

Abstract
Herpes simplex virus 1 (HSV-1) forms replication compartments (RCs), domains in which viral DNA replication, late-gene transcription, and encapsidation take place, in the host cell nucleus. The formation of these domains leads to compression and marginalization of host cell chromatin, which forms a dense layer surrounding the viral RCs and constitutes a potential barrier to viral nuclear egress or primary envelopment at the inner nuclear membrane. Surrounding the chromatin layer is the nuclear lamina, a further host cell barrier to egress. In this study, we describe an additional phase in RC maturation that involves disruption of the host chromatin and nuclear lamina so that the RC can approach the nuclear envelope. During this phase, the structure of the chromatin layer is altered so that it no longer forms a continuous layer around the RCs but instead is fragmented, forming islands between which RCs extend to reach the nuclear periphery. Coincident with these changes, the nuclear lamina components lamin A/C and lamin-associated protein 2 appear to be redistributed via a mechanism involving the UL31 and UL34 gene products. Viruses in which the UL31 or UL34 gene has been deleted are unable to undergo this phase of chromatin reorganization and lamina alterations and instead form RCs which are bounded by an intact host cell chromatin layer and nuclear lamina. We postulate that these defects in chromatin restructuring and lamina reorganization explain the previously documented growth defects of these mutant viruses.
Herpes simplex virus 1 (HSV-1) is a large double-stranded DNA virus which replicates in the nucleus of infected host cells. Viral DNA replication and late gene transcription occur in replication compartments (RCs), nuclear domains which are formed when viral replication successively annexes large portions of the nucleus during the early stages of infection. During the later stages of replication, assembly of viral capsids occurs and DNA packaging takes place in RCs. Following this, capsids move to the inner nuclear membrane (INM), where primary envelopment takes place as they bud into the perinuclear space, acquiring an envelope derived from the INM. Herpesvirus particles are then thought to fuse with the outer nuclear membrane, releasing viral nucleocapsids into the host cell cytosol (17, 28). Finally, the nucleocapsid undergoes secondary envelopment and egress from the cell (24).
During RC formation and annexation of space in the host cell nucleus, cellular chromatin is marginalized and compressed (18, 26). This results in a layer of host cell chromatin surrounding the RC, which potentially constitutes a barrier through which viral capsids must move to reach the INM. Thus, to bud through the INM, viral nucleocapsids must move through the compacted host chromatin and the nuclear lamina. The nuclear lamina, a proteinaceous layer underlying the INM, is composed of integral INM proteins such as lamin-associated protein 2 (LAP2) and lamin B receptor, which interact with lamin proteins A, B, and C (2, 6, 33). Lamins are intermediate filament proteins capable of head-to-tail polymerization and interfilamental interactions, which form a network, thought to have a mesh-like structure, which underlies the INM (1, 9, 32, 33). Lamin C is a splice variant of lamin A (13). This network is proposed to provide mechanical support for the INM, lending stability to nuclear size and shape and allowing the nuclear membrane to withstand forces exerted upon it from cytoplasmic components, including the cytoskeleton (2).
The lamina has recently been proposed to play a role in various other nuclear processes, such as control of gene expression and DNA structure, at various times during the cell cycle (2, 7, 10). INM proteins such as LAP2 contain an LEM (standing for lap2-emerin-MAN1) domain, which interacts with the barrier-to-autointegration factor (BAF) protein, which in turn interacts with chromatin (12, 36). Lamin proteins have also been shown to interact with DNA and DNA-binding proteins (10). Thus, the nuclear lamina bridges the gap between the INM and chromatin, possibly facilitating communication between the exterior and interior of the nucleus and holding the two together. From the viral perspective, it forms a continuous fibrous meshwork which separates the interior of the nucleus, where viral capsids are assembled, from the INM, where the capsids must dock in order to achieve primary envelopment.
The HSV-1 UL31 and UL34 proteins have been implicated in nuclear egress (22, 23, 25). Work by Reynolds et al. and others has shown that these proteins are localized to the nucleus and, when expressed together, exhibit an INM distribution (22, 23, 34). UL31 is thought to interact with components of the nuclear matrix (3). UL34 is a transmembrane protein which localizes to the INM, the outer nuclear membrane, and the endoplasmic reticulum in transfected or infected cells (20, 34). Mutant viruses lacking the gene for either UL31 or UL34 are defective for viral egress and replicate poorly on noncomplementing cell lines (4, 25). Immunofluorescence and electron microscopic studies have shown that the defect appears to occur at the stage of nuclear egress, because viral capsids are observed to accumulate in the nucleus and are apparently unable to bud through the INM into the perinuclear space (25).
Recently, changes in the nuclear lamina have been observed during mouse cytomegalovirus infection (19). This virus has been shown to encode two proteins, M50 and M53, which, when expressed together, localize to the INM (19). They are postulated to facilitate nuclear egress by recruiting the host cell protein kinase C, which phosphorylates host cell lamins. This is proposed to lead to a partial dismantling of the nuclear lamina, allowing viral capsids to move through the lamina and dock at the INM (19). M50 and M53 have been postulated to be analogous to HSV-1 proteins UL34 and UL31, respectively, because their subcellular localization is similar and these proteins have also been implicated in nuclear egress (19, 22, 25). However, whether UL31 and UL34 have effects on the nuclear lamina and whether the mechanisms by which these proteins act are similar to those of M50 and M53 remain to be determined. HSV-1 infection has been reported to affect nuclear lamins. Scott and O'Hare reported that at late times in infection, the lamin B receptor becomes increasingly mobile, partially relocalizing to the endoplasmic reticulum. This implies a breakdown of contacts between this receptor and the nuclear lamina (27).
In this work, we report changes that occur in the structure of the marginalized host chromatin layer during infection with a wild-type HSV-1. These changes appear to represent a new stage in RC development during viral maturation and nuclear egress. We addressed the role of UL31 and UL34 during nuclear egress in HSV-1 infection by using wild-type and mutant viruses lacking the open reading frames encoding UL31 and UL34. We also studied nuclear structures during exogenous expression of UL31 and UL34 from plasmids. Second, we documented the changes that occur in the distribution of nuclear lamina proteins during infection with wild-type virus and provide evidence for a role of UL31 and UL34 in these changes.
MATERIALS AND METHODS
Cells and viruses.
RD14 cells expressing the UL31 gene (4) were the kind gift from B. Roizman. 143/1099E cells expressing the UL34 gene (25), Vero cells, Hep-2 cells, and RD14 cells were maintained in Dulbecco's modified Eagle's medium (Gibco-BRL) supplemented with 10% heat-inactivated fetal calf serum (FCS).
Wild-type HSV-1 KOS strain virus was grown and viral titers were determined in Vero cells. The R5132 deletion mutant virus lacking the UL31 gene and the R5133 rescued virus were kind gifts from B. Roizman (4). The RR1072 deletion mutant virus lacking the UL34 gene and the RR1072Rep rescued virus have been described (25). The R5132 and R5133 viruses were grown and viral titers were determined on the complementing RD14 cell line. The R1072 and R1072Rep viruses were grown and viral titers were determined on the complementing 143/1099E cell line. To ensure that the mutant viruses showed replication-defective phenotypes, as reported previously (4, 25), a single-cycle growth curve experiment was performed in Vero cells, and viral titers were determined on the appropriate complementing cell lines (data not shown). At 24 h postinfection, the mutant viruses were found to replicate to titers significantly lower than those of the rescued viruses: RR1072 replicated to 2 log10 below RR1072Rep, and R5132 replicated to 1 log10 below R5133. These growth defects are similar in magnitude to those reported previously for these viruses (3, 25).
Viral titrations.
Viral titers were determined for viral stocks on Vero cells (wild-type KOS strain), 143/1099E cells (RR1072, RR1072Rep, and KOS), or RD14 cells (R5132, R5133, and KOS) (4, 25). Cells were seeded at 106 cells/well in six-well plates and incubated overnight at 37°C. Samples were serially diluted 1:10 in phosphate-buffered saline (PBS), and 200 μl of each dilution was used to inoculate each well. The cells were incubated at 37°C for 1 h before removal of the inoculum and addition of Dulbecco's modified Eagle's medium plus 1% FCS plus anti-HSV-1 human immunoglobulin, followed by incubation for 48 h at 37°C.
Deletion mutant viruses were found to make very small plaques; therefore, to ensure the accuracy of the titrations, monolayers for all assays of all the viruses were stained specifically for viral proteins with an antiserum to HSV-1. Plaque assays were fixed in 4% formaldehyde in PBS for 20 min at room temperature. The cells were then permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature and incubated in PBS containing 4% FCS overnight at 4°C. Cells were then incubated with primary anti-HSV-1 rabbit serum at 1:200 for 1 h at room temperature. They were then washed three times for 5 min in PBS plus 4% FCS and incubated for 30 min in PBS containing anti-rabbit immunoglobulin antibody conjugated to horseradish peroxidase (1:1,000). The cells were finally washed three times in PBS plus 4% FCS, and after the final wash had been removed, 200 μl of 3,3′,5,5′−tetramethylbenzidine-stabilized substrate for horseradish peroxidase solution (Promega) was added. This colorigenic horseradish peroxidase substrate labeled the viral plaques blue. The plaques were then counted with a dissecting microscope under a 10× ocular objective, and viral titers were calculated as PFU per milliliter. Infections for all experiments were carried out at a multiplicity of infection of 5. Cells were seeded the day before infection and incubated at 37°C overnight. Virus was diluted in PBS containing 0.1% glucose and 1% heat-inactivated bovine serum and applied to cells for 1 h at 37°C. The inoculum was then removed and replaced with Dulbecco's modified Eagle's medium plus 10% FCS, and the cells were incubated at 37°C for the remainder of the experiment.
Plasmids and transfections.
Plasmid pJB233, expressing UL31, and plasmid pJB234, expressing UL34, have been described (22). The day before transfection, Vero cells were seeded into 24-well plates containing glass coverslips for fluorescence experiments. Transfections were carried out with plasmids pJB233 and pJB234 (0.3 μg of each) and Lipofectamine reagent (Gibco-BRL) according to the manufacturer's instructions. Cells were fixed for immunofluorescence at 24 h after transfection.
Antibodies and immunofluorescence.
Antibodies to UL31 and UL34 have been described elsewhere (22). The UL31 antiserum was preabsorbed on RD14 cells fixed in 4% formaldehyde and then permeabilized with 0.2% Triton X-100 (Sigma). Mouse monoclonal antibody to lamin A/C was obtained from Santa Cruz Biotechnology Inc. Mouse monoclonal antibody to LAP2 was obtained from BD Transduction Laboratories. Antibody to histone H1 was obtained from Upstate Biotechnology Inc. The 3-83 antiserum specific for ICP8 has been described (11). Secondary antibodies conjugated to horseradish peroxidase were obtained from Santa Cruz Biotechnology Inc. (rabbit and mouse) and Affinity Bioreagents (chicken). Secondary antibodies conjugated to Alexa 594 and Alexa 488 dyes were obtained from Molecular Probes Inc.
Vero or HEp-2 cells were seeded for immunofluorescence at 2 × 105 cells/well on glass coverslips in 24-well plates and incubated overnight at 37°C before infection or transfection experiments were performed. For infection experiments, HEp-2 cells and Vero cells were used in replicate experiments, although in each case data for only one cell type are shown for the sake of brevity. HEp-2 cells were used to show the relevance of the data to a human cell line. In all experiments, they gave results similar to those obtained with Vero cells (data not shown). Following infection or transfection, cells were fixed in 4% formaldehyde in PBS for 20 min at room temperature. Cells were permeabilized with acetone at −20°C for 2 min. Following several washes in PBS, cells were blocked overnight in IF buffer (PBS containing 4% goat serum [Sigma]). Primary antibodies were diluted appropriately and applied to cells in IF buffer, and the cells were incubated for 1 h at room temperature. UL31 and UL34 antibodies were used at 1:200, LAP2 was used at 1:200, H1 was used at 1:200, lamin A/C was used at 1:10, and ICP8 was used at 1:200. Cells were then washed three times for 5 min at room temperature in IF buffer. Secondary antibodies were applied diluted 1:1,000 in IF buffer for 30 min at room temperature. The cells were then washed three times in IF buffer for 5 min at room temperature, and coverslips were mounted with Prolong Antifade reagent (Molecular Probes Inc.).
Slides were viewed with an Axioplan 2 microscope (Zeiss) with a 100× objective and a 10× ocular objective. Images were collected with the Openlab suite of programs (Improvision) and a Hamamatsu C4742 digital camera. To collect images of representative intensity, exposure settings were equal for viewing each set of samples, the brightest sample in each case being used to set the exposure time. Images were compiled into montages with Adobe Photoshop. For image presentation, single nuclei are shown in most cases in order to show detail. In all figures except Fig. 1, these were collected with the 100× objective and 10× ocular objective, but due to cropping they may not be strictly according to scale. In Fig. 1, images are presented to scale; hence, comparisons of nuclear size are valid in this figure. Digitally enlarged images were produced with Photoshop and are not strictly to scale.
FIG. 1.

Digital images of fluorescence micrographs of mock-infected (i to iv) and HSV-1 KOS-infected (v to xvi) Vero cells. Cells were fixed at 6 h (v to viii), 10 h (ix to xii), or 18 h (xiii to xvi) postinfection (hpi) and stained with antibodies specific for histone H1 (red) and ICP8 (green). Replication compartments were labeled at 6 h (arrowheads, v to viii) and 10 h (arrows, ix to xii) postinfection. Red- and green-channel images were merged to show the staining patterns in relation to each other (iii and iv, vii and viii, xi and xii, and xv and xvi). Images were enlarged digitally in order to show detail (iv, viii, xii, and xvi).
SDS-PAGE and Western blotting.
Vero cells were infected with HSV-1 KOS strain, R5132, R5133, RR1072, or RR1072Rep at a multiplicity of infection of 5 and incubated for 18 h at 37°C. Cells were harvested in Laemmli's sample buffer containing one protease inhibitor cocktail tablet (Roche) per 10 ml on ice, and the samples were immediately incubated in a boiling water bath for 5 min. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrically transferred to a polyvinylidene difluoride membrane with the Bio-Rad Transblot system according to the manufacturer's instructions. Membranes were probed with antisera to UL31 (1:200), UL34 (1:200), lamin A/C (1:200), and LAP2 (1:5,000), washed three times for 5 min in PBS plus 0.2% Tween 20 (Sigma), and incubated with secondary antibodies conjugated to horseradish peroxidase diluted 1:10,000. The horseradish peroxidase signal was detected with enhanced chemiluminescence reagents (Amersham) on standard X-ray film (Kodak).
RESULTS
Late development of RCs to the nuclear periphery.
Coincident with the formation of RCs in the nucleus of HSV-1-infected cells, host chromatin is compressed and marginalized (Fig. 1i to iv) (16). Host chromatin is excluded from viral RCs, which occupy the center of the nucleus, and forms a layer around the nuclear periphery, underlying the nuclear lamina and the INM (16). To investigate how HSV-1 overcomes these barriers to nuclear egress, we used immunofluorescence to study the distributions of chromatin- and nuclear lamina-associated proteins in infected cells. Vero cells were infected with wild-type HSV-1 and incubated for 6, 10, or 18 h at 37°C or were mock infected before incubation. The cells were fixed and processed for immunofluorescence staining of ICP8, an HSV replication protein known to be a major component of RCs (19), along with one of the cellular proteins, histone H1 (H1, Fig. 1), LAP2 (Fig. 2), or lamin A/C (Fig. 3).
FIG. 2.

Digital images of fluorescence micrographs showing mock-infected (i to iv) and HSV-1 KOS-infected (v to viii) Vero cell nuclei. Cells were fixed at 18 h postinfection (hpi) and stained with antibodies specific to LAP2 (red) and ICP8 (green). Red- and green-channel images were merged to show the relationship between the two staining patterns (iii and iv, vii and viii). Certain images were digitally enlarged to show detail (iv and viii).
FIG. 3.

Digital images of fluorescence micrographs showing mock-infected (i to iv) and HSV-1 KOS-infected (v to viii) Vero cell nuclei. Cells were fixed at 18 h postinfection (hpi) and stained with antibodies specific for lamin A/C (red) and ICP8 (green). Red- and green-channel images were merged to show the relationship between the two staining patterns (iii and iv, vii and viii). Certain images were digitally enlarged in order to show detail (iv and viii).
We saw no staining of HSV RCs with the H1 antibody, and HSV DNA is not thought to bind H1 during lytic infection (15). Cells infected for 6 h showed formation of multiple RCs, as detected by staining for ICP8 (Fig. 1v to viii). RCs were small and occupied areas of the nucleus from which H1 staining was excluded (Fig. 1v). In cells fixed at 10 h postinfection, we observed marginalization of host chromatin to form a continuous layer around RCs (Fig. 1ix to xii). The RCs were large and well defined and occupied areas in the central portion of the nucleus which were left unstained by the H1 antibody (Fig. 1v and ix). These findings were similar to those reported previously (18, 26). Mock-infected cells showed no significant ICP8 staining (Fig. 1ii to iv). The H1 nuclear staining pattern in mock-infected cells was mostly homogenous, with slightly brighter staining at the nuclear periphery (Fig. 1i, iii, and iv), the peripheral nuclear staining likely being due to marginal distribution of inactive heterochromatin at the nuclear periphery (8). As reported previously (18), we saw changes in nuclear size and shape at late times during infection, in contrast to mock-infected cell nuclei, which remained roughly constant in size and retained a largely ovoid shape.
By 18 h postinfection, the RCs were no longer confined to the central portion of the nucleus, but extended throughout the nuclear volume, reaching the nuclear periphery (Fig. 1xiv to xvi). The H1 distribution was fragmented and no longer formed a continuous layer around the RC (Fig. 1xiii, xv, and xvi). Channels through the chromatin layer and spaces at the nuclear rim were filled with ICP8 staining (Fig. 1xv and xvi and data not shown). Thus, at late stages of viral infection, the compacted host cell chromatin layer was redistributed, allowing RCs access to the nuclear periphery. This may facilitate nucleocapsid movement from RCs to the INM.
In cells stained for nuclear lamin components, replication compartments extended through the nucleus to the periphery (Fig. 2vi; Fig. 3vi) and through the LAP2 layer, which showed a punctate distribution (Fig. 2v and vii to viii). In an enlarged image (Fig. 2viii), ICP8 staining in RCs was observed to extend into the LAP2-positive nuclear rim, filling some of the discontinuities in LAP2 staining. No colocalization of the two proteins was observed. This provided evidence that RCs extended through breaches in the LAP2 layer. In all mock-infected cells, LAP2 appeared to form a smooth, evenly stained layer, which delineated the nuclear boundary (Fig. 2i to iv). At earlier points during infection (6 and 10 h postinfection), LAP2 staining distribution resembled that in mock-infected cells (data not shown). This indicated that the changes in the structure of the LAP2 layer occurred at later times during infection and thus may have occurred specifically to facilitate primary envelopment.
Staining for lamin A/C gave results similar to those seen with LAP2 staining. In HSV-1-infected cells, staining was punctate, with clear gaps at the nuclear periphery, which contained ICP8 (Fig. 3v to viii). Mock-infected cells showed smooth peripheral staining, with an intranuclear pattern which was mostly homogenous. This intranuclear staining has been described previously as an “intranuclear veil” of lamins (9). Similar to LAP2, at earlier time points during infection, lamin A/C staining distribution resembled that in mock-infected cells (data not shown).
Thus, RC extension late in HSV-1 infection not only breaches the host chromatin layer but also reaches and appears to breach the nuclear lamina. In concert with this, significant rearrangements of host chromatin and two components of the nuclear lamina occur.
Effects of HSV-1 UL31 and UL34 mutant viruses on nuclear structure.
The gene products encoded by the HSV-1 UL31 and UL34 genes have been implicated in nuclear egress (22, 23, 25). Having observed changes induced by wild-type HSV-1 virus in the nuclear lamina and compacted host chromatin layer, we were interested to see whether these gene products played a role in these processes. Vero cells or Hep2 cells were infected with mutant viruses lacking the UL31 or UL34 gene or with rescued viruses in which these genes had been reintroduced. At 18 h postinfection, mock-infected and infected cells were fixed and processed for immunofluorescence staining of ICP8 along with H1, LAP2, or lamin A/C.
In cells infected with the UL34 deletion mutant virus R1072, large RCs were observed at 18 h postinfection, positioned in the central portion of the nucleus and surrounded by a layer of marginalized host chromatin (Fig. 4A, iv to vi). No obvious breaches in this layer were observed in the vast majority of cells. Instead, the chromatin layer was observed to be continuous around RCs, and ICP8 staining was absent from the nuclear periphery and INM (Fig. 4A, v and vi). A rescued virus containing a functional UL34 gene showed a phenotype similar to that shown above for wild-type HSV-1 KOS in that RCs were observed to extend throughout the nucleus, reaching the nuclear periphery in places (Fig. 4A, vii to ix). Concurrent with this, H1 staining showed fragmentation of the host chromatin layer, exposing the periphery at several points (Fig. 4A, ix).
FIG. 4.

Digital images of fluorescence micrographs of mock-infected (A, i to iii; B, i; and C, i) and HSV-1-infected cells. (A) Vero cells were infected with the deletion mutant viruses RR1072 (deletion of UL34; A, iv and vi) or R5132 (deletion of UL31; A, x to xii) or rescued virus RR1072Rep (rescued UL34; A, vii to ix) or R5133 (rescued UL31; A, xiii and xiv). Cells were fixed at 18 h postinfection and stained with antibodies specific for H1 (red) and ICP8 (green). (B) Vero cells were infected with the deletion mutant virus RR1072 or R5132 or with wild-type HSV-1 KOS strain (B, ii and iii). Cells were fixed at 18 h postinfection and stained with antibodies specific for LAP2 (red) and ICP8 (green). (C) Hep2 cells were infected with the deletion mutant virus RR1072 or R5132 or with wild-type HSV-1 KOS strain (C, ii and iii). Cells were fixed at 18 h postinfection and stained with antibodies specific for lamin A/C (red) and ICP8 (green). In all cases, red- and green-channel images were merged to show the relationship between the staining patterns (merge column). Images were digitally enlarged to show detail (zoom column).
A mutant virus lacking the UL31 gene, R5132, showed a phenotype similar to that of the UL34 deletion mutant virus. RCs were large and occupied the central portion of the nucleus but did not closely abut the nuclear rim (Fig. 4A, x to xii). As with the UL34-rescued virus, a UL31-rescued virus showed a wild-type phenotype, with ICP8 staining extending to the nuclear periphery through breaches in the host chromatin layer (Fig. 4A, xiii to xiv). Thus, it appears that UL31 and UL34 are required for rearrangements in the host chromatin layer at late times in viral infection.
The UL31 and UL34 deletion mutant viruses were also used to evaluate the role of these proteins in HSV-1-induced nuclear lamina rearrangements. Cells infected with the mutant viruses lacking UL31 or UL34 protein failed to show significant LAP2 or lamin A/C rearrangements (Fig. 4B, iv to ix; and Fig. 4C, iv to ix). LAP2 and lamin A/C were distributed in a continuous layer around the nuclear periphery, which was unbreached by ICP8 staining. In contrast to this, cells infected with wild-type HSV-1 showed discontinuous LAP2 and lamin A/C staining and extension of RCs to the nuclear rim, as described above (Fig. 1, 2, and 3; Fig. 4B, ii and iii; and Fig. 4C, ii and iii). Rescued viruses which expressed both UL31 and UL34 were also assessed and showed significant LAP2 and lamin A/C rearrangements, similar to those observed with wild-type HSV-1 (data not shown). Therefore, the UL31 and UL34 proteins appear to be involved in facilitating rearrangements of components of the host cell nuclear lamina during infection.
Effects of exogenous expression of UL31 and UL34 on the nuclear lamina.
To further investigate the role of the UL31 and UL34 proteins in the changes in distribution of nuclear lamina proteins during HSV-1 infection, we transfected Vero cells with plasmids encoding these genes. Untransfected cells (Fig. 5A, iv to vi, and B, iv to vi) or cells transfected with a plasmid expressing green fluorescent protein (data not shown) showed similar, normal distributions of chromatin (data not shown) and nuclear lamina proteins.
FIG. 5.

Digital images of fluorescence micrographs of Vero cells either untransfected (A, iv to vi; B, iv to vi) or transfected with plasmids expressing UL31 plus UL34 (A, i to iii; B, i to iii). Cells were fixed at 24 h posttransfection and stained with antibodies specific for lamin A/C (A, ii and iii, v and vi) or LAP2 (B, ii and iii, v and vi) and UL34 (A and B, i, iii, iv, and vi). Red and green channel images were merged to show the relative distribution of staining (merge column).
In cells transfected with both UL31- and UL34-expressing plasmids, the levels of expression of UL31 and UL34 were higher in transfected cells than in infected cells. UL31 was found at the nuclear rim (data not shown), and UL34 showed an even distribution around the nuclear periphery (Fig. 5A, i to iii, and B, i to iii). This distribution was similar to that reported previously for these proteins when exogenously expressed (22). LAP2 staining was reduced in intensity and showed punctate distribution around the nuclear rim (Fig. 5B, ii and iii). No colocalization between the viral proteins and LAP2 was observed (Fig. 5B, iii; and data not shown). Lamin A/C distribution was also altered in cells expressing UL31 and UL34 (Fig. 5A, ii and iii) in that the staining intensity was reduced, and at the nuclear rim the distribution of lamin A/C was punctate (Fig. 5A, ii and iii), in contrast to the smooth staining seen in untransfected cells (Fig. 5A, v and vi).
Thus, in keeping with data collected from cells infected with viruses expressing UL31 and UL34, these proteins, when expressed together, were capable of dramatically disrupting proteins of the nuclear lamina. The distribution of LAP2 and lamin A/C shifted from an even distribution around the nuclear boundary to a punctate distribution with reduced fluorescence intensity. No close colocalization between UL31 or UL34 and LAP2 or lamin A/C was observed. The fact that UL31 and UL34 affect the lamina in the absence of other viral components implies that these two proteins may be directly responsible for the cellular changes observed.
HSV-1 infection did not cause changes in levels of lamina proteins.
Because HSV-1 has been reported to cause a decrease in lamin A/C levels (27), it was possible that the dramatic changes in LAP2 and lamin A/C protein distribution seen during HSV-1 infection were due to degradation of the proteins. To examine the effects of HSV-1 infection on the cellular levels of these proteins, Vero cells were infected with wild-type (HSV-1), deletion mutant (d31 or d34) or rescued (31R or 34R) viruses for 18 h or mock infected (Fig. 6). Infected cell lysates were analyzed by SDS-PAGE and Western blotting. LAP2 antiserum recognized a single polypeptide species corresponding in size to that protein (55 kDa) (Fig. 6i). Lamin A/C antiserum recognized lamin A (70 kDa) and its splice variant lamin C (65 kDa) (Fig. 6ii). The UL31 and UL34 antisera recognized polypeptide species corresponding in size to those reported previously for the proteins (UL31, 34 kDa; UL34, 31 kDa; Fig. 6iii and iv).
FIG. 6.
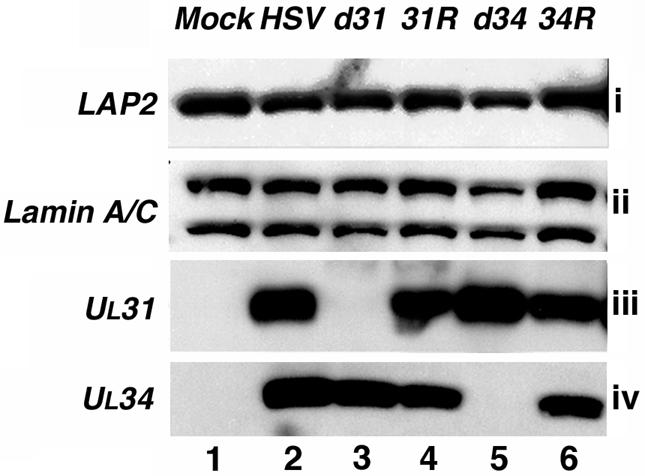
Immunoblot showing cellular levels of LAP2 (i), lamin A/C (ii), UL31 (iii), and UL34 (iv). Vero cells were either mock infected (lane 1) or infected with HSV-1 KOS (lane 2), R5132 (deletion of UL31, lane 3), R5133 (rescued UL31, lane 4), RR1072 (deletion of UL34, lane 5), or RR1072Rep (rescued UL34, lane 6) at a multiplicity of infection of 5 for 18 h. Cells were harvested, subjected to SDS-PAGE and Western blotting, and probed with antibodies specific for the proteins shown.
No significant differences in the levels of lamin A/C or LAP2 polypeptides were seen between mock-infected and wild-type HSV-1-infected cell samples (Fig. 6, i and ii). Rescued viruses which expressed both UL31 and UL34 (31R and 34R) and which exhibited changes in lamin A/C and LAP2 distribution by immunofluorescence also showed no significant changes in lamin A/C or LAP2 expression levels compared to mock-infected cell samples in this assay. Lamin A/C and LAP2 expression levels in samples from cells infected with viruses lacking the UL31 (d31) or UL34 (d34) gene were similar to those seen in mock-infected cell lysates. Thus, the changes in the structure of the nuclear lamina observed by immunofluorescence were not due to degradation of the host cell proteins studied. Rather, it appears that the observed alterations in the staining pattern of lamin A/C and LAP2 may be due to changes in the subcellular localization of components of the nuclear lamina as a result of expression of the virally encoded proteins UL31 and UL34.
DISCUSSION
Late maturation of viral replication compartments.
In this study we observed that HSV RCs mature at late times during infection and breach the marginalized host chromatin layer and the nuclear lamina in a process that requires the HSV UL31 and UL34 gene products. This allows the RCs, in which viral DNA encapsidation occurs (5, 14, 29), to expand to the nuclear envelope, where primary envelopment, the budding of the nucleocapsids through the inner nuclear membrane, takes place. The lack of access of the filled capsids to the INM in cells infected with UL31 and UL34 deletion mutant viruses could explain their previously documented defects in envelopment of nucleocapsids (22, 23, 25). Other studies have postulated the need to disrupt the nuclear lamina for herpesvirus budding (19, 27), but this is the first report to show the disruption of the marginalized host chromatin and breaching of the chromatin and lamina layers by RCs.
RC formation starts with the assembly of HSV proteins and parental viral DNA molecules near ND-10 sites (16, 31) in structures called prereplicative sites (20). As DNA replication occurs, these structures can grow by expansion and by coalescence (30). The resulting globular structures, or RCs (20), are assembled in the central portion of the host cell nucleus and displace host cell chromatin to the periphery of the nucleus (marginalization) (26). We found that this first phase of chromatin marginalization occurred during the initial phase of RC formation. This occurred in all the viruses studied and thus does not require the UL31 and UL34 proteins. In this study we show that, at later times in infection, the RCs penetrate the host chromatin and the nuclear lamina and reach a region of the nucleoplasm close to the INM. In experiments with exogenously expressed UL31 and UL34 proteins, we found that these proteins are not sufficient for fragmentation of host chromatin (data not shown). Therefore, we conclude that the initial marginalization, which we saw in all viruses, and the later fragmentation of the chromatin layer, which we saw only with wild-type and rescued viruses, constitute separate events in infected cells. We propose that marginalization is caused by the growing RC(s) and the later fragmentation is caused by the UL31 and UL34 gene products.
Changes in the distribution of nuclear lamina components.
In this study, changes in the structure of the marginalized host chromatin layer appeared to occur in parallel with changes in the structure of the nuclear lamina. We describe alterations in host cell lamin A/C and LAP2 distribution during infection with wild-type HSV-1. Both of these proteins shifted from a continuous peripheral nuclear staining pattern to a discontinuous distribution. Changes in lamin A/C distribution during HSV-1 infection have been reported previously (27). The changes reported earlier are different from those reported here but were similarly interpreted as being consistent with modification of the lamina to promote viral egress. It may be that the changes in lamin distribution seen during the late stages of HSV-1 infection are cell type specific in that previous experiments used HeLa cells, whereas Vero and HEp-2 cells were used in this study.
The changes in LAP2 distribution reported here represent novel observations for HSV-1 infection or indeed for any other virus. The fact that a transmembrane INM protein is affected along with the lamin layer implies that the structure of the lamina is affected at multiple levels; thus, infection not only affects the distribution of the fibrous layer underlying the INM but also modifies the structure of the INM. Mutant viruses lacking either the UL31 or UL34 gene are unable to alter the distribution of LAP2 and lamin A/C. Thus, during infection, UL31 and UL34 are both required for alteration of the distribution of nuclear lamina components, and in the absence of either protein, the lamina remains unmodified. In cells infected with the deletion mutant viruses, capsids faced two intact cellular barriers to nuclear egress, the compressed chromatin layer and the nuclear lamina. Egress of the deletion viruses was not completely blocked by these barriers; however, viral titers were significantly reduced (5, 25; data not shown). The host cell barriers described here may explain the growth defects documented previously for these viruses (5, 25) and the specific defect in nuclear egress previously documented for the UL34 deletion virus (25).
When expressed in the absence of other viral proteins, the UL31 and UL34 proteins were able to exert effects on the nuclear lamina similar to those documented during wild-type viral infection. Thus, the changes in lamina structure observed during wild-type virus infection may be directly attributable to the activity of these proteins in the nucleus. The effects on lamin A/C and LAP2 were more extreme during exogenous expression of the proteins than during infection, possibly due to overexpression in the transfected cells. Interestingly, alterations in the lamin A/C and LAP2 staining patterns were also seen when UL31 and UL34 were expressed separately (results not shown). This was different from the situation in cells infected with the single deletion mutant viruses, in which lamin A/C and LAP2 distribution was unchanged late during infection. One possibility is that overexpression of the individual proteins enables each of them to have some effect on the nuclear lamina.
Mechanism of altered distribution of nuclear lamina and chromatin.
Here, we describe the effects of HSV-1 infection on both the chromatin and lamina layers. We hypothesize that UL31 and UL34 cause disruption of the nuclear lamina and associated proteins and that this disruption might allow the dispersal of cellular chromatin through altered contacts between chromatin and lamina components.
No reduction in the cellular levels of lamin A/C or LAP2 was seen either during viral infection or during exogenous expression of UL31 or UL34. Thus, lamina components do not appear to be degraded during infection. Nevertheless, it should also be noted that our findings are in contrast to previous studies in which HSV-1 was observed to cause a decrease in lamin A/C expression during infection of COS cells (26). This could reflect a cell type-dependent, virally mediated lamin degradation pathway or possibly a difference in kinetics of lamin degradation in different cell types. The previous findings were conducted at 24 h postinfection, whereas our data were collected at 18 h postinfection. However, we found no alteration in lamin A/C levels at 24 h postinfection in similar experiments in Vero cells (data not shown).
Instead, the apparent change in distribution may be due to redistribution or altered antibody accessibility. The accompanying paper (21) reports that UL31 interacts with lamin A, which could cause depolymerization of lamin filaments and/or obscure a binding site for the mouse monoclonal antibody that we used to stain lamin A/C. We observed that HSV-1 infection also altered the distribution of LAP2; thus, it seems unlikely that the changes in distribution in both proteins studied are due entirely to epitope masking, but at least part of the observed effects are due to redistribution of lamina proteins.
Alternatively, it is possible that the mechanism of lamina restructuring is similar to that previously proposed for murine cytomegalovirus (19). Proteins M50 and M53, postulated to be analogous in function to UL31 and UL34, were shown to recruit the cellular kinase protein kinase C, leading to the phosphorylation of lamins. This was proposed to lead to the depolymerization, redistribution, and breaching of the nuclear lamina in order to facilitate viral egress. In preliminary experiments, we found no evidence by fluorescence to support the idea of recruitment of protein kinase C to the INM during HSV-1 infection (results not shown); however, this result does not rule out a similar mechanism for this virus.
Previously, the mechanisms by which viral nucleocapsids move out of replication compartments and reach the inner nuclear membrane were largely obscure. Here, we document an additional phase of RC maturation that leads to their extension throughout the peripheral nucleoplasm, breaching the cellular chromatin layer and the nuclear lamina to approach the inner nuclear membrane. The marginalized cellular chromatin becomes dispersed during this phase of RC maturation, and lamin A/C and LAP2 staining patterns are altered. Hence, the virus breaches these two cellular barriers to nuclear egress. An interaction between UL31 and lamin A/C appears likely to be involved in these nuclear changes, and the UL31 and UL34 gene products are required for both the chromatin dispersal and lamina rearrangements documented here. The fact that expression of both is required during viral infection supports the previously suggested hypothesis that they interact physically in this context and function as a complex (22). This is also consistent with the previously reported requirement for UL34 expression for UL31 stability (34). Hence, we propose that nucleocapsids, which are assembled in RCs, are able to move directly from these viral bodies to undergo primary envelopment at the inner nuclear membrane. Further studies should elucidate how UL31 and UL34 function to disperse the host chromatin and nuclear lamina and associated proteins to promote primary envelopment of the nucleocapsid.
Acknowledgments
We thank Bernard Roizman for providing the viral strains and cell lines used in this work. We thank R. Milne and T. Dudek for helpful discussion and advice on the manuscript and C. Boehler for excellent technical assistance.
This research was supported by NIH grants AI063106 (D.M.K), AI41478 (R.R.), and GM50740 and AI52341 (J.B.).
REFERENCES
- 1.Aebi, U., J. Cohn, L. Buhle, and L Gerace. 1986. The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323:560-564. [DOI] [PubMed] [Google Scholar]
- 2.Burke, B., and J. Ellenberg. 2002. Remodeling the walls of the nucleus. Nat. Rev. Mol. Cell Biol. 3:487-497. [DOI] [PubMed] [Google Scholar]
- 3.Chang, Y. E., C. Van Sant, P. W. Krug, A. E. Sears, and B. Roizman. 1997. The null mutant of the UL31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J. Virol. 71:8307-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, Y. E., and B. Roizman. 1993. The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J. Virol. 67:6348-6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Bruyn Kops, A., S. L. Uprichard, M. Chen, and D. M. Knipe. 1998. Comparison of the intranuclear distribution of herpes simplex virus proteins involved in various viral functions. Virology 252:162-178. [DOI] [PubMed] [Google Scholar]
- 6.Dechat, T., B. Korbei, O. A. Vaughan, S. Vlcek, C. J. Hutchison, and R. Foisner. 2000. Lamina-associated polypeptide 2α binds intranuclear A-type lamins. J. Cell Sci. 113:3473-3484. [DOI] [PubMed] [Google Scholar]
- 7.Dechat, T., S. Vlcek, and R. Foisner. 2000. Review: lamina-associated polypeptide 2 isoforms and related proteins in cell cycle-dependent nuclear structure dynamics. J. Struct. Biol. 129:335-345. [DOI] [PubMed] [Google Scholar]
- 8.Francastel, C., D. Schubeler, D. I. K. Martin, and M. Groudine. 2000. Nuclear compartmentalization and gene activity. Nat. Rev. Mol. Cell Biol. 1:137-143. [DOI] [PubMed] [Google Scholar]
- 9.Goldman, R. D., Y. Gruenbaum, R. D. Moir, D. K. Schumaker, and T. P. Spann. 2002. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 16:533-547. [DOI] [PubMed] [Google Scholar]
- 10.Hutchison, C. J. 2002. Lamins: building blocks or regulators of gene expression? Nat. Rev. Mol. Cell Biol. 3:848-858. [DOI] [PubMed] [Google Scholar]
- 11.Knipe, D. M., D. Senechek, S. A. Rice, and J. L. Smith. 1987. Stages in the nuclear association of the herpes simplex virus transcriptional activator protein ICP4. J. Virol. 61:276-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laguri, C., B. Gilquin, N. Wolff, R. Romi-Lebrun, K. Courchay, I. Callebaut, H. J. Worman, and S. Zinn-Justin. 2001. Structural characterization of the LEM motif common to three human inner nuclear membrane proteins. Structure 9:503-511. [DOI] [PubMed] [Google Scholar]
- 13.Laliberte, J. F., A. Dagenais, M. Filion, V. Bibor-Hardy, R. Simard, and A. Royal. 1984. Identification of distinct messenger RNAs for nuclear lamin C and a putative precursor of nuclear lamin A. J. Cell Biol. 98:980-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamberti, C., and S. K. Weller. 1998. The herpes simplex virus type 1 cleavage/packaging protein UL32 is involved in efficient localization of capsids to replication compartments. J. Virol. 72:2463-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leinbach, S. S., and W. C. Summers. 1980. The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J. Gen. Virol. 51:45-59. [DOI] [PubMed] [Google Scholar]
- 16.Maul, G. G., A. M. Ishov, and R. D. Everett. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67-75. [DOI] [PubMed] [Google Scholar]
- 17.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monier, K., J. C. Gonzalez Armas, S. Etteldorf, P. Ghazal, and K. F. Sullivan. 2000. Annexation of the interchromosomal space during viral infection. Nat. Cell Biol. 2:661-665. [DOI] [PubMed] [Google Scholar]
- 19.Muranyi, W., J. Haas, M. Wagner, G. Krohne, and U. H. Koszinowski. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854-857. [DOI] [PubMed] [Google Scholar]
- 20.Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857-868. [DOI] [PubMed] [Google Scholar]
- 21.Reynolds, A. E., L. Liang, and J. D. Baines. 2004. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes UL31 and UL34. J. Virol. 78:5564-5575. [DOI] [PMC free article] [PubMed]
- 22.Reynolds, A. E., B. J. Ryckman, J. D. Baines, Y. Zhou, L Liang, and R. Roller. 2001. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803-8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams and Wilkins, Philadelphia, Pa.
- 25.Roller, R. J., Y. Zhou, R. Schnetzer, J Ferguson, and D. DeSalvo. 2000. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 74:117-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz, J., and B. Roizman. 1969. Similarities and differences in the development of laboratory strains and freshly isolated strains of herpes simplex virus in HEp-2 cells: electron microscopy. J. Virol. 4:879-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott, E. S., and P. O'Hare. 2001. Fate of the inner nuclear membrane protein lamin B receptor and nuclear lamins in herpes simplex virus type 1 infection. J. Virol. 75:8818-8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment→deenvelopment→reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taus, N. S., B. Salmon, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115-125. [DOI] [PubMed] [Google Scholar]
- 30.Taylor, T. J., E. E. McNamee, C. Day, and D. M. Knipe. 2003. Herpes simples virus replication compartments can form by coalescence of smaller compartments. Virology 309:232-241. [DOI] [PubMed] [Google Scholar]
- 31.Uprichard, S. L., and D. M. Knipe. 1997. Assembly of herpes simplex virus replication proteins at two distinct intranuclear sites. Virology 229:113-125. [DOI] [PubMed] [Google Scholar]
- 32.Weber, K. 1986. Link between lamins and intermediate filaments. Nature 320:402. [DOI] [PubMed] [Google Scholar]
- 33.Worman, H. J., J. Yuan, G. Blobel, and S. D. Georgatos. 1988. A lamin B receptor in the nuclear envelope. Proc. Natl. Acad. Sci. USA 85:8531-8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamauchi, Y., C. Shiba, F. Goshima, A. Nawa, T Murata, and Y. Nishiyama. 2001. Herpes simplex virus type 2 UL34 protein requires UL31 protein for its relocation to the internal nuclear membrane in transfected cells. J. Gen. Virol. 82:1423-1428. [DOI] [PubMed] [Google Scholar]
- 35.Ye, G.-J., and B. Roizman. 2000. The essential protein encoded by the UL31 gene of herpes simplex virus 1 depends for its stability on the presence of UL34 protein. Proc. Natl. Acad. Sci. USA 97:11002-11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng, R., R. Ghirlando, M. S. Lee, K. Mizuuchi, M. Krause, and R. Craigie. 2000. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA 97:8997-9002. [DOI] [PMC free article] [PubMed] [Google Scholar]