

Abstract
Multivesicular bodies (MVBs) are cholesterol-enriched organelles formed by the endocytic pathway. The topology of vesicle formation in MVBs is identical to that of retroviral budding from the plasma membrane, and budding of human immunodeficiency virus type 1 (HIV-1) into MVBs in macrophages has recently been visualized. The Gag proteins from HIV-1, as well as many other retroviruses, contain short motifs that mediate interactions with MVBs and other endocytic components, suggesting that Gag proteins directly interface with the endocytic pathway. Here, we show that HIV-1 Gag contains an internalization signal that promotes endocytosis of a chimeric transmembrane fusion protein. Mutation of this motif within Gag strongly inhibits virus-like particle production. Moreover, wild-type Gag, but not the internalization-defective mutation, can be induced to accumulate within CD63-positive MVBs by treatment of cells with U18666A, a drug that redistributes cholesterol from the plasma membrane to MVBs. We propose that HIV-1 Gag contains a signal that promotes interaction with the cellular endocytic machinery and that the site of particle production is regulated by the subcellular distribution of cholesterol.
Retroviral assembly occurs via an ordered process. Binding of retroviral Gag proteins to cellular membranes and multimerization of Gag proteins into a submembraneous array drives the formation of enveloped viral particles that bud out from the membrane into the extracellular space. Gag proteins from retroviruses and lentiviruses are necessary and sufficient to drive the formation of virus-like particles (VLPs) in the absence of other viral proteins (12). Particles produced in this manner do not undergo proteolytic processing and maturation, and they resemble immature noninfectious virions (12).
The late stage of virus assembly, which involves the budding and release of particles from the cell, is mediated by the L domain, a short peptide motif within Gag (13). Recently, several proteins involved in endocytic and/or lysosomal degradation pathways, including Tsg101 (11, 42), Nedd4 (16, 37), AIP1 (38), and AP-2 (34), have been shown to bind L domains. All of these proteins are involved in the sorting of cellular proteins into internalization and/or lysosomal degradation pathways. For example, the L domain of human immunodeficiency virus type 1 (HIV-1) Gag consists of a PTAP sequence that binds to Tsg101, a protein required for the recognition and sorting of ubiquitinated membrane proteins into the internal vesicles of multivesicular bodies (MVBs) (3, 6). These vesicles arise from the inward budding of the lumenal membrane of the MVB, a process that is topologically identical to the outward budding of enveloped viruses from the plasma membrane, i.e., away from the cytoplasm (31). Interaction of HIV-1 Gag with Tsg101 and other components of the MVB ESCRT complex is required for efficient particle production (11, 13). In macrophages, HIV-1 Gag is present on and buds from internal membranes that resemble MVBs (28, 32, 33, 35). However, in T cells and most permissive tissue culture cell lines, HIV-1 Gag is primarily localized to the plasma membrane, the site for virus assembly and release (7, 15, 26, 29, 30, 32). It is not known how Gag recruits MVB proteins to the plasma membrane, and it is not understood how Gag is targeted to MVBs in macrophages. We hypothesized that Gag itself may undergo endocytosis as a trafficking step between the plasma membrane and MVBs.
In order to investigate the potential interface between HIV-1 Gag and the endosomal pathway, we tested whether Gag contains endocytosis motifs. A chimeric protein was generated by fusing the extracellular and transmembrane domains of human CD4 to Gag. CD4 contains a well-defined endocytosis signal in its cytoplasmic tail (2, 36). Replacement of this tail with Gag allowed us to test for the presence of trafficking signals within Gag, particularly endocytosis signals. Here, we show that the Gag sequence acts to increase internalization of the fusion protein. The region of Gag responsible for increased internalization was mapped to the C-terminal domain of capsid (CA), the p2 spacer peptide, and nucleocapsid (NC). This region contains a number of motifs that resemble well-characterized endocytosis signals. We generated point mutations of each of these potential signals and identified a dileucine-like motif required not only for the rapid internalization of the fusion protein but also for the assembly and release of VLPs.
MATERIALS AND METHODS
Antibodies and reagents.
Rabbit anti-p24CA antiserum was used to detect Gag. Anti-CD4 monoclonal antibody (MAb) (Leu-3a) was obtained from the Monoclonal Core Facility (Memorial Sloan-Kettering Cancer Center, New York, N.Y.). Anti-EEA1 and anti-CD63 MAbs were obtained from BD Transduction Laboratories (San Diego, Calif.). Texas Red-transferrin was obtained from Molecular Probes (Eugene, Oreg.). Bovine serum albumin (BSA) fraction V was purchased from Fisher Scientific (Pittsburgh, Pa.). 1-Step Slow TMB (3,3′, 5,5′-tetramethylbenzidine) was purchased from Pierce (Rockford, Ill.). Phorbol 12-myristate 13-acetate (PMA) was obtained from Sigma (St. Louis, Mo.). U18666A was obtained from Biomol (Plymouth Meeting, Pa.).
DNA plasmids.
The Rev independent Gag expression vectors pCMV5 Gag and pGagGFP have been discussed elsewhere (15, 41). pEGFP-N2 CD4FL (expressing full-length human CD4) and pCD4T(−).GFP and pCD4T(−).UL11 (expressing fusion proteins of tailless CD4 with GFP or herpesvirus UL11 proteins, respectively) were kind gifts from John Wills (Pennsylvania State University College of Medicine, Hershey, Pa.) (20). pcDNA3 CD4T(−) expresses a truncated CD4 protein lacking an internalization signal. Other CD4 fusion constructs were generated by subcloning PCR products in frame into the BamHI and NotI sites of pCD4T(−).GFP. Point mutations within the Gag sequence were generated by PCR. pSVL dynamin-2 bb myc and pSVL K44E dynamin-2 bb myc were kind gifts of Enrique Rodriguez-Boulan (Weill Graduate School of Medical Sciences, New York, N.Y.).
Cell culture and transfections.
COS-1 cells were maintained in 10% fetal bovine serum in Dulbecco modified Eagle medium. For transfections, COS-1 cells were seeded to approximately 50% density and transfected the following day with 3 to 5 μg of plasmid DNA by using Lipofectamine 2000 (Gibco BRL Life Technologies, Carlsbad, Calif.).
Enzyme-linked immunosorbent assay-based internalization (ELISAint) assay.
One day following transfection, COS-1 cells were seeded onto 24-well trays to approximately 50% density. The following day, the trays were placed on ice, the media were removed, and the cells were washed twice with 1 ml of ice-cold phosphate-buffered saline (PBS) with 3% (wt/vol) BSA (PBS-BSA). The cells were then incubated for 1 h in 200 μl of PBS-BSA containing 0.37 μg of Leu-3a MAb/ml to label surface CD4. After three washes in PBS-BSA, Dulbecco modified Eagle medium plus 10% fetal bovine serum was added and the cells were incubated at 37°C for various lengths of time (either 20 and 60 min or 5, 10, and 20 min). At the indicated times, the trays were put back on ice and washed twice with ice-cold PBS along with a tray that had remained on ice (the 0-min time point). Cells were then fixed for 20 min in PBS containing 3.7% paraformaldehyde on ice. This step was followed by two washes with PBS-BSA. The remainder of the experiment was performed at room temperature. Cells were then incubated for 1 h in 200 μl of PBS-BSA containing horseradish peroxidase-conjugated sheep anti-mouse secondary antibody (Amersham Biosciences, Piscataway, N.J.) to label Leu-3a MAb remaining at the surface. Cells were then washed extensively with PBS. Horseradish peroxidase activity was detected by the addition of 250 μl of TMB to each well and incubation from 10 to 30 min at room temperature or 37°C followed by the addition of 1 N HCl to stop the reaction and to oxidize the detection reagent. The A450 of each sample was measured with a spectrophotometer. For each experiment, a set of cells were transfected with an arbitrary plasmid (usually pEGFP-N1 [BD Biosciences Clontech, Palo Alto, Calif.]) in order to measure nonspecific binding of Leu-3a at the initial time point (t0). The value obtained was subtracted from the other values to calculate specific binding of the antibody, giving a measure of the amount of CD4 present on the cell surface at each time point. The percentage of internalization for each time point (tn) was calculated as 1 − (amount of CD4 at tn/amount of CD4 at t0).
Analysis of VLP production.
VLPs were isolated and analyzed by anti-Gag Western blotting as previously described (18) except that media were clarified by passage through a 45-μm-pore-size filter before they were loaded onto the 20% sucrose cushion. For some experiments, VLP production was measured by the release of 35S-labeled Gag into the culture media as previously described (19).
Fluorescence microscopy.
Transfected COS-1 cells were grown on coverslips and processed 48 h postransfection (15). Cells were incubated with a 1:500 dilution of anti-EEA1 or anti-CD63 MAbs in PBS for 90 min after fixation and permeabilization or with 0.74 μg of Leu-3a anti-CD4 MAb/ml without permeabilization to label surface CD4. Following four 5-min washes in PBS, cells were incubated with or Alexa Fluor 488- or Alexa Fluor 594-conjugated goat anti-mouse antibodies (Molecular Probes) for 45 min. Cells were then washed four times for 5 min each time with PBS and mounted on microscope slides. Enhanced green fluorescent protein fluorescence was visualized directly. For some experiments, cells were pretreated for 16 h with 3 μg of U18666A/ml in culture media prior to fixation and preparation for microscopy. Laser scanning confocal microscopy was performed with a Zeiss LSM510 confocal microscope equipped with an Axiovert 100 M inverted microscope with a ×63, 1.2-numerical-aperture water immersion lens for imaging as previously described (15). Filipin staining and detection was performed according to the method of Tabas et al. (40), with the exception that excitation was achieved with a UV laser (364 nm) and emission was detected with a 385- to 470-nm band-pass filter. Texas Red-transferrin was used according to the manufacturer's specifications. Colocalization was measured by using MetaMorph software (Universal Imaging Corp., Downingtown, Pa.). Image areas were chosen, and the extent of overlap between red and green colors was calculated by the program on a pixel-by-pixel basis weighted by intensity overlap.
Visualization of internalized anti-CD4.
To visualize internalized anti-CD4, cells expressing CD4T(−).Gag and grown on coverslips were fed 0.37 μg of Leu-3a anti-CD4 MAb/ml for 2 h and fixed in paraformaldehyde. The cells were then stained with Alexa Fluor 488 (green) goat anti-mouse before permeabilization. After permeabilization to allow access of the secondary antibody to internalized Leu-3a, the cells were stained with Alexa Fluor 594 (red) goat anti-mouse.
RESULTS
Endocytosis of a CD4-Gag chimera.
In order to determine whether HIV-1 Gag contains an endocytosis signal, we designed a series of CD4-based chimeric proteins. The cytoplasmic tail of CD4 contains an internalization signal that includes essential serine and leucine residues and is activated by phorbol esters (2, 36) (Fig. 1A). This signal was removed by truncating the CD4 cytoplasmic tail [CD4T(−)], and HIV-1 Gag was fused in frame to generate CD4T(−).Gag. An ELISAint assay was developed to measure the internalization of CD4 constructs in transfected COS-1 cells (see Materials and Methods). This assay measures the loss of surface-bound anti-CD4 MAb Leu-3a to internal sites as a function of time. The efficacy of this assay was first verified by using known control proteins. Full-length CD4 was internalized more rapidly than CD4T(−) in the presence of the phorbol ester PMA (Fig. 1B). In addition, the fusion protein CD4T(−).UL11, previously shown to contain an internalization signal (20), was internalized at an enhanced rate similar to the rate of internalization of full-length CD4 in the presence of PMA (Fig. 1B).
FIG. 1.

Internalization of a CD4-Gag fusion protein. (A) A schematic of CD4, tailless CD4 [CD4T(−)], and CD4 fusion [CD4T(−).ORF] proteins. The amino acid sequence of the cytoplasmic tail domain (CT) is shown with the dileucine internalization motif underlined and italicized. EC, extracellular domain; TM, transmembrane; ORF, any sequence appended to CD4T(−). (B) Percentage of surface-bound anti-CD4 internalized over the indicated time course by cells expressing CD4 (filled squares), CD4T(−) (filled diamonds), CD4T(−).UL11 (open circles), or CD4T(−).GFP (open triangles). Data are means ± standard errors of the means for quadruplicate experiments repeated three to five times. For CD4 and CD4T(−), the cells were treated with 50 ng of PMA/ml. (C) Internalization of CD4T(−).Gag (open squares) compared to that of CD4T(−) (filled triangles). Data represent means ± standard errors of the means for five to seven quadruplicate experiments.
Use of the ELISAint assay revealed that CD4T(−).Gag was internalized at a markedly increased rate relative to that for CD4T(−) (Fig. 1C). To ensure that loss of the CD4 signal was due to internalization rather than loss of surface CD4T(−).Gag by incorporation into VLPs, VLPs were purified from the culture media of cells expressing either Gag or the CD4T(−).Gag chimera. Only cells expressing Gag released VLPs into the media at any detectable level over a period of 24 h (Fig. 2A). Increased loss of surface CD4T(−).Gag was not likely due to an altered recycling rate for the chimeric protein, because rapid loss of this protein from the surface was observed at very early time points (within 5 to 10 min [see Fig. 4B]) before recycling back to the cell surface came into play. Monensin, an inhibitor of recycling, also had no effect on the observed rate (not shown). Internalization of CD4T(−).Gag was then directly visualized by selective labeling of surface and internal CD4 epitopes with different fluorophores (see Materials and Methods). Under these conditions, a distinct accumulation of internalized antibody (red) was detected in the perinuclear region, readily distinguishable from the (green) staining of surface-bound antibody (Fig. 2B). Taken together, these data strongly suggest that the CD4-Gag chimera is rapidly endocytosed.
FIG. 2.

(A) Comparison of VLP production by Gag and that by CD4T(−).Gag. Lysates from cells expressing Gag or CD4T(−).Gag were collected, and VLPs were purified from the tissue culture media after 24 h of incubation. Aliquots of each lysate were assayed for the presence of Gag protein by Western blotting with anti-p24CA. The wild-type Gag band is labeled Pr55 Gag. CD4T(−).Gag migrates at ∼110 kDa. The asterisk indicates an anti-p24CA reactive band that is occasionally seen in CD4T(−).Gag cell lysates. This is probably a degradation product of the fusion protein. (B) Internalization of anti-CD4 MAb by COS-1 cells expressing CD4T(−).Gag. After the uptake of anti-CD4, cells were stained with a green fluorophore prior to permeabilization to detect surface MAbs as described in Materials and Methods. After permeabilization, internalized MAbs were stained with a red fluorophore.
FIG. 4.

Surface expression and internalization of CD4 fusion proteins. (A) CD4 fusion proteins on the surfaces of cells were detected by indirect immunofluorescence without permeabilization. (B) Internalization of CD4T(−).DsRed fusion proteins. Filled squares, CD4; filled diamonds, CD4T(−); open squares, CD4T(−).Gag; open triangles, CD4T(−).GFP; open circles, CD4T(−).DsRed (solid lines) and CD4T(−).dbl DsRed (dashed lines). Data are means of the results for two to four quadruplicate experiments.
Identification of an endocytosis signal within Gag.
We next generated chimeras containing selected portions of the Gag sequence in order to map the internalization signal(s). As depicted in Fig. 3A and B, CD4-Gag constructs fell into two clearly distinguishable classes, consisting of those having either fast or slow internalization rates. The minimal domain supporting the increased rate of internalization comprised the C-terminal domain of CA, the p2 spacer peptide, and NC (Gag residues 279 to 432) (Fig. 3C). The same region of the Gag protein, in conjunction with a myristoylation site and a late domain, is the minimal portion of Gag required to form particles (1) and thus can be considered the core assembly domain. This fact indicates a possible relationship between assembly and/or multimerization of the chimera and internalization.
FIG. 3.

Mapping of endocytosis signals within Gag. (A) Internalization of anti-CD4 by cells expressing selected CD4T(−) fusion proteins. Data are means for two to seven quadruplicate experiments. (B) Schematic representation of the set of CD4T(−) fusion proteins used to map internalization determinants within the Gag sequence. The locations of the matrix (MA), CA, (NC, and p6 domains are indicated by shaded boxes. Gag residue numbers for the beginning and end of each fusion construct are shown. A fast rate of internalization is relative to that for CD4T(−). (C) The amino acid sequence of the minimal internalization sequence of Gag, which includes the C-terminal portion of CA, p2, and NC (residues 279 to 432 of Gag). Potential endocytosis signals are underlined and italicized. Residues that have been mutated in this study are indicated by asterisks. (D) Internalization CD4T(−).Gag point mutations. Filled diamonds, CD4T(−); open squares, CD4T(−).Gag; filled triangles, CD4T(−).Gag(Y301K); open triangles, CD4T(−).Gag(LL321,322AA); gray triangles, CD4T(−).Gag(IL-333,334AA). Data represent means ± standard errors of the means for two to seven quadruplicate experiments.
To test the potential connection between multimerization and internalization, we generated chimeric proteins in which tailless CD4 was fused to DsRed, a protein known to form tetramers (4, 44). We detected the surface distribution of CD4 and CD4 fusion proteins by indirect immunofluorescence and confocal microscopy. As shown in Fig. 4A, both wild-type CD4 and the CD4-GFP fusion protein were present in a diffuse pattern on the plasma membrane and on plasma membrane ruffles. CD4T(−).DsRed and CD4T(−).dbl DsRed, a protein containing two tandem DsRed cassettes fused to tailless CD4, formed large aggregates on the plasma membrane, most likely due to multimerization. Similarly, CD4-Gag was present in discrete puncta on the surfaces of cells. However, although CD4-Gag was rapidly internalized, neither of the DsRed fusion proteins exhibited an increased rate of internalization compared to control proteins (Fig. 4B). These data suggest that multimerization alone is insufficient to drive rapid internalization.
An alternative hypothesis is that a specific internalization signal is present within residues 279 to 432 of Gag. These signals are generally tyrosine based (NPXY or YXXφ, where φ is a bulky hydrophobic residue) or leucine based (D/EXXLL or D/EXXXLL; leucines are occasionally replaced with methionine or isoleucine, and acidic residues are occasionally replaced or in conjunction with serine or threonine) (8). Three such sequences are present in the minimal domain (Fig. 3C). Point mutations disrupting each of these potential signals were generated within CD4T(−).Gag, and the internalization rates of these mutations were measured. Mutation of either LL321,322 or IL333,334 within Gag inhibited internalization of the chimera, whereas mutation of Y301 had no effect (Fig. 3D). Thus, potential internalization signals may be present in the dileucine-like sequences. These residues and the acidic and/or hydrophilic residues upstream of them are highly conserved in all clades of HIV-1 as well as in primate lentiviruses.
We next proceeded to determine whether mutations of the LL, IL, and YXXφ motifs affected the ability of Gag to form VLPs (Fig. 5A). The three mutations described above (Y301K, LL321,322AA, and IL-333,334AA) were generated in the context of full-length Gag protein, and the mutated proteins were expressed in COS-1 cells. The Y301K mutation had little or no effect on VLP production compared to that in wild-type Gag. In contrast, disruption of either LL321,322 or IL-333,334 almost completely abolished VLP production. We then tested whether either of the defective mutants could be rescued by coexpression with wild-type Gag protein. Such rescue experiments have been performed successfully with membrane-binding or late-domain-deficient mutants and indicate whether the mutants are still capable of assembly, as opposed to membrane binding or particle release (21, 24). The mutants were expressed in conjunction with wild-type GagGFP, a fusion construct which forms VLPs at an efficiency equivalent to that of wild-type Gag. GagGFP migrates at ∼80 kDa and is readily distinguishable from the 55-kDa Gag mutations on Western blots. The mutant Gag proteins produced increased levels of VLPs when coexpressed with GagGFP (Fig. 5B) but produced few or no VLPs when coexpressed with a GFP control protein. The extent of rescue was similar to that for the nonmyristoylated G2A Gag mutant, which did not produce VLPs when coexpressed with GFP but could be isolated in VLPs at 5 to 10% of the efficiency of wild-type GagGFP when these two proteins were coexpressed (Fig. 5). These results indicate that the mutant proteins are still capable of interacting with wild-type Gag, which complements their assembly defect(s).
FIG. 5.

VLP production of wild-type Gag and Gag with Y301K, LL321,322AA, and IL-333,334AA mutations. (A) Cell lysates and purified VLPs from cells expressing wild-type Gag or the indicated mutations were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting with anti-Gag antibodies. (B) Partial rescue of defective Gag mutations by a wild-type Gag protein. Western blots are of a VLP assay of cells coexpressing Gag point mutations and GagGFP (+) or GFP as a negative control (−). Mutated Gag proteins are 55 kDa (lower arrows). GagGFP is ∼80 kDa (upper arrows). Bands indicated by asterisks are apparently degradation products of GagGFP. (C) Quantification of VLP release. Open bars represent VLP release (ratio of Gag signal from VLP fraction to signal from cell lysate) from cells coexpressing Gag mutations and GFP. Closed bars represent VLP release from cells coexpressing Gag mutants and wild-type GagGFP. Each release value is a percentage of the VLP release of GagGFP. Data are means ± standard errors of the means for two to four duplicate experiments.
To further characterize the assembly defects of the mutant Gag proteins, GagGFP constructs containing the LL321,322 or IL-333,334 mutations were designed. Unlike GagGFP, which is present in punctate structures that represent plasma membrane assembly sites, Gag(LL321,322AA)-GFP appeared to be distributed within the cytosol (Fig. 6A). Membrane flotation assays confirmed that, unlike wild-type Gag, this mutated protein was excluded from the floating membrane fraction (Table 1). Mutation of L321 to alanine has recently been shown to abolish CA dimerization in vitro (9). Thus, the LL321,322AA mutation appears to inhibit Gag-Gag interactions in the context of the full-length Gag precursor protein and thereby inhibit stable membrane binding. In contrast, Gag(IL-333,334AA)-GFP exhibited a punctate plasma membrane distribution similar to that of wild-type GagGFP (Fig. 6A) and fractionated primarily with the floating membrane fraction on sucrose gradients (Table 1). The surface distributions of the CD4-Gag fusion proteins bearing these mutations showed similar phenotypes. CD4T(−).Gag LL321,322AA displayed a diffuse plasma membrane pattern, suggesting a loss of Gag-Gag interactions, whereas CD4T(−).Gag IL-333,334AA was largely punctate like the wild-type Gag fusion protein (Fig. 4A).
FIG. 6.

Distribution of GagGFP and early endosomal markers. (A) GFP fluorescence of cells expressing wild-type GagGFP or the indicated mutations. (B) GagGFP fluorescence is depicted in green (left), endosomal markers are depicted in red (middle), and the overlay of the two images is depicted on the right. Colocalization is visible as yellow. EEA1 was detected by indirect immunofluorescence with an anti-EEA1 MAb. The lower panels represent cells that were incubated with Texas Red-transferrin for 1 h. This treatment labels early endosomes and the endosomal recycling compartment.
TABLE 1.
Membrane binding of Gag and Gag point mutants
| Protein | % Membrane bound (mean ± SEM) | na |
|---|---|---|
| Gag | 75 ± 1 | 4 |
| Gag LL321,322AA | 10 ± 5 | 4 |
| Gag IL333,334AA | 77 ± 8 | 2 |
n, number of experiments.
Colocalization of Gag with endosomal markers.
If wild-type Gag contains a functional internalization signal, one would expect to find a Gag population present on endosomal membranes. There was minimal overlap between wild-type GagGFP and either EEA1 or transferrin, markers of early endosomal compartments (Fig. 6B).
If Gag enters the endocytic pathway by internalization from the plasma membrane, it should be possible to alter the steady-state distribution of Gag by altering the function or composition of specific components of the endocytic system. Coexpression of Gag with dominant-negative (K44E) dynamin had no apparent effect on Gag localization or VLP production (not shown), suggesting that Gag does not internalize via clathrin-coated pits or other dynamin-dependent structures. In addition, treatment of cells with 50 mM NH4Cl, an inhibitor of endosomal acidification, had no effect on VLP production (data not shown). We therefore employed another approach to altering the endocytic pathway by altering the distribution of cholesterol within endocytic compartments. There is ample evidence to support a role for membrane cholesterol in HIV-1 assembly. Most of the cholesterol is concentrated in the plasma membrane. The remainder is asymmetrically distributed in endosomes (14), with particularly high concentrations of cholesterol in MVBs (23). Treatment of cells with the drug U18666A causes marked redistribution of cholesterol from the plasma membrane to the MVBs (22) and results in late endosome/MVB sorting defects (17). When COS-1 cells were treated with U18666A, a striking accumulation of Gag in CD63-positive enlarged MVBs was observed (Fig. 7A and B). In contrast, the Gag(IL-333,334AA)-GFP mutation did not undergo redistribution in response to U18666A, indicating that a functional internalization signal is required for Gag to associate with MVBs. The fluorescent staining pattern of filipin, which detects cholesterol, was shifted to a perinuclear compartment in U18666A-treated cells (Fig. 7C), thereby confirming that the drug induced cholesterol redistribution. However, treatment of cells with U18666A had no apparent effect on the amount of VLP production by wild-type Gag (Fig. 8A and B). Since most of the Gag in U18666A-treated cells is in MVBs and since virus particles are still being produced, these results suggest that the budding of particles into MVBs can occur in COS-1 cells.
FIG.7.

The presence of Gag on CD63-positive late endosomes in cells treated with U18666A. (A) GFP fluorescence of cells expressing GagGFP or Gag(IL-333,334AA)-GFP is depicted in the left panels, anti-CD63 immunofluorescence is depicted in the middle panels, and the overlay of red and green fluorescence is depicted in the right panels with colocalization appearing as yellow. The indicated images are from cells treated with U18666A. (B) Colocalization of GagGFP or Gag(IL-333,334AA)-GFP in control cells (open bars) or U18666A-treated cells (filled bars) as determined by analysis of confocal images of 6 to 18 cells for each condition. Means and standard deviations are depicted. (C) Filipin fluorescence of control and U18666A-treated cells detected by confocal microscopy.
FIG. 8.
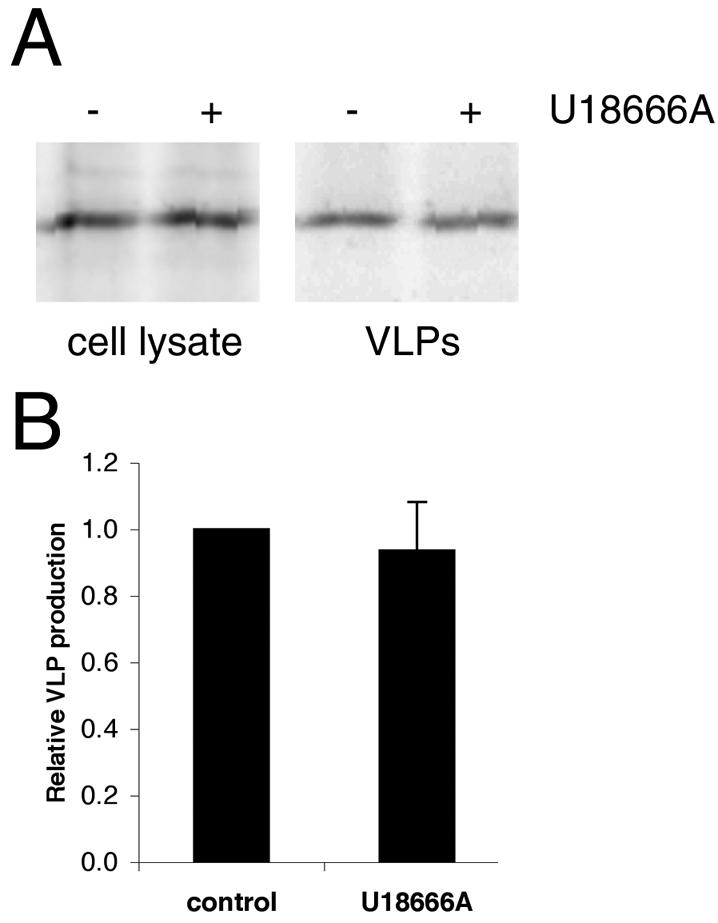
VLP production in cells treated with U18666A. (A) Gag was immunoprecipitated from 35S-labeled cells treated or untreated with U18666A and from VLPs collected after 4 h of labeling. (B) Quantification of results shown in panel A. Data are means ± standard errors of the means for two duplicate experiments.
DISCUSSION
In this study, we used CD4-Gag chimeras to demonstrate that HIV-1 Gag contains a dileucine-like endocytosis signal that promotes rapid internalization. There is now abundant evidence in the literature to indicate that retroviral Gag proteins interact with proteins involved in various stages of endocytosis (16, 38, 42). For example, HIV-1 Gag binds to Tsg101, RSV Gag binds to Nedd4, and EIAV as well as HIV-1 Gag proteins bind to AIP1/ALIX. Moreover, Gag proteins have been shown to localize to and/or bud from endocytic compartments. Moloney murine leukemia virus Gag is present in lysosomes and recycling endosomes, and HIV-1 is localized to and buds from MVBs in macrophages (5, 33, 35). The mechanism whereby Gag becomes localized to endocytic compartments is not known. One possibility is that Gag may first bind to the plasma membrane and then localize to endocytic compartments as a consequence of internalization. Our finding that a mutated Gag protein that lacks a functional endocytosis signal, Gag(IL-333,334AA), does not localize to MVBs supports the latter hypothesis. We propose that the internalization of Gag from the plasma membrane via an endocytic pathway could account for the presence of Gag in endocytic compartments and could facilitate the interactions of Gag proteins with endocytic proteins. Alternatively, newly synthesized Gag may first bind directly to MVBs and then later traffic to the plasma membrane, as has recently been proposed (43). In this case, the IL motif would be a signal for MVB targeting. It is also possible that the effect of the IL mutation on VLP production is distinct from its effect on Gag trafficking.
The region of Gag that promoted rapid internalization of a CD4-Gag chimera contained the C-terminal domain of CA, p2, and NC. This same region also promotes the multimerization of Gag. Experiments performed with CD4T(−) fused to DsRed, a protein with a well-documented ability to form tetramers (4, 44), suggest that multimerization alone is not sufficient to drive rapid internalization of CD4 chimeras. As depicted in Fig. 4A, despite the presence of large aggregates of CD4T(−)DsRed constructs on the cell surface, increased rates of internalization for these chimeras were not observed. It does, however, remain possible that the CD4-DsRed constructs are not themselves multimerized but, rather, are associated with large cellular aggregates. Thus, the possibility that, within the context of Gag, multimerization may play a role in internalization remains.
Gag containing the LL321,322AA mutation was defective for membrane binding, which may in part be due to decreased multimerization. In contrast, membrane flotation analysis as well as confocal imaging indicated that the IL-333,334AA mutant was clearly plasma membrane bound, in the contexts both of Gag itself and a CD4T(−)Gag chimera. Several lines of evidence support the notion that IL-333,334AA Gag is also capable of forming Gag-Gag multimers. This construct could be rescued into VLPs by coexpression with wild-type GagGFP. The extent of rescue was as great as or greater than that observed for the rescue of nonmyristoylated G2A Gag into VLPs by wild-type GagGFP (Fig. 5). Moreover, the localization patterns of IL-333,334AA Gag as well as the CD4T(−)IL-333,334AA Gag chimera were clearly punctate, consistent with the presence of multimerized Gag (19). It remains possible that the IL-333,334AA mutant has a subtle effect on multimerization that cannot be detected by these individual assays. Alternatively, the defect in VLP production exhibited by IL-333,334AA Gag could be due to defective association with components of the endocytic pathway.
There are at least three well-described mechanisms for internalizing proteins and lipids from the plasma membrane, including endocytosis via clathrin-coated pits, caveolae, and rafts. Our finding that coexpression of Gag with dominant-negative dynamin had no effect on Gag localization or VLP production implies that Gag internalization does not proceed via clathrin-coated pits or caveolae. Disruption of lipid rafts by treatment of cells with cyclodextrins is known to inhibit viral particle production (27). However, rafts are required for the HIV-1 assembly process (19, 27), and it is not known whether the cyclodextrin effect is due to the inhibition of raft- and/or non-raft-mediated endocytosis (39), inhibition of Gag-mediated assembly, or both. It is interesting that multiple new pathways for internalization, including dynamin-independent pathways, have been described recently (for example, see reference 25).
One of the unanswered questions in the field of virus assembly is how the location for particle production is chosen. It is well established that, in many cell types, HIV-1 Gag is primarily localized at the plasma membrane. However, recent evidence clearly implicates the MVB as a site for Gag localization and particle production in macrophages. Our data (Fig. 8) strongly suggest that Gag distribution is regulated by the intracellular distribution of cholesterol. Treatment of cells with U18666A had a striking effect on Gag localization, causing a dramatic accumulation of Gag in CD63-positive MVBs. Particle production occurred at levels equivalent to those for untreated cells. These results imply that a COS-1 cell can be induced to behave like a macrophage with respect to virus assembly by redistributing cholesterol from the plasma membrane to the MVB.
Three HIV-1 proteins have been shown to interface with the cellular endocytic machinery: Env, Nef, and Vpu. Env has a tyrosine-based internalization motif (10). In cells that express Env alone, Env internalization is rapid, whereas in HIV-1-infected cells, Env internalization is very slow. The presence of Gag is both necessary and sufficient for the inhibition of Env internalization (10). It is possible that interaction of Gag with endocytic sorting factors reduces the availability of these components for binding to other proteins, resulting in a reduced rate of Env internalization.
In conclusion, the data presented in this paper strongly suggest that HIV-1 Gag contains a dileucine-like signal that mediates the trafficking of Gag from the plasma membrane to the MVB. This signal is independent of the L domain, which has been shown to interact with endosomal proteins. The steady-state distribution of Gag in COS-1 cells is primarily at the plasma membrane, implying that either the rate of Gag endocytosis is slow or the rates of trafficking out of MVBs (recycling to the plasma membrane or entry into lysosomes) are rapid. The situation is different for macrophages, in which Gag is found in MVBs. We propose that the steady-state subcellular localization of Gag, and therefore the site of VLP formation, can be regulated by the relative distribution of cellular cholesterol in a particular cell type. Moreover, these results imply that the internalization of Gag is functionally important, as mutation of the endocytosis signal blocks the ability of Gag to target MVBs and also inhibits VLP production.
Acknowledgments
We thank Mira Perlman and Rajeshwari Valiathan for critical reading of the manuscript, Mira Perlman for assistance with the VLP assays, Raisa Louft-Nisenbaum for expert technical assistance, and Debra Alston for manuscript preparation. We also thank John Wills for the donation of plasmids expressing CD4, CD4T(−).GFP, and CD4T(−).UL11.
This work was supported by NIH grant CA72309. O.W.L. is a Dorris J. Hutchison Graduate Fellow.
REFERENCES
- 1.Accola, M. A., B. Strack, and H. G. Gottlinger. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 74:5395-5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiken, C., J. Konner, N. R. Landau, M. E. Lenburg, and D. Trono. 1994. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76:853-864. [DOI] [PubMed] [Google Scholar]
- 3.Babst, M., G. Odorizzi, E. J. Estepa, and S. D. Emr. 2000. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic 1:248-258. [DOI] [PubMed] [Google Scholar]
- 4.Baird, G. S., D. A. Zacharias, and R. Y. Tsien. 2000. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 97:11984-11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basyuk, E., T. Galli, M. Mougel, J. M. Blanchard, M. Sitbon, and E. Bertrand. 2003. Retroviral genomic RNAs are transported to the plasma membrane by endosomal vesicles. Dev. Cell 5:161-174. [DOI] [PubMed] [Google Scholar]
- 6.Bishop, N., A. Horman, and P. Woodman. 2002. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 157:91-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blom, J., C. Nielsen, and J. M. Rhodes. 1993. An ultrastructural study of HIV-infected human dendritic cells and monocytes/macrophages. APMIS 101:672-680. [DOI] [PubMed] [Google Scholar]
- 8.Bonifacino, J. S., and L. M. Traub. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395-447. [DOI] [PubMed] [Google Scholar]
- 9.del Alamo, M., J. L. Neira, and M. G. Mateu. 2003. Thermodynamic dissection of a low affinity protein-protein interface involved in human immunodeficiency virus assembly. J. Biol. Chem. 278:27923-27929. [DOI] [PubMed] [Google Scholar]
- 10.Egan, M. A., L. M. Carruth, J. F. Rowell, X. Yu, and R. F. Siliciano. 1996. Human immunodeficiency virus type 1 envelope protein endocytosis mediated by a highly conserved intrinsic internalization signal in the cytoplasmic domain of gp41 is suppressed in the presence of the Pr55gag precursor protein. J. Virol. 70:6547-6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrus, J. E., U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka, and W. I. Sundquist. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55-65. [DOI] [PubMed] [Google Scholar]
- 12.Gheysen, D., E. Jacobs, F. de Foresta, C. Thiriart, M. Francotte, D. Thines, and M. De Wilde. 1989. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 59:103-112. [DOI] [PubMed] [Google Scholar]
- 13.Gottlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 88:3195-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao, M., S. X. Lin, O. J. Karylowski, D. Wustner, T. E. McGraw, and F. R. Maxfield. 2002. Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J. Biol. Chem. 277:609-617. [DOI] [PubMed] [Google Scholar]
- 15.Hermida-Matsumoto, L., and M. D. Resh. 2000. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 74:8670-8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kikonyogo, A., F. Bouamr, M. L. Vana, Y. Xiang, A. Aiyar, C. Carter, and J. Leis. 2001. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA 98:11199-11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi, T., M. H. Beuchat, M. Lindsay, S. Frias, R. D. Palmiter, H. Sakuraba, R. G. Parton, and J. Gruenberg. 1999. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1:113-118. [DOI] [PubMed] [Google Scholar]
- 18.Lindwasser, O. W., and M. D. Resh. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75:7913-7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindwasser, O. W., and M. D. Resh. 2002. Myristoylation as a target for inhibiting HIV assembly: unsaturated fatty acids block viral budding. Proc. Natl. Acad. Sci. USA 99:13037-13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loomis, J. S., J. B. Bowzard, R. J. Courtney, and J. W. Wills. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209-12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313-1319. [DOI] [PubMed] [Google Scholar]
- 22.Mayran, N., R. G. Parton, and J. Gruenberg. 2003. Annexin II regulates multivesicular endosome biogenesis in the degradation pathway of animal cells. EMBO J. 22:3242-3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mobius, W., E. van Donselaar, Y. Ohno-Iwashita, Y. Shimada, H. F. Heijnen, J. W. Slot, and H. J. Geuze. 2003. Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 4:222-231. [DOI] [PubMed] [Google Scholar]
- 24.Morikawa, Y., S. Hinata, H. Tomoda, T. Goto, M. Nakai, C. Aizawa, H. Tanaka, and S. Omura. 1996. Complete inhibition of human immunodeficiency virus Gag myristoylation is necessary for inhibition of particle budding. J. Biol. Chem. 271:2868-2873. [DOI] [PubMed] [Google Scholar]
- 25.Naslavsky, N., R. Weigert, and J. G. Donaldson. 2003. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14:417-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nermut, M. V., W. H. Zhang, G. Francis, F. Ciampor, Y. Morikawa, and I. M. Jones. 2003. Time course of Gag protein assembly in HIV-1-infected cells: a study by immunoelectron microscopy. Virology 305:219-227. [DOI] [PubMed] [Google Scholar]
- 27.Ono, A., and E. O. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 98:13925-13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orenstein, J. M., M. S. Meltzer, T. Phipps, and H. E. Gendelman. 1988. Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: an ultrastructural study. J. Virol. 62:2578-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Owens, R. J., J. W. Dubay, E. Hunter, and R. W. Compans. 1991. Human immunodeficiency virus envelope protein determines the site of virus release in polarized epithelial cells. Proc. Natl. Acad. Sci. USA 88:3987-3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer, E., and C. S. Goldsmith. 1988. Ultrastructure of human retroviruses. J. Electron Microsc. Tech. 8:3-15. [DOI] [PubMed] [Google Scholar]
- 31.Patnaik, A., V. Chau, and J. W. Wills. 2000. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 97:13069-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pautrat, G., M. Suzan, D. Salaun, P. Corbeau, C. Allasia, G. Morel, and P. Filippi. 1990. Human immunodeficiency virus type 1 infection of U937 cells promotes cell differentiation and a new pathway of viral assembly. Virology 179:749-758. [DOI] [PubMed] [Google Scholar]
- 33.Pelchen-Matthews, A., B. Kramer, and M. Marsh. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puffer, B. A., S. C. Watkins, and R. C. Montelaro. 1998. Equine infectious anemia virus Gag polyprotein late domain specifically recruits cellular AP-2 adapter protein complexes during virion assembly. J. Virol. 72:10218-10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raposo, G., M. Moore, D. Innes, R. Leijendekker, A. Leigh-Brown, P. Benaroch, and H. Geuze. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic 3:718-729. [DOI] [PubMed] [Google Scholar]
- 36.Shin, J., C. Doyle, Z. Yang, D. Kappes, and J. L. Strominger. 1990. Structural features of the cytoplasmic region of CD4 required for internalization. EMBO J. 9:425-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strack, B., A. Calistri, M. A. Accola, G. Palu, and H. G. Gottlinger. 2000. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 97:13063-13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strack, B., A. Calistri, S. Craig, E. Popova, and H. G. Gottlinger. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689-699. [DOI] [PubMed] [Google Scholar]
- 39.Subtil, A., I. Gaidarov, K. Kobylarz, M. A. Lampson, J. H. Keen, and T. E. McGraw. 1999. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. USA 96:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabas, I., X. Zha, N. Beatini, J. N. Myers, and F. R. Maxfield. 1994. The actin cytoskeleton is important for the stimulation of cholesterol esterification by atherogenic lipoproteins in macrophages. J. Biol. Chem. 269:22547-22556. [PubMed] [Google Scholar]
- 41.Tritel, M., and M. D. Resh. 2000. Kinetic analysis of human immunodeficiency virus type 1 assembly reveals the presence of sequential intermediates. J. Virol. 74:5845-5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.VerPlank, L., F. Bouamr, T. J. LaGrassa, B. Agresta, A. Kikonyogo, J. Leis, and C. A. Carter. 2001. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 98:7724-7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Schwedler, U. K., M. Stuchell, B. Muller, D. M. Ward, H. Y. Chung, E. Morita, H. E. Wang, T. Davis, G. P. He, D. M. Cimbora, A. Scott, H. G. Krausslich, J. Kaplan, S. G. Morham, and W. I. Sundquist. 2003. The protein network of HIV budding. Cell 114:701-713. [DOI] [PubMed] [Google Scholar]
- 44.Wall, M. A., M. Socolich, and R. Ranganathan. 2000. The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat. Struct. Biol. 7:1133-1138. [DOI] [PubMed] [Google Scholar]