier P, Stow M, Gu X, Baribaud F, Shin N, Thomas B, Burn T, Hollis G, Yeleswaram S, Solomon K, Friedman S, Wang A, Xue CB, Newton RC, Scherle P, Vaddi K. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. J Immunol. 2005;175:5370–5378. doi: 10.4049/jimmunol.175.8.5370. [DOI] [PubMed] [Google Scholar]
- 50.Kooi E-J, Prins M, Bajic N, Beliën JA, Gerritsen WH, van Horssen J, Aronica E, van Dam AM, Hoozemans JJ, Francis PT, van der Valk P, Geurts JJ. Cholinergic imbalance in the multiple sclerosis hippocampus. Acta Neuropathol. 2011;122:313–322. doi: 10.1007/s00401-011-0849-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiaravalloti ND, DeLuca J. Cognitive impairment in multiple sclerosis. Lancet Neurol. 2008;7:1139–1151. doi: 10.1016/S1474-4422(08)70259-X. [DOI] [PubMed] [Google Scholar]
- 52.Rao S, Leo G, Bernardin L, Unverzagt F. Cognitive dysfunction in multiple sclerosis. I. frequency, patterns, and prediction. Neurology. 1991;41:685–691. doi: 10.1212/WNL.41.5.685. [DOI] [PubMed] [Google Scholar]
- 53.Rudick R a, Lee J-C, Nakamura K, Fisher E. Gray matter atrophy correlates with MS disability progression measured with MSFC but not EDSS. J Neurol Sci. 2009;282:106–111. doi: 10.1016/j.jns.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher E, Lee J-C, Nakamura K, Rudick R a. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol. 2008;64:255–265. doi: 10.1002/ana.21436. [DOI] [PubMed] [Google Scholar]
- 55.Fisniku LK, Chard DT, Jackson JS, Anderson VM, Altmann DR, Miszkiel KA, Thompson AJ, Miller DH. Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann Neurol. 2008;64:247–254. doi: 10.1002/ana.21423. [DOI] [PubMed] [Google Scholar]
- 56.McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998;86:20–29. doi: 10.1016/S0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- 57.Butt AM, Fern RF, Matute C. Neurotransmitter signaling in white matter. Glia. 2014;ᅟ:1–18. doi: 10.1002/glia.22674. [DOI] [PubMed] [Google Scholar]
- 58.Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J Mol Neurosci. 2012;48:66–76. doi: 10.1007/s12031-012-9773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matyash V, Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Res Rev. 2010;63:2–10. doi: 10.1016/j.brainresrev.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 60.Hewett JA. Determinants of regional and local diversity within the astroglial lineage of the normal central nervous system. J Neurochem. 2009;110:1717–1736. doi: 10.1111/j.1471-4159.2009.06288.x. [DOI] [PubMed] [Google Scholar]
- 61.Goursaud S, Kozlova EN, Maloteaux J-M, Hermans E. Cultured astrocytes derived from corpus callosum or cortical grey matter show distinct glutamate handling properties. J Neurochem. 2009;108:1442–1452. doi: 10.1111/j.1471-4159.2009.05889.x. [DOI] [PubMed] [Google Scholar]
- 62.Thompson WL, Van Eldik LJ. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes. Brain Res. 2009;1287:47–57. doi: 10.1016/j.brainres.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brosnan CF, Cannella B, Battistini L, Raine CS. Cytokine localization in multiple sclerosis lesions: correlation with adhesion molecule expression and reactive nitrogen species. Neurology. 1995;45:6–11. doi: 10.1212/WNL.45.6_Suppl_6.S16. [DOI] [PubMed] [Google Scholar]
- 64.Prins M, Eriksson C, Wierinckx A, Bol JG, Binnekade R, Tilders FJ, Van Dam AM. Interleukin-1β and Interleukin-1 receptor antagonist appear in grey matter additionally to white matter lesions during experimental multiple sclerosis. PLoS One. 2013;8:e83835. doi: 10.1371/journal.pone.0083835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Biber K, Neumann H, Inoue K, Boddeke HWGM. Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 66.Chavarría A, Cárdenas G. Neuronal influence behind the central nervous system regulation of the immune cells. Front Integr Neurosci. 2013;7:64. doi: 10.3389/fnint.2013.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tian L, Rauvala H, Gahmberg CG. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009;30:91–99. doi: 10.1016/j.it.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 68.Roberts TK, Eugenin E a, Lopez L, Romero IA, Weksler BB, Couraud PO, Berman JW. CCL2 disrupts the adherens junction: implications for neuroinflammation. Lab Invest. 2012;92:1213–1233. doi: 10.1038/labinvest.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Horssen J, Brink BP, de Vries HE, van der Valk P, Bø L. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol. 2007;66:321–328. doi: 10.1097/nen.0b013e318040b2de. [DOI] [PubMed] [Google Scholar]
- 70.Simpson J, Rezaie P, Newcombe J, Cuzner ML, Male D, Woodroofe MN. Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol. 2000;108:192–200. doi: 10.1016/S0165-5728(00)00274-5. [DOI] [PubMed] [Google Scholar]
- 71.Eltayeb S, Berg A-L, Lassmann H, Wallström E, Nilsson M, Olsson T, Ericsson-Dahlstrand A, Sunnemark D. Temporal expression and cellular origin of CC chemokine receptors CCR1, CCR2 and CCR5 in the central nervous system: insight into mechanisms of MOG-induced EAE. J Neuroinflammation. 2007;4:14. doi: 10.1186/1742-2094-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boddeke EW, Meigel I, Frentzel S, Gourmala NG, Harrison JK, Buttini M, Spleiss O, Gebicke-Härter P. Cultured rat microglia express functional beta-chemokine receptors. J Neuroimmunol. 1999;98:176–184. doi: 10.1016/S0165-5728(99)00096-X. [DOI] [PubMed] [Google Scholar]
- 73.Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001;101:249–255. doi: 10.1007/s004010000284. [DOI] [PubMed] [Google Scholar]
- 74.Gehrmann J, Banati RB, Kreutzberg GW. Microglia in the immune surveillance of the brain: human microglia constitutively express HLA-DR molecules. J Neuroimmunol. 1993;48:189–198. doi: 10.1016/0165-5728(93)90191-Z. [DOI] [PubMed] [Google Scholar]
- 75.Hart AD, Wyttenbach A, Perry VH, Teeling JL. Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav Immun. 2012;26:754–765. doi: 10.1016/j.bbi.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li T, Pang S, Yu Y, Wu X, Guo J, Zhang S. Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain. 2013;136:3578–3588. doi: 10.1093/brain/awt287. [DOI] [PubMed] [Google Scholar]
- 77.Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik V, Qin S, Rottman J, Sellebjerg F, Strieter RM, Frederiksen JL, Ransohoff RM. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103:807–815. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Furtado GC, Marcondes MC, Tsai J, Wensky A, Lafaille JJ. Swift entry of myelin-specific T Lymphocytes into the central nervous system in Spontaneous Autoimmune Encephalomyelitis. J Immunol. 2008;181:4648–4655. doi: 10.4049/jimmunol.181.7.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 80.Traugott U, Reinherz EL, Raine CS. Multiple sclerosis. distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J Neuroimmunol. 1983;4:201–221. doi: 10.1016/0165-5728(83)90036-X. [DOI] [PubMed] [Google Scholar]
- 81.Weiss H a, Millward JM, Owens T. CD8+ T cells in inflammatory demyelinating disease. J Neuroimmunol. 2007;191:79–85. doi: 10.1016/j.jneuroim.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 82.Howell OW, Reeves C a, Nicholas R, Carassiti D, Radotra B, Gentleman SM, Serafini B, Aloisi F, Roncaroli F, Magliozzi R, Reynolds R. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134:2755–2771. doi: 10.1093/brain/awr182. [DOI] [PubMed] [Google Scholar]
- 83.Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA, Nicholas R, Roncaroli F, Reynolds R. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. 2012;135:2925–2937. doi: 10.1093/brain/aws189. [DOI] [PubMed] [Google Scholar]