

Abstract
We used vesicular stomatitis virus to test the effect of complementation on the relative fitness of a deleterious mutant, monoclonal antibody-resistant mutant (MARM) N, in competition with its wild-type ancestor. We carried out competitions of MARM N and wild-type populations at different multiplicities of infection (MOIs) and initial ratios of the wild type to the mutant and found that the fitness of MARM N relative to that of the wild type is very sensitive to changes in the MOI (i.e., the degree of complementation) but depends little, if at all, on the initial frequencies of MARM N and the wild type. Further, we developed a mathematical model under the assumption that during coinfection both viruses contribute to a common pool of protein products in the infected cell and that they both exploit this common pool equally. Under such conditions, the fitness of all virions that coinfect a cell is the average fitness in the absence of coinfection of that group of virions. In the absence of coinfection, complementation cannot take place and the relative fitness of each competitor is only determined by the selective value of its own products. We found good agreement between our experimental results and the model predictions, which suggests that the wild type and MARM N freely share all of their gene products under coinfection.
RNA viruses form highly polymorphic populations called quasispecies (8, 9, 13). The origin of much of their genetic diversity is high mutational pressure. Most RNA viruses have mutation rates on the order of one miscopied base per genome and generation (10, 11). However, high mutational pressure is not necessarily the only factor promoting genetic diversity. Niche differentiation can allow stable polymorphisms in an infected host. For instance, during a polyclonal immune response, different subpopulations may have different sensitivities to individual antibodies; variation within a host can also be related to differences in the tropism of different viral subpopulations. Virus-virus interactions may also promote stable polymorphisms in an infected host. In cell culture, such interactions readily occur during replication inside a cell and are likely to be an important contributor to the maintenance of variation. When two or more virions coinfect the same cell, complementation can take place. Not every function can be complemented in trans. In positive-stranded viruses, translation and replication are coupled (25); therefore, many functions need to be provided in cis and cannot be rescued by complementation (33, 37). Other proteins can freely interact with heterologous genomes or replicons, even from different viral species (18, 21). In the absence of compartmentalization or other limitations to the diffusion of viral products, soluble proteins can interact with any genome inside the cell, potentially changing the phenotype of the virions and masking targets for natural selection to operate. A remarkable example of this phenomenon is phenotypic mixing and hiding, particularly for monoclonal antibody (MAb)-resistant mutants (MARM) (20, 43).
Complementation obviously relies on coinfection, that is, on the density of the infecting population (also the multiplicity of infection [MOI]). If the MOI is too low and coinfection is infrequent, complementation cannot operate. Thus, a change in density can be viewed as a change in the environment and as a factor of evolutionary processes. Indeed, work with foot-and-mouth disease virus showed adaptation to replication at a high MOI (34). Subsequently, Turner and Chao demonstrated in phage φ6 that strains adapted to low-MOI replication and strains adapted to high-MOI replication behaved as “cooperators” and “defectors,” respectively, during intrahost competition (40) and that the payoff matrix of competitions conformed to the prisoner's dilemma (41).
We have used vesicular stomatitis virus (VSV) to determine the effect of complementation on the relative fitness of a deleterious mutant compared to that of its wild-type (wt) ancestor. In contrast to the studies done with foot-and-mouth disease virus or phage φ6, in this study we have used VSV populations without a previous history of selection for cooperation or defection. VSV, the prototype of the Rhabdoviridae family, is an enveloped, nonsegmented, negative-stranded virus (30). It has a genome of approximately 11 kb with five open reading frames that encode at least five viral proteins. The N protein encapsidates the viral genome, while the L and P proteins constitute the RNA-dependent RNA polymerase that directs transcription and replication. Of the two structural proteins, M provides a link between the nucleocapsid and the membrane, and it is implicated in cytopathology (1). The G protein is the only external protein in the virion; it provides receptor-mediated recognition during entry into host cells. VSV can infect a variety of mammalian and insect cells. Replication in the mammalian cells is typically cytolytic, and high titers of virus are produced. Insects and insect cells are persistently infected, with little or no obvious cytopathology and shedding of virus at low titers. Mutation rates in VSV are comparable to those in other RNA viruses (11). The virus is effectively asexual, as there is no significant amount of recombination (3, 28, 29). VSV has been used as a model to study general principles of evolutionary genetics and the particular rules governing the evolution of RNA viruses (reviewed in reference 8).
Our results showed that despite the lack of previous selection for defection, the relative fitness of the deleterious mutant was higher at a high MOI, demonstrating that it could draw a benefit from coinfection with the wt virus. The overall results were in good agreement with a mathematical model based on averaging of the relative fitness of the two competitors when they are allowed to coinfect cells.
MATERIALS AND METHODS
Cells and viruses.
The experiments were carried out with VSV, Indiana serotype (Mudd-Summers strain). The wt population is clonal in origin and has a history of limited replication in BHK-21 cells at a low MOI. The fitness of the wt was arbitrarily set to 1.0. MARM N is a low-fitness mutant resistant to MAb I1 that has been previously described (26, 27). For the present study, it was amplified once in BHK-21 cells at a low MOI. The fitness of MARM N obtained from 24-h competition passages was 0.43 ± 0.02. All of the infections were done with BHK-21 cells obtained from John Holland's laboratory. The monolayers were grown in minimal essential medium supplemented with 7% bovine calf serum and 0.06% Proteose Peptone 3 to a density of 0.8 × 105 to 1 × 105 cells/cm2. MAb I1 was produced in large batches from a hybridoma kindly provided by Douglas Lyles (24).
Fitness assays.
Fitness was determined by competition of wt and MARM N populations. Our protocol was similar to the one previously described in detail (12, 19), but we incorporated some modifications. The range of MOIs tested was 0.1 to 100, and the sizes of infecting populations were between 1.75 × 104 and 1.75 × 108 PFU. W24 well plates (1.75 cm2 of surface per well) were inoculated with 14 μl of the desired amount of a mixture of the wt and MARM N. For T175 flasks (175 cm2), we used 1.4 ml of the mixture. Original mixtures and viral yields harvested at 9 h postinfection were titrated in T25 flasks. Competitions were limited to a 9-h period (instead of following the typical 24-h period) to avoid differences in the number of rounds of replication between low-MOI and high-MOI infections. The choice of this time was based on growth curves, and its relationship with 24-h competitions is consistent with titers and with the behavior of fitness changes for this pair of competitors (47). All fitness assays were limited to a single competition passage, to avoid accumulation of defective interfering particles in infections at a high MOI (6, 7). In order to get an accurate determination of wt/MARM N ratios, titrations were done with T25 flasks by triplicate plaque assay in the presence and absence of MAb I1. Because we carried out all of our fitness determinations by doing a single competition passage, we could calculate fitness as the ratio of the wt/MARM N ratios before and after the competition passage. These values are comparable to those obtained by calculating the slope of the fit during multiple-passage competitions in previous work (19, 26, 27). Each fitness value was the average of 6 to 16 determinations.
RESULTS
MARM N fitness is density dependent.
Our first aim was to determine whether changes in virus density have an effect on the fitness value of MARM N. We tested density dependence by inoculating T175 flasks and W24 well plates with mixtures of the wt and MARM N at a 1:1 ratio and a total population size of 1.75 × 106 PFU. This population size is equivalent to an MOI of 10 in W24 well plates and 0.1 in T175 flasks. In order to determine whether the absolute population size has an effect on fitness, we included W24 well plates inoculated at an MOI of 0.1 (i.e., with 1.75 × 104 PFU) and T175 flasks inoculated at an MOI of 10 (i.e., with 1.75 × 108 PFU). The results of these determinations showed statistically significant differences between competitions carried out at a high MOI and competitions carried out at a low MOI (Table 1). In contrast, changes in virus population size of up to 100-fold did not have a significant effect on the fitness value of MARM N (Table 1).
TABLE 1.
Relative fitness of MARM N for different MOIs and population sizes
| Plaque size | MOI = 0.1 | MOI = 10 | Pa |
|---|---|---|---|
| T175 | 0.54 ± 0.03 | 0.79 ± 0.06 | 8 × 10−4 |
| W24 | 0.48 ± 0.05 | 0.89 ± 0.05 | 10−5 |
| Pa | 0.28 | 0.26 | NAb |
Probability of fitness values being the same according to a two-sample t test.
NA, not applicable.
We extended our initial studies by testing MARM N fitness at a range of MOIs of 0.1 to 100 by using mixtures of the wt and MARM N at a 1:1 initial ratio. Since we had previously demonstrated that populations size does not have a significant impact on the results, all of the competitions were done by changing the size of the inoculum on W24 well plates. We found that MARM N fitness was lowest at an MOI of 0.1 and highest at an MOI of 100. At the latter MOI, MARM N had almost reached a fitness of 1; that is, MARM N was selectively neutral in comparison to the wt. For intermediate MOIs, MARM N fitness was interpolated smoothly between the two extremes (Fig. 1).
FIG. 1.

Relative fitness of MARM N as a function of the MOI. The solid line indicates the prediction from our model, the final equation in the Appendix, with s = 0.552.
Model of density dependence based on complementation.
Our findings on the density dependence of MARM N fitness prompted us to devise a simple model based on the assumption that MARM N and the wt share all protein products equally in coinfected cells, that is, that MARM N and the wt each act as both a defector and a cooperator with respect to the other strain. We chose this model because MARM N approaches neutrality at a high MOI. If MARM N were a successful overall defector, then it could presumably reach a fitness higher than 1 at a high MOI. Likewise, if the wt were an overall defector, or if for some reason MARM N could not take advantage of the wt's protein products as effectively as the wt does, then MARM N fitness would remain significantly below 1, even at a high MOI. Our model has a single free parameter, the fitness disadvantage s of MARM N at a low MOI, and rests on the assumption that complementation is determined by the MOI. The mathematical details of the model are given in the Appendix. The model is similar to one used by Turner et al. (39) to study the limit of coinfection in bacteriophage φ6. We fitted the model prediction of how MARM N fitness should change with the MOI to the experimental data. For fitting the data to the model, we minimized χ2 as a function of s: If xi is a set of data points, with respective errors σi, and yi(s) is a set of model predictions for xi at a given s, then χ2 is defined as
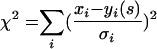 |
The fit gave a selective disadvantage of s = 0.552 for MARM N (χ2 = 2.0, 4 degrees of freedom, P = 0.74, where P indicates the probability with which data consistent with the model exceed the observed χ2; that is, the lower P is, the less likely it is that the model and the data agree.) A selective disadvantage of s = 0.552 translates into a relative fitness of MARM N of 1 − s = 0.448. Figure 1 shows that with this choice of s, the model and data agree very well, consistent with the result of the χ2 test.
MARM N fitness is, at most, weakly frequency dependent.
The model not only makes predictions of how MARM N fitness should change with MOI, but it also predicts that the fitness should depend only weakly on MARM N frequency.
We tested this prediction with additional competitions of the wt and MARM N at an MOI of 1 and an MOI of 10, starting with diverse initial ratios of the two competitors. The results are shown in Fig. 2. At both MOIs, frequency dependence is very weak, if at all existent. The measured data points agree very well with the model at an MOI of 10 (χ2 = 0.82, 3 degrees of freedom, P = 0.85). The fit of the data at an MOI of 1 is quantitatively poor (χ2 = 55.3, 5 degrees of freedom, P ≪ 10−5) but qualitatively good in that there is no clear overall frequency dependence (correlation coefficient r = −0.007).
FIG. 2.

Relative fitness of MARM N as a function of the initial frequency for an MOI of 1 (diamonds) and an MOI of 10 (circles). The solid lines indicate the predictions from our model, the final equation in the Appendix, with s = 0.552.
A third test of frequency dependence is to carry out repeated 24-h passages. For all of the measurements reported in the previous paragraphs, we extracted virus after 9 h, in order to prohibit a second round of replication when the initial infection is done at a low MOI. In 24-h passages at a nominally low MOI, a second round of replication takes place at a high MOI. Complementation is not expected during the first round of low-MOI infection, since there is virtually no coinfection, but it will have an effect during the second, high-MOI, round of replication. Thus, any significant frequency-dependent selection should be apparent (14, 47). In repeated passages, this frequency dependence would translate into a deviation from a straight exponential decay of mutant frequency. Figure 3 shows that MARM N decays exponentially, again in agreement with our model predictions of little or no frequency-dependent selection (χ2 = 2.6, 3 degrees of freedom, P = 0.46).
FIG. 3.

Decay of MARM N concentration in standard 24-h passages. The line represents an exponential fit to the data, exp(−0.36 − 0.59t), where t indicates the passage number.
DISCUSSION
Unlike in previous reports (34, 40, 41, 42), the competitors described here had not been previously subjected to selection at different MOIs. Nevertheless, fitness clearly depended on the density, but not the size, of the infecting population. While density dependence can be predicted and indeed operates independently of previous selection for cooperation or defection, a major difference can be expected depending on the history of the competitors. If competitors have been selected for cooperation or defection, a significant level of frequency-dependent selection is expected (41, 42).
In the absence of previous selection for cooperation or defection, frequency dependence should be minimal, and its contribution to fitness differences is expected to be less than the variation typically observed among replicas of the same experiment. For example, for s = 0.552 at an MOI of 1, our model predicts a change in fitness from 0.51 at a 100:1 wt-to-MARM N ratio to 0.57 at a 1:100 ratio (a change of approximately 10% over a 10,000-fold change in ratios). We found similar (or even smaller) amounts of frequency dependence at other MOIs. Overall, our experimental results agree very well with this prediction. Both the results of single-passage competitions at an MOI of 10 and the multiple-passage competitions done with an initial MOI of 0.1 are in excellent agreement with the quantitative predictions of the model. The only quantitative (but not qualitative) disagreement with the predictions of the model was observed in the single-passage competitions at an MOI of 1. It is worth noting that most of the contribution to the departure originated from two subsets of data. These were a group of 8 determinations at an initial ratio 10:1 (of a total of 16 determinations) and the 6 determinations at an initial ratio 16:1. For an overall χ2 of 57 for the complete data set, the contributions of these values were 24.3 and 22, respectively (thus, the contribution of the remaining data is 10.8, which corresponds to P = 0.38 at 10 degrees of freedom). Each subset of competitions was done on a single day, and therefore any bias would affect all of the values in a subset in the same direction and thus translate into underestimated errors. Moreover, there was no clear tendency among the results to higher-than-expected or lower-than-expected values, suggesting that experimental variation was responsible for the observed departure, rather than any additional biological phenomena that we failed to consider. For instance, gene dosage correlates with the degree of coinfections, but it is not a likely explanation of the observed behavior, since any alteration of the cell metabolism would be equal for both competitors. Our results are in general agreement with those of Holland and coworkers, showing MARM VSV hiding during high-MOI infections (but not during low-MOI infections) in the presence of neutralizing MAbs (20).
In situations in which viral products are totally free to diffuse through the cytoplasm, the present model should hold for any pair of competitors that satisfies the following two conditions. (i) The difference in fitness is not a consequence of mutations involved in cooperation or defection. Otherwise, frequency-dependent selection should play an important role during competition (14, 41, 42), and other models need to be considered (47). (ii) The differences in fitness are located in elements subject to trans complementation. If the fitness disadvantage maps into functions that are carried in the virion (for instance, entry), complementation cannot operate during the first competition passage, but it will have an effect during further passages (46). If the fitness disadvantage is in some cis-acting elements, such as encapsidation signals, complementation may never occur and there will be neither density-dependent nor frequency-dependent selection.
Viral products do not always diffuse freely inside infected cells. Replication of positive-stranded viruses takes place when they are bound to cell membranes, which limits the potential for individual proteins to trans complement. A typical example are the nonstructural proteins in this group of viruses, which need to be provided in cis (33, 37, 38). Comparison of the model predictions and the experimental data suggests that for the pair of mutants we have studied, our two assumptions are accurate, so that coinfection is the main (and possibly the only) determinant of the degree of complementation, and interactions among viral products suffer no limitations.
Any environmental factor affecting coinfection should have an effect on the degree of complementation and, thus, on the survival of a deleterious mutant. Viruses like measles virus carry on their surface glycoproteins that promote cell-to-cell fusion (2, 45); cell-to-cell fusion of infected cells can be seen as an effective increase of the MOI. On the other hand, many viruses, including VSV, have mechanisms leading to superinfection exclusion (44), limiting the number of virions that coinfect the same cell. Under superinfection exclusion, complementation may not work efficiently. In our experimental setting, superinfection exclusion does not seem to have an important effect. This observation is consistent with the known mechanisms that limit secondary infections in VSV, which are not operational until at least 2 h postinfection (35). Superinfection exclusion can limit, but not always abolish, coinfection. For instance, human immunodeficiency virus type 1 induces superinfection exclusion by sequestering the CD4 receptor (5, 23), but the average number of integrated genomes in an infected cell is three (22).
We have shown that the fate of a nonneutral mutation, excluding those described above, will depend not only on its selective value but also on the density at which the competitors are replicating. Thus, replication at a constantly high MOI will slow down the fixation of any advantageous mutations, in agreement with the predictions of Frank (15). For instance, if we consider MARM N as extinct when the wt/MARM ratio goes beyond 100:1, replication at a low MOI would lead to extinction after only six passages. The same competition at an MOI of 10 would require more than 40 passages until extinction is reached. Unfortunately, repeated passages at a high MOI cannot be carried out for long periods of time with VSV, since defective interfering particles are expected to accumulate and will act as an additional selective force (6, 7). Interestingly, a reduction in the fixation rate of advantageous mutations similar to that caused by complementation is expected from clonal interference (16), even though the underlying biological mechanisms are completely unrelated. Density dependence relies only on the sharing of viral products during coinfection, while clonal interference is due to competition among multiple beneficial mutations during the replication of large populations (17).
Complementation may have a direct impact during antiviral treatment. Escape from drugs or antibodies often involves a genetic tradeoff (4), so that resistant strains become dominant only after exposure to the selective pressure. After treatment cessation, resistant strains may incorporate additional compensatory mutations and remain resistant (reference 36 and references therein) or may revert to the wt. However, during reversion, complementation may contribute to the maintenance of resistant mutants and cause quick rebounding of the viral load if a second period of antiviral treatment is applied. Overall, the results of complementation are similar to those predicted from quasispecies memory (31, 32), and it is possible that memory and complementation are related phenomena (46). It is likely that if complementation has an effect on viral memory, it will operate more efficiently in rhabdoviruses than in picornaviruses. In a more general scenario, complementation may allow the maintenance of mutations that are deleterious under the conditions prevailing at one point but beneficial in a different environment, possibly contributing to viral adaptability.
Acknowledgments
Bonnie Ebendick provided excellent technical assistance.
Work at the Medical College of Ohio was funded by NIAID (NIH) grant R01-AI45686 to I.S.N., and C.O.W. received support from NSF contract DEB-9981397.
APPENDIX
We are considering the coinfection of cells by wt virions and virions that carry a deleterious mutation. We assume that the MOI determines the level of coinfection. We further assume that the wt, when infecting a cell by itself, produces M offspring virions, while the mutant produces M(1 − s) offspring virions when infecting a cell by itself. In other words, the fitness of the mutant relative to that of the wt is 1 − s. For the case in which the wt and mutant coinfect, we assume that both contribute equally to the overall virion production, so that the amount of offspring virions produced by a cell is given by the average number of offspring virions expected from the infecting particles. For example, if a cell is infected by n particles, of which k are wt and n − k are mutant virions, then the overall number of particles produced is M[1 − s(n − k)/n]. We assume further that both the wt and mutant genomes are equally likely to get encapsidated, so that the fractions of wt and mutant offspring virions are proportional to the respective fractions of infecting particles. For the above example of a cell infected by k wt virions and n − k mutant virions, a fraction, k/n, of the offspring virions has the wt genotype and the remaining fraction, (n − k)/n, has the mutant genotype.
Let the fraction of wt virions in the population be x and the fraction of mutant virions be y = 1 − x, and consider a cell that is infected by n particles. The probability that k of these particles are wt is
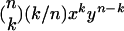 |
Therefore, on average, cells infected by n virions produce p(n) wt virions, with p(n) given by
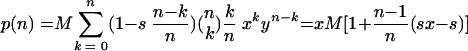 |
Likewise, the average number of mutant virions produced is
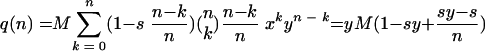 |
The expressions p(n) and q(n) give us the expected number of wt and mutant offspring virions produced by cells infected by exactly n particles. However, not all cells are infected by the same number of particles. The number of particles per cell is Poisson distributed, and the parameter λ of the Poisson distribution corresponds to the MOI. Therefore, in order to calculate the overall expected number of wt offspring virions, P, we have to average p(n) over all n:
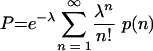 |
After taking the sum, we find
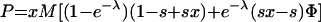 |
Here, Φ is given by
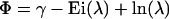 |
where γ ≈ 0.577216 is the Euler constant and Ei(z) is the exponential integral
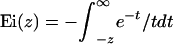 |
Similarly, averaging over q(n) leads to the overall expected number of mutant offspring, Q:
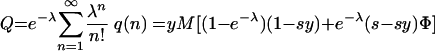 |
The fitness, w, of the mutant relative to that of the wt is given by the ratio of the final fractions, Q/P, divided by the ratio of the initial fractions, y/x. We find
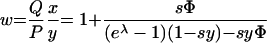 |
REFERENCES
- 1.Blondel, D., G. G. Harmison, and M. Schubert. 1990. Role of matrix protein in cytopathogenesis of vesicular stomatitis virus. J. Virol. 64:1716-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cattaneo, R., and J. K. Rose. 1993. Cell-fusion by the envelope glycoproteins of persistent measles viruses which caused lethal human brain disease. J. Virol. 67:1493-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chare, E. R., E. A. Gould, and E. C. Holmes. 2003. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 84:2691-2703. [DOI] [PubMed] [Google Scholar]
- 4.Coffin, J. M. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483-489. [DOI] [PubMed] [Google Scholar]
- 5.Crise, B., L. Buonocore, and J. K. Rose. 1990. CD4 is retained in the endoplasmic reticulum by the human immunodeficiency virus type 1 glycoprotein precursor. J. Virol. 64:5585-5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DePolo, N. J., C. Giachetti, and J. J. Holland. 1987. Continuing coevolution of virus and defective interfering particles and of viral genome sequences during undiluted passages: virus mutants exhibiting nearly complete resistance to formerly dominant defective interfering particles. J. Virol. 61:454-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePolo, N. J., and J. J. Holland. 1986. Very rapid generation/amplification of defective interfering particles by vesicular stomatitis virus variants isolated from persistent infection. J. Gen. Virol. 67:1195-1198. [DOI] [PubMed] [Google Scholar]
- 8.Domingo, E., C. K. Biebricher, M. Eigen, and J. J. Holland. 2001. Quasispecies and RNA virus evolution: principles and consequences. Landes Bioscience, Georgetown, Tex.
- 9.Domingo, E., and J. J. Holland. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51:151-178. [DOI] [PubMed] [Google Scholar]
- 10.Drake, J. W. 1993. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 90:4171-4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake, J. W., and J. J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duarte, E. A., I. S. Novella, S. Ledesma, D. K. Clarke, A. Moya, S. F. Elena, E. Domingo, and J. J. Holland. 1994. Subclonal components of consensus fitness in an RNA virus clone. J. Virol. 68:4295-4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eigen, M., J. McCaskill, and P. Schuster. 1989. The molecular quasi-species. Adv. Chem. Phys. 75:149-263. [Google Scholar]
- 14.Elena, S. F., R. Miralles, and A. Moya. 1997. Frequency-dependent selection in a mammalian RNA virus. Evolution 51:984-987. [DOI] [PubMed] [Google Scholar]
- 15.Frank, S. A. 2001. Multiplicity of infection and the evolution of hybrid incompatibility in segmented viruses. Heredity 87:522-529. [DOI] [PubMed] [Google Scholar]
- 16.Gerrish, P. J., and R. E. Lenski. 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102/103:127-144. [PubMed] [Google Scholar]
- 17.Hill, W. G., and A. Robertson. 1966. The effect of linkage on limits to artificial selection. Genet. Res. 8:269-294. [PubMed] [Google Scholar]
- 18.Holland, J. J., and C. E. Cords. 1964. Maturation of poliovirus RNA with capsid protein coded by heterologous enteroviruses. Proc. Natl. Acad. Sci. USA 51:1082-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holland, J. J., J. C. de la Torre, D. K. Clarke, and E. A. Duarte. 1991. Quantitation of relative fitness and great adaptability of clonal populations of RNA viruses. J. Virol. 65:2960-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holland, J. J., J. C. de la Torre, D. A. Steinhauer, D. Clarke, E. Duarte, and E. Domingo. 1989. Virus mutation frequencies can be greatly underestimated by monoclonal antibody neutralization of virions. J. Virol. 63:5030-5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia, X.-Y., M. van Eden, M. G. Busch, E. Ehrenfeld, and D. F. Summers. 1998. trans-encapsidation of a poliovirus replicon by different picornavirus capsid proteins. J. Virol. 72:7972-7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung, A., R. Maier, J. P. Vartanian, G. Bocharov, V. Jung, U. Fischer, E. Meese, S. Wain-Hobson, and A. Meyerhans. 2002. Recombination: multiply infected spleen cells in HIV patients. Nature 418:144. [DOI] [PubMed] [Google Scholar]
- 23.Kawamura, I., Y. Koga, N. Ohhori, K. Onodera, G. Kimura, and K. Nomoto. 1989. Depletion of the surface CD4 molecule by the envelope protein of human immunodeficiency virus expressed in a human CD4+ monocytoid cell line. J. Virol. 63:3748-3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lefrancois, L., and D. S. Lyles. 1982. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121:157-167. [PubMed] [Google Scholar]
- 25.Novak, J. E., and K. Kirkegaard. 1994. Coupling between genome translation and replication in an RNA virus. Genes Dev. 8:1726-1737. [DOI] [PubMed] [Google Scholar]
- 26.Novella, I. S., E. A. Duarte, S. F. Elena, A. Moya, E. Domingo, and J. J. Holland. 1995. Exponential increases of RNA virus fitness during large population transmissions. Proc. Natl. Acad. Sci. USA 92:5841-5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novella, I. S., S. F. Elena, A. Moya, E. Domingo, and J. J. Holland. 1995. Size of genetic bottlenecks leading to virus fitness loss is determined by mean initial population fitness. J. Virol. 69:2869-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pringle, C. R. 1982. The genetics of vesiculoviruses. Arch. Virol. 72:1-34. [DOI] [PubMed] [Google Scholar]
- 29.Pringle, C. R., and A. J. Easton. 1997. Monopartite negative strand RNA genomes. Semin. Virol. 8:49-57. [Google Scholar]
- 30.Rose, J. K., and M. A. Whitt. 2001. Rhabdoviridae: the viruses and their replication, p. 1221-1244. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 1. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 31.Ruíz-Jarabo, C. M., A. Arias, E. Baranowski, C. Escarmís, and E. Domingo. 2000. Memory in viral quasispecies. J. Virol. 74:3543-3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruíz-Jarabo, C. M., A. Arias, C. Molina-París, C. Briones, E. Baranowski, C. Escarmís, and E. Domingo. 2002. Duration and fitness dependence of quasispecies memory. J. Mol. Biol. 315:285-296. [DOI] [PubMed] [Google Scholar]
- 33.Sawicki, D. L., and S. G. Sawicki. 1985. Functional analysis of the A complementation group mutants of Sindbis HR virus. Virology 144:20-34. [DOI] [PubMed] [Google Scholar]
- 34.Sevilla, N., C. M. Ruíz-Jarabo, G. Gómez-Mariano, E. Baranowski, and E. Domingo. 1998. An RNA virus can adapt to the multiplicity of infection. J. Gen. Virol. 79:2971-2980. [DOI] [PubMed] [Google Scholar]
- 35.Simon, K. O., J. J. Cardamone, Jr., P. A. Whitaker-Dowling, J. S. Youngner, and C. C. Widnell. 1990. Cellular mechanisms in the superinfection exclusion of vesicular stomatitis virus. Virology 177:375-379. [DOI] [PubMed] [Google Scholar]
- 36.Simon, V., N. Padte, D. Murray, J. Vanderhoeven, T. Wrin, N. Parkin, M. Di Mascio, and M. Markowitz. 2003. Infectivity and replication capacity of drug-resistant human immunodeficiency virus type 1 variants isolated during primary infection. J. Virol. 77:7736-7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strauss, J. H., and E. G. Strauss. 1994. The alphaviruses: gene-expression, replication, and evolution. Microbiol. Rev. 58:491-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teterina, N. L., and W. D. Zhou, and W. M. Cho, and E. Ehrenfeld. 1995. Inefficient complementation activity of poliovirus 2C and 3D proteins for rescue of lethal mutations. J. Virol. 69:4245-4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner, P. E., C. L. Burch, K. A. Hanley, and L. Chao. 1999. Hybrid frequencies confirm limit to coinfection in the RNA bacteriophage φ6. J. Virol. 73:2420-2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner, P. E., and L. Chao. 1998. Sex and the evolution of intrahost competition in RNA virus φ6. Genetics 150:523-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner, P. E., and L. Chao. 1999. Prisoner's dilemma in an RNA virus. Nature 398:441-443. [DOI] [PubMed] [Google Scholar]
- 42.Turner, P. E., and L. Chao. 2003. Escape from prisoner's dilemma in RNA phage φ6. Am. Nat. 161:497-505. [DOI] [PubMed] [Google Scholar]
- 43.Valcárcel, J., and J. Ortín. 1989. Phenotypic hiding: the carryover of mutations in RNA viruses as shown by detection of mar mutants in influenza virus. J. Virol. 63:4107-4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitaker-Dowling, P., J. S. Youngner, C. C. Widnell, and D. K. Wilcox. 1983. Superinfection exclusion by vesicular stomatitis virus. Virology 131:137-143. [DOI] [PubMed] [Google Scholar]
- 45.Wild, T. F., E. Malvoisin, and R. Buckland. 1991. Measles-virus—both the hemagglutinin and fusion glycoproteins are required for fusion. J. Gen. Virol. 72:439-442. [DOI] [PubMed] [Google Scholar]
- 46.Wilke, C. O., and I. S. Novella. 2003. Phenotypic mixing and hiding may contribute to memory in viral quasispecies. BMC Microbiol. 3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilke, C. O., D. D. Reissig, and I. S. Novella. 2004. Replication at periodically changing multiplicity of infection promotes stable coexistence of competing viral populations. Evolution 58:900-905. [DOI] [PubMed]