

Abstract
The diketo acids are potent inhibitors of human immunodeficiency virus (HIV) integrase (IN). Mutations in IN, T66I, S153Y, and M154I, as well as T66I-S153Y and T66I-M154I double mutations, confer resistance to diketo acids (D. J. Hazuda et al., Science 287:646-650, 2000). The effects of these IN mutations on viral replication, enzymatic activity, and susceptibility to other HIV inhibitors are reported herein. By immunofluorescence assay and real-time PCR, all mutant viruses demonstrated a modest delay in viral spread compared to that of reference HIV. These viruses also showed a statistically significant defect in integration without defects in reverse transcription. Recombinant IN containing S153Y, T66I, and M154I-T66I mutations had an approximately twofold decrease in both disintegration and 3′-end-processing-strand transfer activities in vitro. In contrast, IN containing M154I demonstrated a greater than twofold increase in specific activity in both reactions. All mutant HIVs were resistant to l-chicoric acid, a dicaffeoyltartaric acid IN inhibitor, both in tissue culture and in biochemical assays, yet remained susceptible to the reverse transcriptase inhibitors zidovudine and nevirapine. Thus, IN mutations conferring resistance to the diketo acids can yield integration defects, attenuated catalysis in vitro, and cross-resistance to l-chicoric acid.
After reverse transcription of the viral RNA to a double-stranded cDNA, the human immunodeficiency virus (HIV) integrase (IN) catalyzes the covalent incorporation of the cDNA into the host chromosome (reviewed in reference 13). Integration occurs in several steps: first, IN catalyzes 3′-end processing, in which the terminal 2 nucleotides (nt) from each 3′ end of the viral long terminal repeats (LTRs) are removed, exposing a CpA dinucleotide, which is absolutely conserved (19, 25). Next, IN catalyzes strand transfer, a concerted cleavage-ligation reaction in which the free hydroxyl at each 3′ end of the LTR undergoes nucleophilic attack on both strands of the host chromosome. Finally, the resulting gaps in the DNA are repaired, a step likely mediated by host DNA double-stranded break repair enzymes (16, 17, 68). This yields the fully integrated provirus flanked by 5-bp direct repeats (63, 64).
The IN protein of HIV is composed of 288 amino acids and is divided into three separate functional domains (20). The catalytic core domain spans amino acids 50 to 212 and contains the catalytic triad Asp 64, Asp 116, and Glu 152 (21). This triad coordinates a divalent metal ion; either magnesium or manganese is required for catalytic activity and is absolutely conserved among INs from retroviruses and retrotransposons (38). The N-terminal domain is believed to be involved in protein multimerization (40) and stabilization of protein folding and contains a His-His-Cys-Cys zinc finger-like motif, which coordinates zinc (6, 7). The C-terminal domain of IN has nonspecific DNA binding activity; thus, it is thought to play a role in binding to host DNA (23).
IN is the only viral protein necessary to carry out the integration reaction (15, 33, 54). Moreover, the enzymatic activity of recombinant IN can be studied in vitro using oligonucleotide substrates that mimic the viral LTR ends, IN, and a divalent metal cation (Mg2+ or Mn2+) (10, 54). In addition to the 3′-end-processing and strand transfer reactions, recombinant IN can also catalyze the reversal of the integration reaction, termed disintegration. In disintegration, IN is able to correctly resolve into its respective parts an oligonucleotide resembling viral DNA joined to host DNA (12). While the full-length IN protein is required for the 3′-end processing and strand transfer reactions (60), the catalytic core domain of IN is sufficient for disintegration (7). Whether the disintegration reaction occurs in vivo is unknown.
Integration is absolutely required for stable and productive infection by HIV (39). Furthermore, there is no mammalian homologue for IN (41), and the gene is highly conserved among HIV type 1 (HIV-1) clinical isolates (9). Thus, it is an attractive target for anti-HIV therapeutics. Though several IN inhibitors have been described to date (reviewed in references 46 and 50), only two IN inhibitors are in clinical trials, one of which is a derivative of the diketo acid family of molecules. The diketo acids are selective inhibitors of the strand transfer reaction (30). The most potent of the originally described diketo acids, l-731,988, is reported to inhibit HIV replication by 50% (the 50% effective concentration [EC50]) at 1 to 2 μM in tissue culture (30). It also inhibits strand transfer, with a 50% inhibitory concentration (IC50) of 0.1 μM in the presence of recombinant IN, but has little to no effect on 3′-end processing or disintegration (IC50 = 6 or 20 μM, respectively) (30). Furthermore, several clinical isolates of HIV-1 with up to 15% amino acid divergence from reference IN were susceptible to l-731,988 (47). In order to confirm the site of action of the diketo acids, inhibitor-resistant HIVs were raised to two compounds, l-731,988 and l-708,906, and resistance was mapped to mutations within the IN gene (30). Several mutations were reported, T66I, S153Y, and M154I, as well as double mutations T66I-M154I and S153Y-T66I. Although their resistance phenotype has been described, the effect of these mutations on protein activity and viral replication has not been extensively studied.
l-Chicoric acid (l-CA) is a dicaffeoyltartaric acid, which inhibits HIV replication in tissue culture (EC50 = 0.2 to 1 μM) and integration in vitro (IC50 = 200 to 400 nM [53]). A glycine-140-to-serine point mutation within HIV IN confers complete resistance to l-CA in tissue culture (37), and recombinant IN containing this mutation is resistant in vitro (35). This point mutation also confers cross-resistance to l-731,988, both in tissue culture and in vitro (35). Computer modeling of l-CA into the crystal structure of the catalytic core of IN has identified a putative binding pocket involving amino acids D64, C65, T66, H67, E92, D116, N117, Q148, E152, K156, and K159 (53, 56). Based on the location of resistance mutations for the diketo acids, as well as cross-resistance data for IN containing the G140S mutation, it is hypothesized that the diketo acids and l-CA may bind to a similar site on IN.
To test this hypothesis, the replication kinetics of HIV containing single and double mutations conferring diketo acid resistance were determined. Furthermore, the site of any replication defects was identified using SYBR Green-I real-time PCR, which is a highly quantitative measure of HIV replication and can identify the site of a replication defect (35, 61). IN proteins containing each mutation were also evaluated for enzymatic activity in the disintegration and 3′-end-processing-strand transfer reactions. Each mutant HIV and protein were evaluated for cross-resistance to l-CA. Finally, the susceptibilities of each mutant HIV to the reverse transcriptase (RT) inhibitors zidovudine (ZDV) and nevirapine (NVP) were determined. Elucidating the effect that diketo acid resistance mutations have on HIV replication, on IN activity, and on the susceptibility to other classes of IN inhibitors may aid in our understanding of the mechanism of resistance to the diketo acids as well as to other classes of IN inhibitors. Such an understanding may aid the design of more potent, clinically useful IN inhibitors.
MATERIALS AND METHODS
Inhibitors.
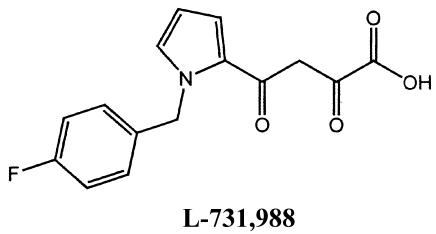
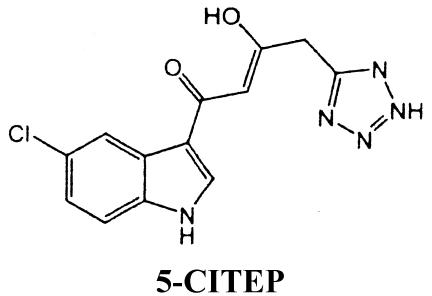
l-731,988 and l-CA (see structures below) were generously provided by Manfred G. Reinecke (Texas Christian University, Fort Worth). Both compounds were >99% pure by nuclear magnetic resonance spectroscopy. Lyophilized l-731,988 was reconstituted to a final 5 mM concentration in H2O; lyophilized l-CA was reconstituted to a concentration of 2 mM in 20% ethanol. ZDV (Sigma) was reconstituted to a final 1 mM concentration in H2O. NVP was a generous gift from William M. Mitchell (Vanderbilt University) and was prepared as described by Essey et al. (26). The structures of l-CA, l-731,988, and 5-CITEP [1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H-tetrazol-5-yl)-propenone] are as follows: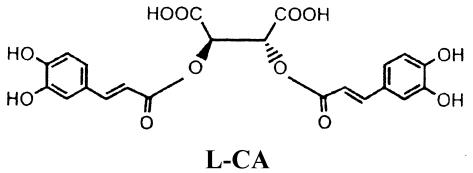
Generation of mutant IN genes.
A pCRScript plasmid containing the IN gene from HIVNL4-3 (accession no. M19921) was cloned and used for mutagenesis via the QuikChange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. Plus- and minus-strand primers were designed to introduce each single mutation (underlined) as follows: T66I+, 5′-GCCCAGGAATATGGCAGCTAGATTGTATACATTTAGAAGG-3′, nt 4399 to 4438; T66I−, 5′-CCTTCTAAATGTATACAATCTAGCTGCCATATTCCTGGGC-3′, nt 4438 to 4399; S153Y+, 5′-CCCCAAAGTCAAGGAGTAATAGAATATATGAATAAAGAATTAAAG-3′, nt 4662 to 4706; S153Y−, 5′-CTTTAATTCTTTATTCATATATTCTATTACTCCTTGACTTTGGGG-3′, nt 4706 to 4662; M154I+, 5′-CCCCAAAGTCAAGGAGTAATAGAATCTATTAATAAAGAATTAAAG-3′, nt 4662 to 4706; M154I−, 5′-CTTTAATTCTTTATTAATAGATTCTATTACTCCTTGACTTTGGGG-3′, nt 4706 to 4662; C280S+, 5′-GGCAGGTGATGATTCCGTGGCAAGTAGACAGG-3′, nt 5049 to 5081; and C280S−, 5′-CCTGTCTACTTGCCACGGAATCATCACCTGCC-3′, nt 5081 to 5049. The entire plasmid was then amplified using the thermostable, proofreading polymerase PfuTurbo (Stratagene). Following amplification, PCR products were digested using DpnI (New England Biolabs) for 2 h at 37°C, and approximately 5 μl was transformed into DH5α competent cells. Sequencing of the resulting plasmids confirmed the presence of each mutation and the absence of additional mutations.
Generation of mutant HIV.
IN genes generated by PCR site-directed mutagenesis were amplified using Pfu polymerase (Stratagene) and primers that allow the IN gene to be inserted into HIVNL4-3 containing XbaI and SacII engineered restriction enzyme sites (37). This results in replacement of the HIVNL4-3 IN gene by mutant IN (35, 37, 47, 48).
Viruses and cell culture.
H9 cells, a CD4+, human T-lymphoblastoid cell line; MT-2 cells, a human T-lymphotropic virus type 1-transformed cell line; and CEM-SS cells, a T-lymphoblastoid cell line, were obtained from the AIDS Research and Reference Reagent Program. Cells were cultured at 37°C in RPMI 1640 containing 25 mM HEPES and supplemented with 11.5% fetal bovine serum (Gemini Biosciences) and 2 mM l-glutamine. Infectious molecular clones were transfected by electroporation into H9 cells. Cultures were cleared of cells via low-speed centrifugation followed by filtration through 0.45-μm-pore-size cellulose acetate filters when the cells became 100% HIV antigen positive by immunofluorescence assay (IFA) (52). The virus stocks were stored at −80°C for use in replication studies.
Quantification of viruses.
HIV was quantified by RT activity from virions in culture supernatant as described previously (45, 52). Briefly, 750 μl of culture supernatant was cleared of cells by low-speed centrifugation and precipitated overnight using 30% polyethylene glycol. Virions were lysed, and the RT activity was determined by quantifying [3H]dTTP incorporation into a poly(rA)-oligo(dT) template.
Viral spread.
Infection and spread were monitored by IFA for expression of HIV antigens as described previously (52). Equal amounts of virus as determined by RT activity were inoculated onto CEM-SS or H9 cells, and at various times beginning 24 h postinoculation, cells were fixed in acetone-methanol (1:1) and labeled with anti-HIV immune globulin followed by fluorescein-conjugated goat anti-human immunoglobulin G, as previously described (52).
Replication by real-time PCR.
In triplicate infections, equal amounts of each virus as determined by RT activity were inoculated onto H9 cells. At 4, 8, 12, 24, 48, and 72 h postinoculation, 106 cells were lysed by addition of 50 μl each of solution A (100 mM KCl, 10 mM Tris-HCl [pH 8.3], 2.5 mM MgCl2) and solution B (10 mM Tris-HCl [pH 8.3], 2.5 mM MgCl2, 1% Tween 20, 1% NP-40, and 20 μg of proteinase K) as described previously (34, 49, 61). The samples were incubated at 60°C for 1 h and heat inactivated at 95°C for 15 min. DNA from each sample was amplified by SYBR Green-I PCR as previously described (35, 49, 61). Primers used were AA55 and M667, which amplify minus-strand strong-stop DNA (69); M661 and M667, which amplify products of complete cDNA synthesis (69); and MH535 and MH536, which amplify two-LTR-circle DNA, the products of failed integration (8). Primers and PCR conditions used were previously published (61). The products were quantified based on an average standard curve ranging from 20 to 20,000 infected-cell equivalents of H9 cells chronically infected with HIV-1LAI, a laboratory-adapted HIV strain obtained from the National Institutes of Health AIDS Research and Reference Reagents program. To quantify integrated HIV DNA, a modification of a nested Alu PCR first described by O'Doherty et al. was performed (43). DNA from each standard and each cell lysate sample was amplified first with Alu (5′-GCCTCCCAAAGTGCTGGGATTACAG-3′) (8) and M661 (69) primers. PCR conditions were as follows: initial denaturation at 94°C for 2 min, followed by 20 cycles of 94°C for 30 s, 63.8°C for 30 s, and 72°C for 1 min 15 s. The resulting PCR product was diluted in an equal volume of H9 cell lysate (106 cells/ml), and 2 μl of the diluted PCR product was amplified in the presence of SYBR Green-I with the AA55 and M667 primers as previously described (35, 49, 61).
EC50.
The susceptibility of each virus to IN or RT inhibitors was determined in MT-2 cells as described previously (42, 49). Triplicate samples of l-CA, l-731,988, ZDV, or NVP were serially diluted 128-fold in 96-well plates. For IN inhibitors, equal RT activities of each virus were preincubated with each inhibitor for 1 h, and then MT-2 cells were added to each well. For RT inhibitors, cells were preincubated for 1 h with drug prior to addition of each virus at equal RT activities. Approximately 5 days later, when control infections showed HIV-induced cytopathic effect, cells were transferred to poly-l-lysine-coated plates and stained with Finter's neutral red dye. Viable cells were quantified on a microcolorimeter by A540. Percent viable cells were determined based on eight uninfected-cell controls (100% viable) and eight replicates of cells plus HIV (virus control, 0% viable). EC50s and 95% confidence intervals were determined using CalcuSyn for Windows.
Generation of mutant IN proteins.
Mutant IN genes were amplified with oligonucleotide primers containing NdeI and HindIII restriction sites (37) and then cloned into a pET-based protein expression vector (62). PCR conditions were as follows: initial denaturation for 2 min at 94°C, followed by 25 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 75 s. Recombinant IN proteins were expressed in BL21 pLysS cells (Stratagene), as previously described (36). Cells were lysed by sonication and ultracentrifugation. The lysate was dialyzed overnight against buffer A (20 mM HEPES, 1 M NaCl, 0.1% NP-40, 10% glycerol, and 5 mM β-mercaptoethanol). Five milliliters of each dialysate was purified via its N-terminal six-His tag with Ni2+ affinity column chromatography (36). Protein purity was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and protein concentration was determined using the Coomassie G-250 protein assay (Pierce). The purified protein was dialyzed against storage buffer (20 mM HEPES [pH 7.5], 0.1 mM EDTA [pH 8.0], 0.3 M NaCl, 20% glycerol, 10 mM dithiothreitol [DTT], and 0.3% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]), aliquoted, and stored at −80°C.
Enzymatic activity of mutant IN proteins.
The specific activity of each IN protein in 3′-end processing-strand transfer and disintegration assays was determined using in vitro assays as described previously (10, 11). The specific activities of each protein were compared to those of reference IN from HIVNL4-3 as described previously (35, 48). For the disintegration reaction, the self-annealing “dumbbell” substrate dBY-1 (5′-TGCTAGTTCTAGCAGGCCCTTGGGCCGGCGCTTGCGCC-3′) was used, whereas 3′-end processing-strand transfer reactions were performed with the complementary V1 and V2 primers (V1, 5′-ATGTGGAAAATCTCTAGCAGT-3′; V2, 5′-ACTGCTAGAGATTTTCCACAT-3′). V1 and dBY-1 substrates were 5′ end labeled with 32P as previously described (10). Briefly, increasing concentrations of IN (5 to 330 nM) in triplicate reactions were incubated with 0.1 pmol of oligonucleotide substrate in a 20-μl reaction mixture containing 20 mM HEPES (pH 7.5), 10 mM DTT, 0.05% NP-40, 7.5% dimethyl sulfoxide, and 10 mM MnCl2. Following 1 h of incubation at 37°C, reactions were stopped by addition of EDTA (pH 8.0) to a final concentration of 18 mM and 7 μl of gel loading buffer. Products were separated by 17% denaturing PAGE and quantified by phosphorimager analysis, and specific activities were determined using SigmaPlot 7.0 linear regression analysis through the linear range of the activity curve.
IC50.
The susceptibility of each mutant IN to l-CA or l-731,988 was determined essentially as previously described (36, 37, 49). Briefly, in triplicate, IN was incubated with V1-V2 as described above in the presence of 0.3 to 10.0 μM inhibitor. For l-CA, the IC50 was determined by quantifying percent conversion of substrate to both 3′-end-processing and strand transfer products compared to that for control enzyme reactions (no inhibitor) with CalcuSyn for Windows. For l-731,988, only strand transfer products were quantified for IC50 analysis since l-731,988 has no effect on 3′-end processing at concentrations used in these studies (30).
RESULTS
HIVs containing the T66I and/or S153Y mutation within IN are delayed for viral spread.
Each mutant HIV was inoculated onto H9 cells at equal RT activities and monitored for viral spread by IFA and RT activity (Fig. 1). By IFA, all mutant viruses except HIV containing the C280S mutation showed a slight delay in viral spread at day 4 (Fig. 1A). In addition to the slight delay at day 4, viruses containing the S153Y, T66I-S153Y, and T66I-M154I mutations within IN were further attenuated compared to HIVNL4-3 (Fig. 1A). The T66I-S153Y double mutation conferred an approximately 1-day delay for spread, while S153Y and T66I-M154I mutant viruses were delayed by approximately 2 days (Fig. 1A). As a control, HIV containing a C280S mutation within IN was inoculated onto H9 cells (Fig. 1A). This virus showed no defect in spread, consistent with previous reports (4). Results by RT paralleled those seen by IFA (Fig. 1B). Similar results were obtained for viruses inoculated onto CEM-SS cells (data not shown).
FIG. 1.

Replication kinetics of mutant HIV determined by IFA and with RT. Equal amounts of HIV as determined by RT activity were inoculated onto H9 cells in triplicate reactions. (A) HIV antigen synthesis was monitored by IFA, and the percentage of HIV antigen-positive cells was measured. (B) Viral supernatant from each culture was cleared of cells, and RT activity was measured from lysed virions by quantifying [3H]dTTP incorporation into a poly(rA)-oligo(dT) template. Points are the means of triplicate infections. Error bars are ±1 standard deviation.
In order to ensure that spread of mutant HIV through culture was not due to reversion events, virions from each culture were isolated and lysed, and the IN genes were amplified using RT-PCR. In each case, IN genes retained the appropriate mutations, with no additional mutations, indicating that the viral spread phenotype observed was not due to reversion or the acquisition of compensatory mutations within IN; however, the T66I-S153Y virus did display, under some conditions, a tendency to revert to S153Y (data not shown). Furthermore, when viruses were reinoculated on cells they demonstrated similar delays in replication (data not shown).
Delayed replication of mutant HIV is due to an integration defect.
In order to determine the site of each replication defect that resulted in delayed viral spread, each mutant HIV was inoculated in triplicate onto H9 cells and HIV replication was monitored by SYBR Green-I quantitative, real-time PCR, as previously described (35, 49, 61). At 4, 8, 12, 24, 48, and 72 h postinoculation, cells were lysed for real-time PCR analysis to detect minus-strand strong-stop DNA (Fig. 2A), completely synthesized HIV cDNA (Fig. 2B), or two-LTR-circle DNA (a measure of failed integration [8, 30] [Fig. 2C and D]). Products were then quantified as infected-cell equivalents based on a standard curve of cell lysates from 20 to 20,000 H9 cells chronically infected with HIVLAI. Standards were linear (r2 = 0.999, 0.999, and 0.997) (insets of Fig. 2A, B, and C, respectively). Representative data are shown for HIVNL4-3 and HIV containing either M154I or T66I-M154I mutations (Fig. 2). Data for all mutant viruses are summarized in Tables 1 and 2. Since the amount of PCR product reached a plateau between 24 and 48 h postinoculation, the first round of replication was completed for each virus by approximately 24 h postinoculation (Fig. 2) (61). Over the first 24 h, equal amounts of minus-strand strong-stop DNA (AA55-M667 product) were present for each virus, indicating that (i) equal amounts of input virus were used to infect each culture, (ii) viral entry was not affected by the IN mutation(s), and (iii) early steps in reverse transcription were also unaffected (Fig. 2A). Similarly, there were no significant differences in the amount of HIV cDNA, indicating that reverse transcription was unaffected by the IN mutation(s) (Fig. 2B). As summarized in Table 1, when minus-strand strong-stop DNA was normalized to cDNA for each virus, no statistically significant difference was seen in the ratio of minus-strand strong-stop DNA to cDNA at 24 h postinoculation for any virus, further confirming that reverse transcription was unaffected by mutations within the IN gene.
FIG. 2.

Replication kinetics of mutant HIV determined by real-time PCR. Equal amounts of HIVNL4-3 (solid circles), NL4-3IN:M154I (open inverted triangles), and NL4-3IN:T66I-M154I (solid squares) were inoculated onto H9 cells in triplicate reactions. Next, 106 cells were lysed and real-time PCR was performed at each time point. (A) Infected-cell equivalents of minus-strand strong-stop DNA (AA55-M667) with standard curve (inset); (B) completely synthesized HIV cDNA (M661-M667) with standard curve (inset); (C) two-LTR-circle DNA (MH535-MH536) with standard curve (inset); (D) mean ratio of two-LTR-circle DNA to cDNA. Each point is the mean of triplicate infections, and error bars are ±1 standard deviation. The number of replicates for each standard curve is indicated in each insert.
TABLE 1.
Effect of IN mutations on HIV reverse transcription 24 h postinoculation
| HIVNL4-3 clone | AA55-M667/cDNA
|
|
|---|---|---|
| Ratioa | Pc | |
| NL4-3 | 1.20b | |
| T66I | 1.44 | 0.377 |
| S153Y | 1.56 | 0.386 |
| M154I | 0.75 | 0.130 |
| T66I-M154I | 1.72 | 0.382 |
| T66I-S153Y | 1.22 | 0.950 |
| C280S | 0.96 | 0.448 |
Minus-strand strong-stop DNA was quantified by the AA55-M667 primer pair, and completely synthesized HIV cDNA was determined using the M661-M667 primer pair. The average ratio was calculated from triplicate infections.
Value is the average of nine replicates.
Statistical significance was determined for the ratio of minus-strand strong-stop DNA to cDNA between the indicated mutant virus and HIVNL4-3. The P value was calculated using Student's two-tailed t test assuming equal variances.
TABLE 2.
Effect of IN mutations on HIV integration 24 h postinoculation
| HIVNL4-3 clone | Two-LTR-circle
|
Integrated/total
|
||||
|---|---|---|---|---|---|---|
| cDNA ratioa | SD | Pb | cDNA Ratioa | SD | Pb | |
| NL4-3 | 0.818 | 0.426 | 0.219 | 0.042 | ||
| T661 | 1.438 | 0.750 | <0.05 | 0.076 | 0.052 | <0.05 |
| S153Y | 2.007 | 1.086 | <0.005 | 0.047 | 0.019 | <0.005 |
| M154I | 1.026 | 0.709 | 0.409 | 0.131 | 0.003 | <0.05 |
| T66I-M1541 | 3.071 | 0.828 | <0.00001 | 0.056 | 0.030 | <0.005 |
| T66I-S153Y | 3.153 | 1.225 | <0.00001 | 0.084 | 0.069 | <0.05 |
| C280S | 1.142 | 0.449 | 0.107 | 0.207 | 0.070 | 0.40 |
Each value is the average of triplicate infections.
Statistical significance was determined for the ratio of integrated DNA or two-LTR-circle DNA to cDNA between the indicated mutant virus and HIVNL4-3. P was calculated using Student's two-tailed t test assuming equal variances.
In contrast to levels of minus-strand strong-stop DNA and cDNA at 24 h postinoculation, levels of two-LTR-circle DNA were increased for all mutant viruses (Fig. 2C and Table 2) except for the C280S mutation. Furthermore, all viruses except for that containing M154I and C280S mutations showed statistically significant increases in the ratio of two-LTR-circle DNA to cDNA (Fig. 2D and Table 2). This failure of integration led to decreased spread, resulting in lower levels of minus-strand strong-stop DNA (Fig. 2A) and cDNA (Fig. 2B) at times after 24 h. Thus, although the level of two-LTR-circle DNA was roughly equivalent for all viruses at 48 h, the ratio of two-LTR-circle DNA to cDNA was increased for mutants after 24 h (Fig. 2D).
To confirm that an increase in the ratio of two-LTR-circle DNA to cDNA is a result of decreased integration, the levels of integrated cDNA were directly quantified for each virus (Fig. 3 and Table 2). Because of the frequency of Alu sequences in the human genome, integrated HIV can be amplified using primers and oligonucleotides that hybridize to Alu sequences and within gag (Fig. 3A) (8, 43). Cell lysates from each time point were first amplified with Alu and M661 primers. The resulting product was subjected to SYBR Green-I real-time PCR with AA55 and M667 primers. Infected-cell equivalents of integrated DNA were calculated based on a standard curve (Fig. 3B, inset). In contrast to levels of minus-strand strong-stop DNA and cDNA at 24 h postinoculation, levels of integrated viral DNA were decreased for all mutant viruses (Fig. 3B and Table 2), except for HIV containing the C280S mutation (Table 2). Furthermore, all viruses except for that containing C280S showed statistically significant decreases in the ratio of integrated viral DNA to cDNA (Table 2). Thus, in general, the viruses with the greatest defects in integration as measured by increased ratio of two-LTR-circle DNA to cDNA also demonstrated the lowest amounts of integrated viral cDNA (Table 2). Therefore, an increase in the ratio of two-LTR-circle DNA to cDNA correlates with a decrease in integrated HIV cDNA. These data demonstrate that real-time PCR results correlate with IFA and RT results in that viruses most delayed for viral spread also had the highest incidence of failed integration and thus the fewest integrated proviruses at 24 h (Fig. 1 and Table 2).
FIG. 3.

HIV containing T66I-S153Y is defective for integration. Integrated HIV cDNA was amplified from cellular lysates of triplicate infections with Alu and M661 primers followed by real-time PCR with the AA55 and M667 primers. (A) Nested PCR. In the first round of amplification, Alu primers bind to human Alu sequences and M661 binds within HIV gag. Twenty cycles of PCR in primer excess results in linear amplification of integrated HIV. The PCR products are of differing sizes dependent upon how close to Alu a virus integrates (Nn). In the second round, performed under real-time conditions, internal primers that amplify HIV DNA, AA55 and M667, generate products of a single size, 140 bp, which can be detected using SYBR Green I. (B) Infected-cell equivalents of integrated HIV DNA from HIVNL4-3 (closed circles) and NL4-3IN:T66I-S153Y (open circles) with standard curve (inset). Each point in panel B is the mean of triplicate infections. The Alu amplification to generate the standard curve was performed three separate times; n is the number of AA55-M667 replicates, and dashed lines are 95% confidence intervals. Error bars are ±1 standard deviation.
Mutations within IN affect the catalytic activity of recombinant IN in vitro.
Recombinant IN proteins containing each of the mutations were generated as previously described (37, 48). Each purified recombinant IN was analyzed by SDS-PAGE and stained with Coomassie blue (Fig. 4). Each protein was greater than 90% pure, and all mutant IN proteins migrated similarly to IN from HIVNL4-3. Proteins containing the M154I single or double mutations were obtained in concentrations higher than those of reference IN but similar to those of protein containing the C280S mutation. Previous studies reported that the C280S mutation confers increased protein solubility (4). Despite several attempts, IN containing the T66I-S153Y double mutation was unable to be expressed and purified. Previous studies had reported that IN containing this double mutation was severely attenuated for catalytic activity (30). Thus, we hypothesize that this protein is either insoluble or misfolded.
FIG. 4.

SDS-PAGE analysis of recombinant HIV IN. Recombinant IN was transformed into BL21 pLysS cells, induced with 0.3 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h, and purified over a Ni2+ affinity column. Lanes: M, markers; 1, IN from HIVNL4-3; 2 to 6, IN containing mutation(s) T66I (lane 2), S153Y (lane 3), M154I (lane 4), T66I-M154I (lane 5), and C280S (lane 6). An approximately 32-kDa band corresponding to purified IN was recovered for each protein (arrow). Numbers at left are molecular masses in kilodaltons.
The specific activities in the disintegration and 3′-end-processing-strand transfer reactions for each mutant IN protein were determined. Schematic representations of the dBY-1 dumbbell substrate and disintegration reaction (Fig. 5A) as well as the V1-V2 substrate and 3′-end-processing-strand transfer reactions (Fig. 5D) are shown. Figure 5 shows representative results for reference IN and IN containing T66I-M154I. Compared to reference IN, IN containing T66I-M154I was attenuated in the disintegration reaction at all enzyme concentrations tested (Fig. 5B and C). Furthermore, 3′-end-processing-strand transfer products were decreased for the double mutant compared to reference IN, even though the protein concentration range used was higher for IN containing T66I-M154I mutations (Fig. 5E and F). Representative enzyme activity plots used to calculate the specific activity of each protein are shown in Fig. 6 for IN containing T66I-M154I and M154I compared to reference IN in the disintegration (Fig. 6A) and 3′-end-processing-strand transfer (Fig. 6B) reactions. Similar to IN containing T66I-M154I, proteins containing C280S, T66I, and S153Y single mutations were attenuated for enzymatic activity with an approximately 1.5- to 2-fold reduction in disintegration specific activity compared with that of reference IN and a two- to threefold decrease in 3′-end-processing-strand transfer activities (Table 3). The IN protein which contained M154I showed a 1.5- to 2-fold increase in specific activity compared to reference IN in both disintegration and 3′-end processing-strand transfer (Table 3). No studies to date have described the effects of an M154I change on IN enzymatic activity. However, we hypothesize that the protein may have increased solubility, given that IN containing this mutation was purified at concentrations similar to those of IN containing C280S (Fig. 4), a mutation that increased IN solubility (4).
FIG. 5.

Enzymatic activities of recombinant IN proteins. Increasing amounts of recombinant IN were incubated in the presence of oligonucleotide substrates for 1 h at 37°C in triplicate reactions. Products were separated by denaturing PAGE. (A) dBY-1 dumbbell disintegration substrate is comprised of viral DNA and target DNA components which, upon addition of IN, are resolved into their respective parts; (B and C) representative gels from disintegration assays are shown for reference IN (B) and IN containing T66I-M154I (C); (D) V1-V2, the 21-mer corresponding to the viral LTR, undergoes 3′-end processing and strand transfer in the presence of IN; (E) reference IN; (F) IN containing T66I-M154I. Numbers at the top of each lane indicate nanomolar enzyme concentrations. S, substrate control; *, substrate; →, disintegration product; −2→, 3′-end processing product; S.T.P., strand transfer products.
FIG. 6.

Specific activities of recombinant IN proteins. Panels show linear regression analysis of representative enzyme activity assays used to determine specific activity (picomoles of product per hour per picomole of IN). Solid circles, reference IN; open triangles, INM154I; open squares, INT66I-M154I. (A) Disintegration; (B) 3′-end-processing-strand transfer assays. Points are the means of triplicate reactions, and error bars are ±1 standard deviation.
TABLE 3.
Specific activities (picomoles of product per picomole of IN per hour) of mutant IN proteins
| IN | Disintegration | r2 | 3′-end processing-strand transfer | r2 |
|---|---|---|---|---|
| Reference | 0.10 | 0.97 | 0.07 | 0.99 |
| C280S | 0.05 | 0.95 | 0.02 | 0.99 |
| T66I | 0.06 | 0.91 | 0.03 | 0.91 |
| S153Y | 0.04 | 0.92 | 0.02 | 0.93 |
| M154I | 0.23 | 0.98 | 0.11 | 0.97 |
| T66I-M154I | 0.06 | 0.99 | 0.03 | 0.99 |
| T66I-S153Y | NDa | ND | ND | ND |
ND, not determined.
Results from disintegration specific activity parallel that from 3′-end processing-strand transfer. Specifically, mutations conferring decreased activity in one reaction conferred attenuation in the other reaction, and mutations leading to increased in vitro activity yielded increased activity in both reactions. Moreover, the level of attenuation for each mutant IN was analogous in disintegration and 3′-end-processing-strand transfer assays. Thus, inhibitor resistance mutations within IN can result in proteins with increased or decreased enzymatic activities.
The decreased specific activity of IN containing the C280S mutation was unexpected given previous reports which demonstrated that 3′-end-processing-strand transfer activity was equivalent to or even better than that of reference IN at a 100 nM protein concentration (4). To reconcile this seeming discrepancy, all mutant IN proteins were evaluated for activity at a 100 nM concentration in both disintegration (Fig. 7A) and 3′-end processing-strand transfer (Fig. 7B). This concentration was two- to threefold higher than the highest concentrations used to determine the specific activities of each protein. At 100 nM, all mutant INs displayed 90 to 100% activity in disintegration compared to reference IN (Fig. 7A). Similarly, IN containing M154I or C280S mutations had slightly increased activity compared to that of reference IN in 3′-end processing-strand transfer (Fig. 7B), consistent with previous reports (4). However, IN containing S153Y, T66I, or T66I-M154I demonstrated only 40 to 60% of reference 3′-end-processing-strand transfer activity (Fig. 7B).
FIG. 7.

Activities of recombinant IN proteins at 100 nM. A 100 nM concentration of each recombinant IN was incubated in the presence of: dBY-1 (A) or V1-V2 (B) substrates for 1 h at 37°C in triplicate reactions. Products were separated by denaturing PAGE. Lanes: 1, INC280S; 2, INT66I-M154I; 3, INM154I; 4, INS153Y; 5, INT66I; 6, reference IN. S, substrate control; *, substrate; →, disintegration product; −2→, 3′-end-processing product; S.T.P., strand transfer products.
Resistance to l-731,988 confers cross-resistance to l-CA in vitro.
Previous studies have shown that resistance to l-CA can confer cross-resistance to other IN inhibitors (35, 36). Given that mutations within IN were shown to confer resistance to the diketo acid class of IN inhibitors (30), we evaluated whether these mutations conferred cross-resistance to l-CA. We first determined the IC50 of both l-731,988 and l-CA against each IN protein (Fig. 8 and Table 4). Since l-731,988 predominantly inhibits strand transfer (30), the IC50 of l-731,988 was determined by quantifying strand transfer products. Representative data and their analyses are shown for IN containing M154-T66I mutations compared to reference IN (Fig. 8). The data were quantified, and the percent inhibition at each inhibitor concentration was plotted (Fig. 8C and F). As previously reported (30), resistant proteins were less susceptible to l-731,988, with resistance ranging from approximately 2-fold (M154I-IN and S153Y-IN) to greater than 30-fold (M154I-T66I) (Table 4) compared to that of IN from HIVNL4-3. In contrast, l-731,988 failed to inhibit 3′-end processing by any protein (Fig. 8A or B). These data are consistent with the reported mechanism of action of the diketo acids as competitive inhibitors of strand transfer. Moreover, the resistance phenotypes are consistent with those reported by Hazuda et al., in which the point mutations conferred 2-fold (S153Y), 5-fold (T66I), 6-fold (M154I), and 15-fold (T66I-M154I) resistance in assays of in vitro strand transfer in which IN is preassembled onto immobilized DNA substrate (30).
FIG. 8.

IN containing T66I-M154I mutations is resistant to l-731,988 and l-CA. Recombinant IN protein was incubated for 1 h at 37°C in triplicate reactions in the presence of oligonucleotide substrate and an 0.03 to 10.0 μM concentration of either l-731,988 or l-CA. Representative results used to determine each IC50 (Table 4) are shown. (A and B) 3′-end processing-strand transfer for IN from HIVNL4-3 (A) or INT66I-M154I (B) in the presence of l-731,988. (C) Quantification of data from triplicate experiments shown in panels A and B. Inset shows semilog plots used to derive IC50s. (D and E) 3′-end processing-strand transfer for reference IN (D) or INT66I-M154I (E) in the presence of l-CA. (F) Quantification of data from triplicate experiments shown in panels D and E. Inset shows semilog plots used to derive IC50s. +, 25 μM l-CA; −, 25 μM l-tartaric acid; E, substrate with enzyme; S, substrate only. Products were resolved by denaturing PAGE and quantified by phosphorimager analysis. *, substrate; →, −2 products; S.T.P., strand transfer products. Numbers above the lanes are the concentrations (micromolar) of each inhibitor.
TABLE 4.
Susceptibilities of IN proteins to l-731,988 and l-CA
| IN protein |
l-CA
|
l-731,988
|
||
|---|---|---|---|---|
| IC50 (nM)a | Fold change | IC50 (nM)b | Fold change | |
| Reference | 206 | 346 | ||
| G140S | 385 | 1.9 | 3,009 | 9 |
| M154I | 237 | 1.2 | 618 | 1.8 |
| S153Y | 384 | 1.9 | 657 | 1.9 |
| T66I | 391 | 1.9 | 1,533 | 4.4 |
| M154I-T66I | 1,014 | 5.0 | 16,000 | 46 |
| S153Y-T66I | NDc | ND | ||
IC50 in the end-processing-strand transfer reaction.
IC50 in the strand transfer reaction.
ND, not determined.
Resistance to l-731,988 having been confirmed, the susceptibility of each mutant protein to l-CA was determined. The double mutation T66I-M154I conferred a fivefold increase in IC50 of l-CA for 3′-end processing and strand transfer (Fig. 8D and E and Table 4). The difference is most evident at high concentrations of inhibitor, particularly for the 3′-end-processing reaction (Fig. 8D and E). IN containing single point mutations showed cross-resistance to l-CA, with an approximately twofold decrease in susceptibility (IC50 = 382 and 391 nM, respectively) for IN containing S153Y and T66I (Table 4). In contrast, the M154I mutation did not appear to confer resistance in enzymatic assays compared to reference IN (237 versus 206 nM, respectively; 1.2-fold). Thus, in summary, most mutations that confer resistance to the diketo acid IN inhibitors confer cross-resistance to l-CA, a dicaffeoyltartaric acid IN inhibitor.
Diketo acid-resistant HIV is cross-resistant to l-CA.
Since it was determined that IN proteins can be cross-resistant, the susceptibilities of viruses containing mutations for resistance to l-CA and l-731,988 were determined. With the exception of the virus containing the S153Y mutation, these point mutations and double mutations conferred complete cross-resistance in antiviral assays (Table 5). HIVNL4-3 containing a G140S mutation in IN, which confers complete resistance to l-CA (EC50 of >62.5 μM [36]), yielded an EC50 for l-731,988 of >15 μM compared to reference HIVNL4-3 (EC50 = 1 μM) (Table 5). Similarly, mutations that confer resistance to l-731,988 conferred complete resistance to l-CA (EC50 of >13.25 μM) (Table 5). Although the EC50 of l-CA against NL4-3IN:S153Y is also >13.25 μM, these data are somewhat misleading. l-CA did confer 30% protection at some concentrations around 3 μM. However, protection declined and thus an EC50 could not be calculated (data not shown). Thus, virus containing S153Y was not completely resistant to l-CA; these results parallel the susceptibility of NL4-3IN:S153Y to l-731,988. Overall, these data suggest that mutations that confer resistance to one inhibitor confer cross-resistance to the other.
TABLE 5.
Mutations within IN render HIVNL4-3 resistant to l-CA and l-731,988
IN inhibitor-resistant HIV remains susceptible to RT inhibitors.
Once an anti-HIV drug is used clinically, the presence of HIV resistant to the agent occurs (58, 67). Thus, we evaluated the susceptibility of each IN inhibitor-resistant virus to two RT inhibitors with distinct mechanisms of action: ZDV, a nucleoside analog RT inhibitor, and NVP, a nonnucleoside RT inhibitor. All viruses remained susceptible to both inhibitors (Table 6). Indeed, some viruses even appeared to have slightly increased susceptibility to RT inhibitors, similar to results obtained by others (18, 26, 35). Although there was a trend toward increased susceptibilities of IN inhibitor-resistant HIV to RT inhibitors, i.e., T66I, M154I, T66I-M154I, and T66I-S153Y, only some achieved statistical significance. These included ZDV against M154I, T66I-M154I, and T66I-S153Y and NVP against T66I and S153Y. However, the C280S mutation yielded a statistically significant decrease in susceptibility to ZDV (Table 6).
TABLE 6.
Susceptibilities of IN inhibitor-resistant HIV to RT inhibitors
| Virus | EC50 (nM)a
|
|
|---|---|---|
| ZDV | NVP | |
| NL4-3 | 39 (31-50) | 83 (68-101) |
| C280S | 110 (93-130) | 90 (64-127) |
| T66I | 28 (21-36) | 53 (48-67) |
| S153Y | 33 (27-41) | 125 (121-130) |
| M154I | 19 (15-24) | 62 (46-83) |
| T66I-M154I | 10 (6-17) | 58 (48-71) |
| T66I-S153Y | 14 (12-17) | 106 (98-114) |
The EC50 was determined against each clone using a cell viability-based assay (42). Numbers are the means of triplicate infections; the ranges are the 95% confidence intervals.
DISCUSSION
The effects of mutations at various conserved amino acids within the IN gene have been the subject of several previous studies (9, 21, 24, 57, 59). In addition to mutations leading to defects in the in vitro integration reaction and delays in viral replication in tissue culture, mutations within IN can have pleiotropic effects on the life cycle. These include effects on reverse transcription, nuclear localization, proteolytic processing, and virion morphology (1, 22, 55, 57). In the studies presented here, viruses containing mutations within IN, with the exception of HIV containing C280S, showed at least a slight delay in spread by IFA (Fig. 1). Viruses containing double mutations were more severely delayed than those containing T66I or M154I single mutations (Fig. 1). Results obtained by IFA and RT complemented real-time PCR results. When replication was evaluated using SYBR Green-I real-time PCR, the delay in viral spread was due to a defect in integration. Each inhibitor-resistant virus conferred a statistically significant defect at integration as measured by the decreased ratio of integrated viral DNA to cDNA. Furthermore, except for M154I, each inhibitor-resistant virus displayed a statistically significant increase in the ratio of two-LTR-circle DNA to HIV cDNA; the M154I virus was least defective for integration as measured by the ratio of integrated DNA to cDNA. In contrast, no defects in viral entry or reverse transcription were observed for any mutant. As defects were seen as early as the first round of infection, the delay in replication cannot be attributed to defects in packaging or budding. Thus, each inhibitor resistance mutation conferred a defect in integration with no effect on other steps of the viral life cycle.
When recombinant IN containing each mutation was evaluated for in vitro catalytic activities, results for IN containing the S153Y and T66I mutations complemented data on HIV containing these mutations. T66I and T66I-M154I conferred an approximately twofold decrease in specific activity compared to reference IN, confirming that the T66I mutation attenuates catalytic activity (Table 3). A two-domain crystal of HIV-1 IN demonstrates that a phosphate ion is coordinated by the side chains of T66, H67, and K159, approximately 7 Å from the active site of IN (66); thus, these residues are hypothesized to be involved in binding of viral DNA. Indeed, photo-cross-linking studies have implicated K159 in binding viral DNA (32). Furthermore, data from molecular dynamics simulations with the IN inhibitor 5-CITEP and the catalytic core domain of IN containing the T66I-M154I double mutation indicate that the T66I mutation disrupts the hydrogen bonding interactions observed for reference IN (2). For reference IN, over the course of this simulation, T66 transiently interacted first with I73, N155, and H67 and next with K159. In contrast, in the context of the T66I-M154I double mutation, I66 interacted only with I73 throughout the course of the study (2). In addition to I66 preventing normal hydrogen bonding interactions, in the context of the double mutation T66I-M154I, the side chain of catalytic core residue E152 is oriented toward D64 and D116, whereas it is oriented away in reference IN. The hydrogen bonding interactions of E152 are also altered (2). Therefore, based on crystal structure and molecular dynamics data, in combination with viral and enzymatic data presented here, we hypothesize that both proteins are attenuated for catalytic activity due to (i) disrupted interaction of the viral DNA ends with the catalytic site of IN and (ii) altered orientation of active-site residues.
Similarly, recombinant IN containing S153Y was attenuated in vitro in agreement with viral kinetic data. Furthermore, given that IN containing the double mutation T66I-S153Y was not expressed and purified despite several attempts, we hypothesize that the S153Y mutation may decrease protein solubility or may render the protein susceptible to misfolding. Our data indicating that IN containing S153Y is attenuated for catalytic activity are consistent with previous studies in which recombinant INs containing T66I-S153Y mutations were reported to have only 5% enzymatic activity compared to reference IN (30).
In contrast to data on the T66I and S153Y mutations, IN containing M154I demonstrated increased specific activity in vitro. By IFA, only a slight delay in viral replication was detected, and by real-time PCR, a statistically significant defect in integration was observed. However, recombinant M154I IN was almost three times more active in disintegration and almost twofold more active in 3′-end-processing-strand transfer reactions than was reference IN (Table 3). This increased specific activity may be due to increased protein solubility, based on similarities between IN containing M154I and IN containing the C280S mutation. Recombinant IN containing the M154I substitution was purified at concentrations similar to those of C280S and at concentrations two- to threefold higher than that obtained for reference IN (Fig. 4 and data not shown). Furthermore, 100 nM IN containing the M154I or C280S mutation had 3′-end-processing-strand transfer activity equivalent to or slightly better than that of reference IN (Fig. 7). The IN with the double mutation T66I-M154I had specific activities in both disintegration and 3′-end-processing assays similar to those obtained for T66I alone (Fig. 5 and 6 and Table 3).
Inhibitor resistance mutations can result in different phenotypes. For example, the replicative fitness of HIV resistant to RT and protease (PR) inhibitors is quite varied (14, 18, 31). Multiple resistance mutations to nonnucleoside RT inhibitors may confer little to no delay in viral spread (14) or RT release (31) while some single point mutations within either RT render HIV severely defective for spread and RT release (31). Furthermore, the presence of some mutations within RT may help compensate for defects conferred by other resistance mutations (18, 31). A similar variety of phenotypes have been described for HIVs resistant to PR inhibitors (18). Results obtained here for IN inhibitor-resistant HIV and proteins demonstrate defects similar to those described for resistance to PR and RT inhibitors (14, 31). In particular, the viral spread results obtained for IN inhibitor-resistant viruses are analogous to those seen for some HIVs containing triple or even quadruple mutations within RT (14). Nine of 11 HIVs displayed wild-type spread, as measured by peak p24 production, while 2 of 11 had a 1-day delay for peak p24 production (14). Despite similar kinetics of p24 release, in competition experiments all resistant viruses demonstrated a lower replicative capacity than that of the wild-type virus (14). In addition, similar to the increased susceptibility of IN inhibitor-resistant HIV to RT inhibitors described herein (Table 6), resistance to one inhibitor can lead to increased susceptibility to different inhibitors either of the same class (31) or of different classes (18, 26, 35, 37).
Each diketo acid resistance mutation conferred cross-resistance to l-CA in both in vitro and tissue culture assays (Fig. 8 and Tables 4 and 5). These data complement recent findings by King et al. demonstrating that HIV or IN containing a G140S mutation within IN was impaired for integration and was cross-resistant to l-731,988 (35). G140S was isolated as a resistance mutation to l-CA (37). Suballelic reconstruction of reference HIV containing IN with the G140S substitution yielded HIV that was completely resistant to l-CA in tissue culture (37). HIV containing G140S also demonstrates resistance to l-CA analogs in tissue culture (36). Recombinant IN containing this substitution was impaired for catalysis and demonstrated eightfold resistance to l-CA and nearly fivefold resistance to l-731,988 in the strand transfer reaction (35). Based on those data on the G140S mutation, it was hypothesized that l-CA and l-731,988 may share a similar binding pocket on IN (35). The data reported in this study provide additional evidence that l-CA and l-731,988 share a common binding pocket and thus common resistance mutations. It should be noted that previous work by Pluymers et al. had suggested that l-CA acts principally at entry, rather than integration (44). The in vitro cross-resistance data presented herein demonstrate that l-CA resistance maps to IN.
l-CA and l-731,988 are both small molecules that contain aromatic rings as well as carbonyl groups and at least one free carboxylic acid moiety. l-CA requires one free carboxylic acid for activity (37), while l-731,988 requires both its free carboxylic acid and adjacent carbonyl groups (29), although data have demonstrated that the active conformation for l-731,988 in vivo is the enol and not the diketo state (65). No cocrystals of either inhibitor with the catalytic core domain of IN have been reported; however, l-CA has been modeled into the active site of IN in several independent studies (51, 56). These computer modeling studies identified amino acids on IN including D64, C65, T66, H67, E92, D116, D117, Q148, E152, K156, and K159 (51, 56) as being involved in l-CA binding.
The data on cross-resistance between the diketo acids and dicaffeoyltartaric acids are intriguing given the cocrystal studies published for the IN inhibitor 5-CITEP with the catalytic core of IN (27). No computer modeling studies with l-731,988 have been published to date; however, given the structural similarities between 5-CITEP and l-731,988 (56) (see structures above) and the similarity of binding by 5-CITEP and l-CA into an inhibitor-binding pocket, it is reasonable to hypothesize that all three may bind a similar pocket. Indeed, mapping of resistance mutations at positions T66, S153, and M154 and the data presented here support the interpretation that l-731,988, like l-CA, interacts near T66 and E152 and perhaps K156 and K159. Indeed, cocrystals of 5-CITEP and HIV IN implicated amino acids T66, Q148, E152, N155, K156, and K159, which match several amino acids identified in l-CA computer modeling studies as well as amino acids important for inhibitor cross-resistance. Additional computer modeling studies on various cinnamoyl IN inhibitors also identified common amino acids involved in binding (5). Thus, the binding pocket occupied by both l-CA and l-731,988 may in fact represent a common binding site for several classes of IN inhibitors.
While the binding pockets for the two compounds may be similar, the mechanism of resistance for IN containing the G140S mutation may differ from that for T66I-M154I. Mutations of G140A, G149A, or both G140A and G149A impair the mobility of a disordered loop spanning residues G140 to G149, leading to impaired catalysis (28). The G140S may also impair loop mobility, as evident by the mutant's impaired viral integration and enzymatic activities (35). In contrast, by molecular dynamics simulations, T66I-M154I conferred increased loop flexibility compared to reference IN (2). Indeed, the flexibility of the double mutant in the presence of 5-CITEP matched that of reference IN alone and was substantially greater than that observed for reference IN with inhibitor (2). Thus, while both G140S and T66I-M154I resistance mutations affect the mobility of the same region and lead to resistance to both the dicaffeoyltartaric acids and diketo acids, they may confer resistance via two distinct mechanisms. The end product may be the same, however, as impaired loop mobility resulting from a G140 mutation could impair hydrogen bonding of E152 in a manner similar to that hypothesized for T66I-M154I (2).
Though the identification of inhibitors of HIV IN has been a subject of intense research, the lack of a full-length IN crystal structure has hindered the identification and development of clinically useful IN inhibitors. The data presented here identify amino acids that may play an important role in inhibitor binding and evaluate the effects that mutations at these amino acid residues have on HIV replication and susceptibility to inhibitors of HIV replication. Elucidation of the exact interactions between amino acids on IN and the functional groups of IN inhibitors would facilitate the design of new, more potent IN inhibitors. Such studies would certainly aid our understanding of the requirements for inhibition of IN and for inhibitor resistance. To date, resistance mutations result in impaired HIV replication and integration. Perhaps most favorable to the clinical use of IN inhibitors is the finding that IN inhibitor-resistant HIVs remain susceptible to or may even have increased susceptibility to other HIV inhibitors (Table 6). Such results are similar to those already published for IN inhibitor-resistant HIV (3, 35, 37) and for PR inhibitor-resistant HIV (18). Thus, combination and/or sequential therapy including drugs targeted against IN may prove a viable treatment option in HIV infection.
Acknowledgments
We are indebted to Brenda McDougall for technical assistance. We thank Manfred G. Reinecke (Texas Christian University) for l-CA and l-731,988. We also acknowledge Hung Fan, Bert Semler, Roz Sandri-Goldin, and Richard Chamberlin for thoughtful comments and insights and Joseph Victoria, Belle Herrera, and Dustin Gerth for critical reviews of the manuscript.
This work was supported in part by grants from the Public Health Service, 5RO1-AI41360 (W.E.R.) and 5T32-GM08620 (D.J.L.), and from the Burroughs Wellcome Fund (#99-2609) (W.E.R.). W.E.R. is a Burroughs Wellcome Fund Clinical Scientist in Translational Research.
REFERENCES
- 1.Ansari-Lari, M. A., L. A. Donehower, and R. A. Gibbs. 1995. Analysis of human immunodeficiency virus type 1 integrase mutants. Virology 211:332-335. [DOI] [PubMed] [Google Scholar]
- 2.Barreca, M. L., K. W. Lee, A. Chimirri, and J. M. Briggs. 2003. Molecular dynamics studies of the wild-type and double mutant HIV-1 integrase complexed with the 5CITEP inhibitor: mechanism for inhibition and drug resistance. Biophys. J. 84:1450-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beale, K., and W. E. Robinson, Jr. 2000. Combinations of reverse transcriptase, protease, and integrase inhibitors can be synergistic in vitro against drug-sensitive and RT inhibitor-resistant molecular clones of HIV-1. Antivir. Res. 46:223-232. [DOI] [PubMed] [Google Scholar]
- 4.Bischerour, J., H. Leh, E. Deprez, J. C. Brochon, and J. F. Mouscadet. 2003. Disulfide-linked oligomers involving C280 residues are formed in vitro and in vivo but are not essential for human immunodeficiency virus replication. J. Virol. 77:135-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buolamwini, J. K., and H. Assefa. 2002. CoMFA and CoMSIA 3D QSAR and docking studies on conformationally-restrained cinnamoyl HIV-1 integrase inhibitors: exploration of a binding mode at the active site. J. Med. Chem. 45:841-852. [DOI] [PubMed] [Google Scholar]
- 6.Burke, C. J., G. Sanyal, M. W. Bruner, J. A. Ryan, R. L. LaFemina, H. L. Robbins, A. S. Zeft, C. R. Middaugh, and M. G. Cordingley. 1992. Structural implications of spectroscopic characterization of a putative zinc finger peptide from HIV-1 integrase. J. Biol. Chem. 267:9639-9644. [PubMed] [Google Scholar]
- 7.Bushman, F. D., A. Engelman, I. Palmer, P. Wingfield, and R. Craigie. 1993. Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding. Proc. Natl. Acad. Sci. USA 90:3428-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butler, S. L., M. S. T. Hansen, and F. D. Bushman. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631-634. [DOI] [PubMed] [Google Scholar]
- 9.Cannon, P. M., W. Wilson, E. Byles, S. M. Kingsman, and A. J. Kingsman. 1994. Human immunodeficiency virus type 1 integrase: effect on viral replication of mutations at highly conserved residues. J. Virol. 68:4768-4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chow, S. A. 1997. In vitro assays for activities of retroviral integrase. Methods 12:306-317. [DOI] [PubMed] [Google Scholar]
- 11.Chow, S. A., and P. O. Brown. 1994. Juxtaposition of two viral DNA ends in a bimolecular disintegration reaction mediated by multimers of human immunodeficiency virus type 1 or murine leukemia virus integrase. J. Virol. 68:7869-7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow, S. A., K. A. Vincent, V. Ellison, and P. O. Brown. 1992. Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science 255:723-726. [DOI] [PubMed] [Google Scholar]
- 13.Coffin, J. M., S. H. Hughes, and H. E. Varmus (ed.). 1997. Retroviruses, 1st ed. Cold Spring Harbor Laboratory Press, Plainview, N.Y. [PubMed]
- 14.Collins, J. A., M. G. Thompson, E. Paintsil, M. Ricketts, J. Gedzior, and L. Alexander. 2004. Competitive fitness of nevirapine-resistant human immunodeficiency virus type 1 mutants. J. Virol. 78:603-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craigie, R., T. Fujiwara, and F. Bushman. 1990. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 62:829-837. [DOI] [PubMed] [Google Scholar]
- 16.Daniel, R., G. Kao, K. Taganov, J. G. Greger, O. Favorova, G. Merkel, T. J. Yen, R. A. Katz, and A. M. Skalka. 2003. Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc. Natl. Acad. Sci. USA 100:4778-4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daniel, R., R. A. Katz, and A. M. Skalka. 1999. A role for DNA-PK in retroviral DNA integration. Science 284:644-647. [DOI] [PubMed] [Google Scholar]
- 18.de la Carriere, L. C., S. Paulous, F. Clavel, and F. Mammano. 1999. Effects of human immunodeficiency virus type 1 resistance to protease inhibitors on reverse transcriptase processing, activity, and drug sensitivity. J. Virol. 73:3455-3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellison, V., H. Abrams, T. Roe, J. Liffson, and P. Brown. 1990. Human immunodeficiency virus integration in a cell-free system. J. Virol. 64:2711-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelman, A., F. D. Bushman, and R. Craigie. 1993. Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J. 12:3269-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engelman, A., and R. Craigie. 1992. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 66:6361-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelman, A., G. Englund, J. M. Orenstein, M. A. Martin, and R. Craigie. 1995. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 69:2729-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engelman, A., A. B. Hickman, and R. Craigie. 1994. The core and carboxyl-terminal domains of the integrase protein of human immunodeficiency virus type 1 each contribute to nonspecific DNA binding. J. Virol. 68:5911-5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelman, A., Y. Liu, H. Chen, M. Farzan, and F. Dyda. 1997. Structure-based mutagenesis of the catalytic domain of human immunodeficiency virus type 1 integrase. J. Virol. 71:3507-3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelman, A., K. Mizuuchi, and R. Craigie. 1991. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67:1211-1221. [DOI] [PubMed] [Google Scholar]
- 26.Essey, R. J., B. R. McDougall, and W. E. Robinson, Jr. 2001. Mismatched double-stranded RNA (polyI-polyC12U) is synergistic with multiple anti-HIV drugs and is active against drug-sensitive and drug-resistant HIV-1 in vitro. Antivir. Res. 52:189-202. [DOI] [PubMed] [Google Scholar]
- 27.Goldgur, Y., R. Craigie, G. H. Cohen, T. Fujiwara, Y. Tomokazu, T. Fujishita, H. Sugimoto, T. Endo, H. Murai, and D. R. Davies. 1999. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl. Acad. Sci. USA 96:13040-13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenwald, J., V. Le, S. L. Butler, F. D. Bushman, and S. Choe. 1999. The mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry 38:8892-8898. [DOI] [PubMed] [Google Scholar]
- 29.Grobler, J. A., K. Stillmock, B. Hu, M. Witmer, P. Felock, A. S. Espeseth, A. Wolfe, M. Egbertson, M. Bourgeois, J. Melamed, J. S. Wai, S. Young, J. Vacca, and D. J. Hazuda. 2001. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. USA 99:6661-6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hazuda, D. J., P. Pelock, M. Witmer, A. Wolfe, K. Stillmock, J. A. Grobler, A. Espeseth, L. Gabryelski, W. Schleif, C. Blau, and M. D. Miller. 2000. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 287:646-650. [DOI] [PubMed] [Google Scholar]
- 31.Huang, W., A. Gamarnik, K. Limoli, C. J. Petropoulos, and J. M. Whitcomb. 2003. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J. Virol. 77:1512-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins, T. M., D. Esposito, A. Engelman, and R. Craigie. 1997. Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 16:6849-6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katz, R. A., G. Merkel, J. Kulkosky, J. Leis, and A. M. Skalka. 1990. The avian retroviral IN protein is both necessary and sufficient for integrative recombination in vitro. Cell 63:87-95. [DOI] [PubMed] [Google Scholar]
- 34.Kellogg, D. E., and S. Kwok. 1990. Detection of human immunodeficiency virus, p. 337-347. In M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (ed.), PCR protocols: a guide to methods and applications. Academic Press, Inc., San Diego, Calif.
- 35.King, P. J., D. J. Lee, R. A. Reinke, J. G. Victoria, K. Beale, and W. E. Robinson, Jr. 2003. Human immunodeficiency virus type 1 integrase containing a glycine to serine mutation at position 140 is attenuated for catalysis and resistant to integrase inhibitors. Virology 306:147-161. [DOI] [PubMed] [Google Scholar]
- 36.King, P. J., G. Ma, W. Miao, Q. Jia, B. R. McDougall, M. G. Reinecke, C. Cornell, J. Kuan, T. R. Kim, and W. E. Robinson, Jr. 1999. Structure-activity relationships: analogues of the dicaffeoylquinic and dicaffeoyltartaric acids as potent inhibitors of human immunodeficiency virus type 1 integrase and replication. J. Med. Chem. 42:497-509. [DOI] [PubMed] [Google Scholar]
- 37.King, P. J., and W. E. Robinson, Jr. 1998. Resistance to the anti-human immunodeficiency virus type 1 compound l-chicoric acid results from a single mutation at amino acid 140 of integrase. J. Virol. 72:8420-8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kulkosky, J., K. S. Jones, R. A. Katz, J. P. G. Mack, and A. M. Skalka. 1992. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 12:2331-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LaFemina, R. L., C. L. Schneider, H. L. Robbins, P. L. Callahan, K. LeGrow, E. Roth, W. A. Schleif, and E. A. Emini. 1992. Requirement of active human immunodeficiency virus type 1 integrase enzyme for productive infection of human T-lymphoid cells. J. Virol. 66:7414-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee, S. P., and M. K. Han. 1996. Zinc stimulates Mg2+-dependent 3′-processing activity of human immunodeficiency virus type 1 integrase in vitro. Biochemistry 35:3837-3844. [DOI] [PubMed] [Google Scholar]
- 41.Melek, M., J. M. Jones, M. H. O'Dea, G. Pais, T. R. Burke, Jr., Y. Pommier, N. Neamati, and M. Gellert. 2001. Effect of HIV integrase inhibitors on the RAG1/2 recombinase. Proc. Natl. Acad. Sci. USA 99:134-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montefiori, D. C., J. W. E. Robinson, S. S. Schuffman, and W. M. Mitchell. 1988. Evaluation of antiviral drugs and neutralizing antibodies against human immunodeficiency virus by a rapid and sensitive microtiter infection assay. J. Clin. Microbiol. 26:231-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Doherty, U., W. J. Swiggard, D. Jeyakumar, D. McGain, and M. H. Malim. 2002. A sensitive, quantitative assay for human immunodeficiency virus type 1 integration. J. Virol. 76:10942-10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pluymers, W., N. Neamati, C. Pannecouque, V. Fikkert, C. Marchand, T. R. Burke, Jr., Y. Pommier, D. Schols, E. De Clercq, Z. Debyser, and M. Witvrouw. 2000. Viral entry as the primary target for the anti-HIV activity of chicoric acid and its tetra-acetyl esters. Mol. Pharmacol. 58:641-648. [DOI] [PubMed] [Google Scholar]
- 45.Poiesz, B. J., F. W. Ruscetti, A. F. Gazder, B. A. Bunn, J. D. Minna, and R. C. Gallo. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 77:7415-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pommier, Y., and N. Neamati. 1999. Inhibitors of human immunodeficiency virus integrase. Adv. Virus Res. 52:427-458. [DOI] [PubMed] [Google Scholar]
- 47.Reinke, R., D. J. Lee, and W. E. Robinson, Jr. 2002. Inhibition of human immunodeficiency virus type 1 isolates by the integrase inhibitor l-731,988, a diketo acid. Antimicrob. Agents Chemother. 46:3301-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reinke, R., N. R. Steffen, and W. E. Robinson, Jr. 2001. Natural selection results in conservation of HIV-1 integrase activity despite sequence variability. AIDS 15:823-830. [DOI] [PubMed] [Google Scholar]
- 49.Reinke, R. A., P. J. King, J. G. Victoria, B. R. McDougall, G. Ma, Y. Mao, M. G. Reinecke, and W. E. Robinson, Jr. 2002. Dicaffeoyltartaric acid analogues inhibit human immunodeficiency virus type 1 (HIV-1) integrase and HIV-1 replication at non-toxic concentrations. J. Med. Chem. 45:3669-3683. [DOI] [PubMed] [Google Scholar]
- 50.Robinson, W. E., Jr. 1998. HIV integrase: the next target? Infect. Med. 15:129-137. [Google Scholar]
- 51.Robinson, W. E., Jr., M. Cordeiro, S. Abdel-Malek, Q. Jia, S. A. Chow, M. G. Reinecke, and W. M. Mitchell. 1996. Dicaffeoylquinic acid inhibitors of human immunodeficiency virus integrase: inhibition of the core catalytic domain of human immunodeficiency virus integrase. Mol. Pharmacol. 50:846-855. [PubMed] [Google Scholar]
- 52.Robinson, W. E., Jr., D. C. Montefiori, D. H. Gillespie, and W. M. Mitchell. 1989. Complement-mediated, antibody-dependent enhancement of HIV-1 infection in vitro is characterized by increased protein and RNA syntheses and infectious virus release. J. Acquir. Immune Defic. Syndr. 2:33-42. [PubMed] [Google Scholar]
- 53.Robinson, W. E., Jr., M. G. Reinecke, S. Abdel-Malek, Q. Jia, and S. A. Chow. 1996. Inhibitors of HIV-1 replication that inhibit HIV integrase. Proc. Natl. Acad. Sci. USA 93:6326-6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sherman, P. A., and J. A. Fyfe. 1990. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl. Acad. Sci. USA 87:5119-5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin, C. G., B. Taddeo, W. A. Haseltine, and C. M. Farnet. 1994. Genetic analysis of the human immunodeficiency virus type 1 integrase protein. J. Virol. 68:1633-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sotriffer, C. A., H. Ni, and A. McCammon. 2000. Active site binding modes of HIV-1 integrase inhibitors. J. Med. Chem. 43:4109-4117. [DOI] [PubMed] [Google Scholar]
- 57.Taddeo, B., W. A. Haseltine, and C. M. Farnet. 1994. Integrase mutants of human immunodeficiency virus type 1 with a specific defect in integration. J. Virol. 68:8401-8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.UK Collaborative Group on Monitoring the Transmission of HIV Drug Resistance. 2001. Analysis of prevalence of HIV-1 drug resistance in primary infections in the United Kingdom. Br. Med. J. 322:1087-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Gent, D. C., A. A. Groeneger, and R. H. Plasterk. 1992. Mutational analysis of the integrase protein of human immunodeficiency virus type 2. Proc. Natl. Acad. Sci. USA 89:9598-9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Gent, D. C., C. Vink, A. A. Groeneger, and R. H. Plasterk. 1993. Complementation between HIV integrase proteins mutated in different domains. EMBO J. 12:3261-3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Victoria, J. G., D. J. Lee, B. R. McDougall, and W. E. Robinson, Jr. 2003. Replication kinetics for divergent type 1 human immunodeficiency viruses using quantitative SYBR green I real-time polymerase chain reaction. AIDS Res. Hum. Retrovir. 19:865-874. [DOI] [PubMed] [Google Scholar]
- 62.Vincent, K. A., V. Ellison, S. A. Chow, and P. O. Brown. 1993. Characterization of human immunodeficiency virus type 1 integrase expressed in Escherichia coli and analysis of variants with amino-terminal mutations. J. Virol. 67:425-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vincent, K. A., D. York-Higgins, M. Quiroga, and P. O. Brown. 1990. Host sequences flanking the HIV provirus. Nucleic Acids Res. 18:6045-6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vink, C., M. Groenink, Y. Elgersma, R. A. Fouchier, M. Tersmette, and R. H. Plasterk. 1990. Analysis of the junctions between human immunodeficiency virus type 1 proviral DNA and human DNA. J. Virol. 64:5626-5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wai, J. S., M. S. Egbertson, L. S. Payne, T. E. Fisher, M. W. Embrey, L. O. Tran, J. Y. Melamed, H. M. Langford, J. P. Guare, Jr., L. Zhuang, V. E. Grey, J. P. Vacca, M. K. Holloway, A. M. Naylor-Olsen, D. J. Hazuda, P. J. Felock, A. L. Wolfe, K. A. Stillmock, W. A. Schleif, L. J. Gabryelski, and S. D. Young. 2000. 4-Aryl-2,4-dioxobutanoic acid inhibitors of HIV-1 integrase and viral replication in cells. J. Med. Chem. 43:4923-4926. [DOI] [PubMed] [Google Scholar]
- 66.Wang, J. Y., H. Ling, W. Yang, and R. Craigie. 2001. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 20:7333-7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wegner, S. A., S. K. Brodine, J. R. Mascola, S. A. Tasker, R. A. Shaffer, M. J. Starkey, A. Barile, G. J. Martin, N. Aronson, W. W. Emmons, K. Stephan, S. Bloor, J. Vingerhoets, K. Hertogs, and B. Larder. 2000. Prevalence of genotypic and phenotypic resistance to anti-retroviral drugs in a cohort of therapy-naive HIV-1 infected US military personnel. AIDS 14:1009-1015. [DOI] [PubMed] [Google Scholar]
- 68.Yoder, K. E., and F. D. Bushman. 2000. Repair of gaps in retroviral DNA integration intermediates. J. Virol. 74:11191-11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Y. Chen. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213-222. [DOI] [PubMed] [Google Scholar]