

Abstract
Many enveloped viruses encode late assembly domains, or L domains, that facilitate virion egress. PTAP-type L domains act by recruiting the ESCRT-I (endosomal sorting complex required for transport I) component Tsg101, and YPXL/LXXLF-type L domains recruit AIP-1/ALIX, both of which are class E vacuolar protein sorting (VPS) factors, normally required for the generation of vesicles within endosomes. The binding cofactors for PPXY-type L domains have not been unambiguously resolved but may include Nedd4-like ubiquitin ligases. Largely because they act as autonomous binding sites for host factors, L domains are generally transferable and active in a context-independent manner. Ebola virus matrix protein (EbVP40) contains two overlapping L-domain motifs within the sequence ILPTAPPEYMEA. Here, we show that both motifs are required for efficient EbVP40 budding. However, upon transplantation into two different retroviral contexts, the relative contributions of the PTAP and PPEY motifs differ markedly. In a murine leukemia virus carrying the EbVP40 sequence, both motifs contributed to overall L domain activity, and budding proceeded in a partly Tsg101-independent manner. Conversely, when transplanted into the context of human immunodeficiency virus type 1 (HIV-1), EbVP40 L-domain activity was entirely due to a PTAP-Tsg101 interaction. In fact, a number of PPXY-type L domains were inactive in the context of HIV-1. Surprisingly, PTAP and YPXL-type L domains that simulated HIV-1 budding reduced the amount of ubiquitin conjugated to Gag, while inactive PPXY-type L domains increased Gag ubiquitination. These observations suggest that active L domains recruit deubiquitinating enzymes as a consequence of class E VPS factor recruitment. Moreover, context-dependent L-domain function may reflect distinct requirements for host functions during the morphogenesis of different viral particles or the underlying presence of additional, as yet undiscovered L domains.
Many enveloped viruses, including all retroviruses that have been examined (8, 20, 36, 48-51) as well as at least some rhabdoviruses (12), filoviruses (11, 25, 38, 40), arenaviruses (41), and, possibly, orthomyxoviruses (13) encode late-assembly or L domains. These are sequences within viral structural proteins that are required for the membrane fusion event that separates viral and cell membranes and thereby releases nascent virion particles. Thus far, three classes of L domains have been described, and each encodes either PT/SAP, PPXY, or YPXL/LXXLF peptide motifs. The fact that viral L domains are functionally interchangeable, in many cases, and can function when placed in various positions within a retroviral Gag protein (21, 25, 31, 51) suggests that they facilitate budding by broadly similar mechanisms involving the recruitment of host factors.
Indeed, host proteins that bind to the PTAP and YPXL/LXXLF motifs have been identified (7, 24, 39, 45, 47) and demonstrated to be required for the function of these L domains (7, 24-26, 39). PTAP motifs act by recruiting ESCRT-I (endosomal sorting complex required for transport I) via a direct interaction with the ESCRT-I component Tsg101. In addition, both the YPXL motif encoded within equine infectious anemia virus p9 and the LXXLF motif encoded within human immunodeficiency virus type 1 (HIV-1) p6 recruit AIP-1/ALIX. Both of these factors are orthologues of proteins found in Saccharomyces cerevisiae that are required for the sorting of ubiquitinated membrane proteins and the formation of vesicles that bud into the lumen of multivesicular bodies. Like other class E vacuolar protein-sorting (VPS) factors, these proteins participate in the formation of protein complexes, and the three complexes, termed ESCRT-I, -II, and -III (2, 3, 17), are linked by a series of interactions between their constituent components and other class E VPS factors, including AIP1/ALIX (16, 24, 34, 39, 46, 47). While the function of PPXY and YPDL L domains does not require ESCRT-I (7, 24, 26), a shared requirement for at least some class E VPS factors is indicated by the observation that expression of dominant negative forms of some ESCRT-III components (24, 39, 47) or the class E VPS ATPase VPS4 blocks retroviral budding mediated by each of the known L-domain motifs (7, 26, 42).
The physiological role of the class E factors appears to be the sorting of ubiquitinated plasma membrane proteins into budding vesicles that are delivered to the interior of maturing endosomes (18, 35). It is believed that the sequential recruitment of ESCRT-I, -II and -III complexes, initiated by ubiquitin (2, 3, 17), is required for this sorting and budding process. Moreover, studies in S. cerevisiae suggest that ESCRT-III-associated deubiquitinating enzyme Doa4 removes the ubiquitin from the cargoes before or concurrent with sorting into invaginating vesicles (1).
In addition to interactions with and a functional requirement for class E VPS factors, viral L domains are also thought to exploit the cellular ubiquitination machinery. Indeed, several observations suggest that ubiquitin plays a central role in viral budding, although the precise mechanism of its participation remains poorly understood. Proteasome inhibitors block the release of certain rhabdoviruses (10) and some but not all retroviruses (29, 30, 32, 33, 37, 38), perhaps by causing the depletion of free ubiquitin. Importantly, these inhibitors induce a defective viral assembly phenotype that resembles that observed in the absence of a functional L domain. Moreover, PPXY motifs in the L domains of Rous sarcoma virus (19), human T-cell leukemia virus (5), Ebola virus (11), and vesicular stomatitis virus (12)have been reported to bind to Nedd4-like E3 ubiquitin ligases and can also induce the ubiquitination of a minimal HIV-1 Gag protein (38, 40). Although a definitive role for ubiquitin ligases in retrovirus release is yet to be established, perhaps the most compelling evidence that they facilitate viral budding is derived from the observation that a Nedd4 binding peptide sequence found in a cellular protein can exhibit viral L-domain activity (38). However, while PTAP motifs are not known to bind any ubiquitin ligase, they have also been reported to induce retroviral Gag ubiquitination (38, 40).
A number of viral L domains, including that present in the Ebola virus matrix (EbVp40) protein (ILPTAPPEYMEA), contain both PT/SAP and PPXY motifs. EbVp40 is essential for viral particle assembly, and expression of this single protein in mammalian cells induces the formation of filamentous virus-like particles that exhibit a density and morphology that correspond to those of virions released by Ebola virus-infected cells (14, 27, 43). Some studies have suggested that the PTAP and PPXY motifs are redundant (22), while our own previous experiments suggested that the PTAP motif plays a dominant role in L-domain function when the aforementioned EbVP40 peptide motif is transplanted to the context of HIV-1 (25).
In this study, we found that both PTAP and PPXY motifs are essential for efficient particle release in the context of EbVp40. Surprisingly, however, we found that PPXY motifs from EbVp40 and other viral L domains are not functional when placed into the context of a full-length HIV-1 Gag protein. In murine leukemia virus (MLV) Gag, either PTAP or PPXY motifs appear to be able to mediate efficient particle release. These results indicate that the activity of PPXY type L domains is highly context dependent, and this may reflect different cofactor requirements for budding among different enveloped viruses. Despite the inability of the PPXY motifs to mediate HIV-1 particle release, they are clearly capable of inducing HIV-1 Gag ubiquitination, indicating that this modification is insufficient to induce efficient particle release in at least some contexts. Surprisingly, PTAP- and YPDL-containing L domains reduce the amount of ubiquitin that is conjugated to HIV-1 Gag in the presence or absence of a PPXY motif. This suggests that recruitment of a deubiquitinating activity might be a secondary consequence of class E VPS factor recruitment during viral release from the cell.
MATERIALS AND METHODS
Plasmid construction and mutagenesis.
A cDNA encoding the EbVp40 protein was inserted into modified versions of pCR3.1 (Invitrogen) so that the expressed protein included either a Myc epitope tag or green fluorescent protein (GFP) at its amino terminus. Single-amino-acid mutations in the EbVP40 L domain, P7L, P11L, and Y13A, were introduced by PCR. For yeast two-hybrid analyses, the wild-type and mutant EbVp40 proteins fused to a VP16 activation domain were expressed with the expression vector pVP16/HA (4), and Tsg101 fused to a Gal4 DNA binding domain was expressed with the expression vector pGBKT7 (Clontech).
The MLV proviral plasmids pNCS (MLV) and pNCS P6-PY (MLV/human PTAP [hPTAP]) (51) were a gift from Stephen Goff. The MLV/ePTAP (Ebola virus PTAP) proviral plasmid and mutants thereof were derived from pNCS by recombinant PCR methods. Plasmids expressing dominant negative forms of Tsg101, pCR3.1/YFP-1-157 and pCR3.1/YFP-303-360, have been described previously (26).
The HIV-1 proviral plasmids are based on pNL/HXB. An XhoI site was generated underlying codons 5 and 6 of p6 to facilitate mutagenesis, and this construct was named WT (25). The P7L and P11L mutants as well as HIV/ePTAP and HIV/mlvPY were derived from the wild-type proviral plasmid. The trans-complementing Gag fusion protein expression plasmids pGagδp6/p6, pGagδp6/p6(L−), pGagδp6/p9, pGagδp6/p2, pGagδp6/p12, pGagδp6/p12/hPTAP, pGagδp6/p12/δPY, and pGagδp6/ePTAP were generated by insertion of PCR products or synthetic oligonucleotides into plasmid pGagδp6, which expresses a truncated form of HIV-1 Gag in which p6 has been deleted and replaced with a multicloning site for insertion of fusion partners (25).
HIV-1 and EbVP40 particle formation assays.
293T cells were transfected with EbVP40 expression vectors or HIV proviral plasmids with Lipofectamine Plus (Invitrogen), and supernatants were harvested at 24 and 48 h after transfection for experiments involving EbVP40 and HIV-1, respectively. In both cases, the culture supernatants were clarified by low-speed centrifugation, and viral particles were harvested by centrifugation through a 20% sucrose cushion at 100,000 × g for 1.5 h. Viral proteins in cell and viral lysates were analyzed by Western blotting.
Yeast two-hybrid assays.
S. cerevisiae Y190 cells were transformed with Gal4 and VP16 fusion proteins plasmids, and protein-protein interactions were measured by β-galactosidase reporter activity as previously described (25).
Western blot analysis.
Virion and cell lysates were separated on 10 or 12% acrylamide gels, and proteins were transferred to nitrocellulose membranes. The blots were probed with primary antibodies against HIV p24 (183-H12-5C), the Myc tag (9E10), or the hemagglutinin (HA) tag (HA.11; Covance). The blots were probed with a peroxidase-conjugated secondary antibody and developed with chemiluminescent substrate reagents (Pierce).
Microscopy.
HOS cells were plated on coverslips and transfected with 400 ng of the wild type or mutant forms of pCR3.1/GFP-EbVp40. 24 h after transfection, the cells were fixed with paraformaldehyde and nuclei were stained with Hoechst 33258. The images were collected and deconvolved with a Deltavision microscope and software (Applied Precision).
MLV and HIV-1 infectivity assays.
The cell line HeLa P4/R5, which expresses CD4 and has an integrated HIV-1 long terminal repeat-lacZ reporter construct, was used as a target cell line for both MLV and HIV-1 infectivity assays. In each case, culture supernatants were harvested 48 h after transfection and clarified by low-speed centrifugation before inoculation of target cells. The β-galactosidase activity in cell lysates was determined with chemoluminescent detection reagents (Galactostar; Tropix) 48 h after infection. In experiments where HIV-1 L-domain defects were complemented in trans, 293T cells were transfected with proviral constructs containing point mutations in the PTAP motif (pNL[LTAL]) (25) or completely lacking p6 (pNLδp6) (23) along with a Gagδp6-derived complementing plasmid.
For experiments involving MLV and dominant negative Tsg101 fragments, 293T cells were transfected with the MLV proviral plasmid, a vesicular stomatitis virus G expression plasmid, and pMSCV/Tat, and pCR3.1/YFP, pCR3.1/YFP-1-157, or pCR3.1/YFP-303-360 (26). Alternatively, a GFP-expressing retroviral vector was used in place of pMSCV/Tat. In this case, Mus dunni cells were used as the target cells, and the infectivity was measured by flow cytometry. In some cases, inhibition of viral production by short interfering RNAs (siRNAs) was measured. In these instances, Lipofectamine 2000 was used for transfection and 20 pmol of siRNA duplex was included in the transfection mixture. The target sequences for the Tsg101-specific and luciferase control siRNAs were CCUCCAGUCUUCUCUGGUC (7) and CUGCCUGCGUGAGAUUCUC, respectively.
Determination of L-domain effects on HIV-1 Gag ubiquitination.
Fusion proteins consisting of a truncated form of HIV-1 Gag lacking p6 (Gagδp6) linked to various L-domain proteins or peptide motifs were expressed by transfection of 293T cells. The L-domain proteins included HIV p6 (and a mutant lacking the PTAP motif [LTAL]), equine infectious anemia virus p9, and MLV p12 (and a mutant lacking the PPPY motif [δPY]). Peptide motifs, including PTAP- and/or PPXY-containing sequences from HIV-1 p6 (hPTAP; PEPTAPPEES), EbVp40 (ePTAP; ILPTAPPEYMEA), Rous sarcoma virus p2b (TSAPPPPYVG), and the epithelial sodium transporter ENaC (TAPPPAYATLG). An HA-tagged form of ubiquitin (44) was simultaneously coexpressed. A trace amount of GFP expression plasmid was included in each transfection to monitor efficiency which was almost identical within each experiment. The cells were harvested 48 h posttransfection and lysed in a buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, 5% glycerol, 1% NP-40, 0.1% sodium dodecyl sulfate, 5 mM N-ethylmaleimide, and a protease inhibitor cocktail (Roche). The lysates were immunoprecipitated with immunoglobulins from an HIV-positive individual coupled to protein G-Sepharose. Similar experiments were done by expressing MLV Gag-GST fusion proteins in which the PPPY motif within p12 was intact or deleted (δPY). In this case, MLV Gag-GST and ubiquitinated conjugates were precipitated with glutathione-agarose beads. In both cases, bead-bound proteins were eluted by boiling in sodium dodecyl sulfate sample buffer and analyzed by Western blotting with anti-Gag, anti-GST, or anti-HA monoclonal antibodies.
RESULTS
Both PTAP and PPEY motifs are required for budding of EbVP40 particles.
We have previously shown that a 12-residue peptide sequence motif, I5LPTAPPEYMEA16, encoded within the EbVP40 matrix protein acts as a transferable L domain that can mediate the budding of HIV-1 particles when inserted into one of at least two alternative locations in the HIV-1 Gag protein (25). The integrity of the PTAP tetrapeptide, which is necessary for interaction with Tsg101, was required for L-domain function in this context. Conversely, others have previously implicated ubiquitin ligase recruitment by the overlapping PPEY motif as being important for Ebola virus L-domain function (11).
To resolve this apparent anomaly, we constructed a series of single-amino-acid substitution mutants of EbVP40, shown in Fig. 1A, in which the PTAP motif or the PPEY motif was selectively altered. As shown in Fig. 1B, mutation of either the PTAP motif (P7L) or the PPEY motif (P11L and Y13A) caused a profound defect in the release of EbVP40 into the extracellular medium as pelletable virus-like particles. To exclude the possibility that the mutations induced drastic conformational changes in EbVP40 that precluded assembly into virus-like particles, we examined the subcellular localization of GFP-EbVP40 fusion proteins bearing each of the mutations. As shown in Fig. 1C, the wild-type GFP-EbVP40 protein assembled into long filamentous structures that projected from the cell into the surrounding medium. The filaments were highly heterogeneous in length and were sometimes branched, much as has been observed with authentic Ebola virus particles. Each of the mutant GFP-EbVP40 proteins assembled into filamentous projections in the same way as did the wild-type GFP-EbVP40 protein. This result effectively excludes mislocalization or gross assembly defects as the cause of the virus-like particle release deficiency that resulted from the introduction of the mutations and defines the mutations as inducing late assembly defects.
FIG.1.

Tsg101-PTAP interaction is necessary but not sufficient to mediate the late stages of EbVP40 virus-like particle release. (A) Alignment of HIV-1 p6 and EbVP40 L-domain sequences, showing conservation of the PTAP but not PPXY motifs (boxed residues). Residues targeted for mutagenesis that selectively eliminate the PTAP motif (P7L) or the PPXY motif (P11L and Y13A) are underlined. The methionine residue in the EbVP40 sequence represents the amino terminus of the protein. (B) Western blot analysis of cell lysates and extracellular virus-like particles upon expression of Myc epitope-tagged wild-type (WT) and mutant EbVP40 proteins in 293T cells. (C) Subcellular localization and filament formation by wild-type and L-domain mutant forms of a GFP-EbVP40 fusion protein. (D) Yeast two-hybrid analysis of EbVP40-Tsg101 interaction. The chart shows β-galactosidase activity (in optical density [OD] units) in lysates of S. cerevisiae Y190 cells expressing GAL4-Tsg101 and the indicated wild-type or mutant form of VP16-EbVP40 fusion protein.
Because the PPEY motif lies in immediate proximity to, and in fact overlaps, the PTAP motif that we have previously shown to be critical for the interaction with Tsg101, we next tested whether the PPEY motif mutants affected EbVp40-Tsg101 binding. As shown in Fig. 1D, a yeast two-hybrid analysis revealed that neither of the PPEY-targeted mutations (P11L and Y13A) affected the ability of EbVP40 to bind to Tsg101. In contrast, and as we have shown previously (25), mutation of the PTAP motif (P7L) completely eliminated the ability of EbVP40 to bind to Tsg101. Thus, we conclude from these experiments that neither the PTAP nor the PPEY motif is in and of itself sufficient to mediate efficient progression of late steps in EbVP40 budding and that Tsg101 recruitment is necessary but not sufficient to mediate budding induced by the EbVP40 L domain in its natural context. These results apparently contradict those obtained by Licata et al. (22), who reported that the PTAP and PPEY motifs in EbVP40 were largely redundant. At present, the reasons for this are unclear, but we have found that the effects of L domains on EbVP40 budding are much less pronounced, with significant levels of viral protein release in the absence of an L domain at the later time points used by Licata et al.
Both PTAP and PPEY motifs in the EbVp40 L domain function in a heterologous context.
To address the contributions of the PTAP and PPEY motifs to the overall L-domain activity in a context where both would be predicted to be functional, we constructed a recombinant MLV-derived proviral plasmid in which the natural L domain was replaced with an EbVP40 derived 12-residue peptide sequence (Fig. 2A). Ordinarily, MLV encodes a PPPY motif that is responsible for L-domain activity, but, as has been reported previously, this can be functionally replaced by an HIV-1-derived PTAP-containing peptide sequence (Fig. 2A) (51). As shown in Fig. 2B, the MLV proviral plasmid encoding the HIV-1-derived PTAP sequence (MLV/hPTAP) generated high levels of infectious virus, but upon mutation of the PTAP motif (P7L), infectious virion production was reduced by approximately 10-fold, presumably due to a reduced affinity of interaction with Tsg101. The MLV proviral plasmid encoding the EbVP40-derived L-domain (MLV/ePTAP) generated levels of infectious virions similar to those generated by MLV/hPTAP, but in this case, mutation of the analogous proline residue reduced infectious virion output only by approximately threefold (Fig. 2B). Thus, in the context of an MLV Gag protein, the PTAP motif within the EbVP40 L domain is not absolutely required for function. Similarly, selective mutation of the PPEY motif (Y13A) in MLV/ePTAP resulted in an approximately two- to threefold reduction in the yield of infectious virions, indicating that the PPEY motif was also not absolutely required in this context. Mutation of both the PTAP and PPEY motifs (P7L Y13A) resulted in virion production that was 10-fold less than that of the parental MLV/ePTAP construct and similar to that of MLV/hPTAP(P7L) (Fig. 2B). Thus, in the MLV context, while neither PTAP nor PPEY motifs are absolutely required, both contribute to overall L-domain activity, and their effects are approximately additive.
FIG. 2.

L-domain activity of PTAP and PPXY motifs from the EbVP40 L domain in the context of MLV Gag. (A) Schematic representation of the MLV/hPTAP and MLV/ePTAP proviral constructs. The endogenous PPPY motif in MLV is replaced by residues from the HIV-1 PTAP L domain or the EbVP40 L domain, as indicated. Residues that were mutated in these constructs are underlined. In both cases P7L refers to the first proline residue in the PTAP motif. Coincidentally, this residue is the seventh residue from the N terminus of both HIV-1 p6 and EbVP40. (B) Effect of mutations in the transplanted HIV-1 and EbVP40 L domains on MLV virion formation. Infectious virus production by cells transfected with a GFP-expressing MLV vector, a vesicular stomatitis virus G expression vector, and the indicated wild-type (WT) or mutant MLV/hPTAP or MLV/ePTAP proviral plasmids. Infectious virions were measured by flow cytometry analysis of cells inoculated with culture supernatants and are given in infectious units (i.u.).
Role of Tsg101 in EbVP40 L-domain activity.
We have previously shown that replacement of the MLV PPPY motif with the HIV-1-derived PTAP-type L domain confers sensitivity to dominant negative forms of Tsg101 (26). To determine the contribution of Tsg101 to EbVP40 L-domain activity, we cotransfected the various MLV-derived proviral plasmids with plasmids expressing dominant negative forms of Tsg101 or Tsg101-targeted siRNAs and measured infectious virus output. As shown in Fig. 3A, Tsg101 siRNAs had only a marginal effect on wild-type MLV production. Therefore, although MLV MA does contain a PTAP motif, its potential ability to recruit Tsg101 appears to be neither necessary nor sufficient to mediate viral budding. In contrast, Tsg101 siRNAs inhibited MLV/hPTAP production by almost 100-fold. However, Tsg101 siRNAs reduced MLV/ePTAP production by only three- to fivefold, suggesting that EbVP40 L-domain activity is only partly dependent on Tsg101.
FIG. 3.
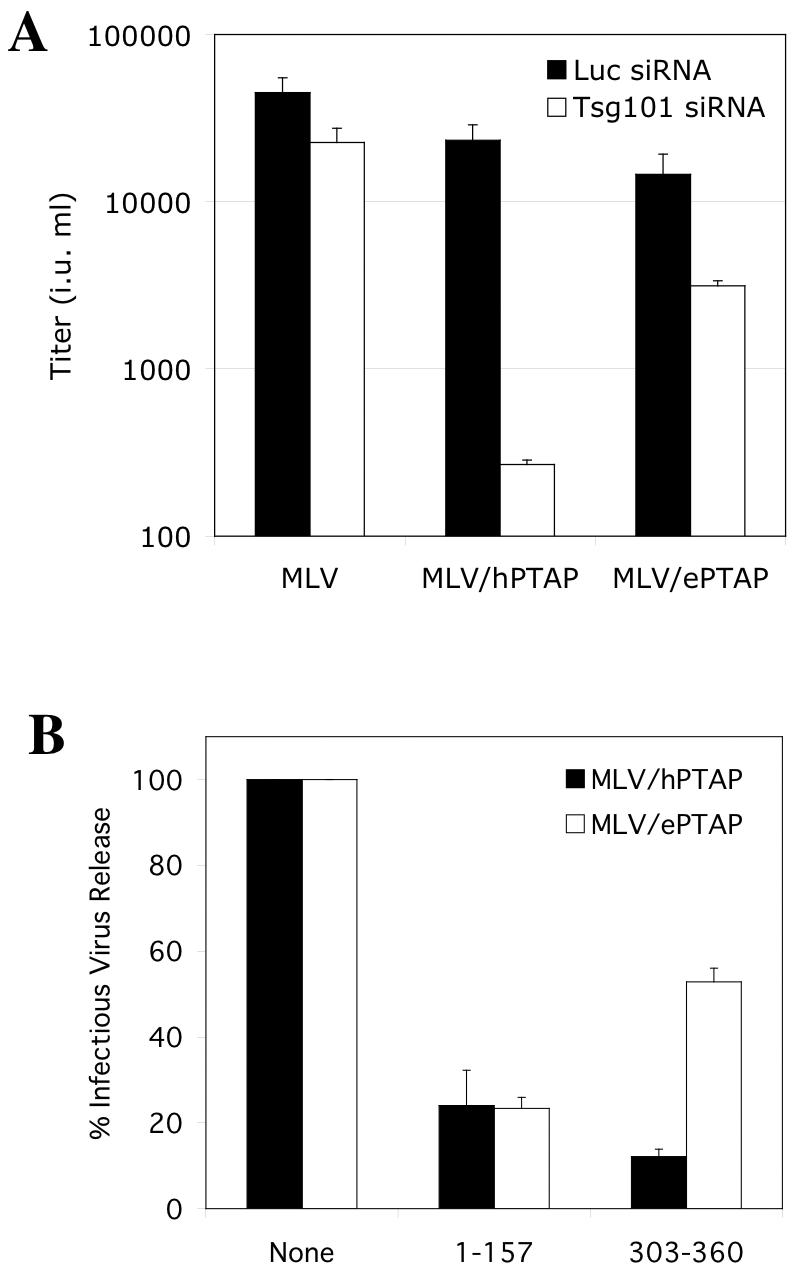
EbVP40 L-domain function is partly Tsg101 independent in the context of MLV Gag. (A) 293T cells were transfected with a GFP-expressing MLV vector, a vesicular stomatitis virus G expression plasmid, and the indicated proviral plasmids, and infectious virion production was measured as in Fig. 2. Also included in the transfection mixture were siRNA duplexes directed at firefly luciferase (Luc, as a control) or Tsg101, as indicated. (B) 293T cells were transfected with an HIV-1 Tat-expressing MLV vector, a vesicular stomatitis virus G expression plasmid, and the indicated proviral plasmids, and infectious virion production was measured by activation of β-galactosidase expression following inoculation of HeLa P4/R5 cells. Plasmids expressing dominant negative fragments of Tsg101 that inhibit PTAP-dependent HIV-1 budding were included in some transfections, and the yield of infectious virions is given as a percentage of that obtained in their absence.
Similarly, and as shown in Fig. 3B, a minimal multimerizing fragment of Tsg101 (residues 303 to 360), which acts as a dominant inhibitor of PTAP L domain function by disrupting the ESCRT-I complex (26), reduced MLV/hPTAP production by approximately eightfold and MLV/ePTAP production by only twofold. Each of these observations suggests that the PPEY motif acts independently of Tsg101 by recruiting other factors, not by influencing the context of the PTAP motif, to facilitate Tsg101 binding. Conversely, a PTAP binding Tsg101 fragment (residues 1 to 157) reduced both MLV/hPTAP and MLV/ePTAP production by approximately fivefold (Fig. 3B). Since Tsg101 (1 to 157) almost certainly acts by binding to PTAP and competitively inhibiting endogenous Tsg101 binding (6), this observation suggests that occupation of the PTAP motif in MLV/ePTAP occludes the overlapping PPEY motif and prevents the recruitment of factors that normally function by binding to it.
PPXY-type L domains cannot functionally replace a PTAP L domain in the context of an HIV-1 provirus.
Previously, we reported that the activity of the EbVP40 L-domain sequence ILPTAPPEYMEA appeared solely due to the interaction of the PTAP motif with Tsg101. This conclusion was based on experiments with a chimeric HIV-1 proviral plasmid (HIV/ePTAP) om which the HIV-1 PTAP motif was replaced with the EbVP40 L domain (25) (Fig. 4A). As shown in Fig. 4B, the chimeric HIV/ePTAP proviral plasmid generated viral particles at a level similar to that of its wild-type HIV-1 counterpart. Mutation of the PTAP motif alone (P7L) dramatically and equivalently attenuated HIV-1 virion release by both wild-type and HIV/ePTAP constructs, as we have reported previously (25). In contrast, the selective mutation of the PPXY motif (P11L) in HIV-1/ePTAP did not affect particle production (Fig. 4B), suggesting that the PPXY motif in the EbVP40 L domain does not contribute to L-domain activity in the context of an HIV-1 provirus. This was surprising, given that the same motif clearly contributes to overall L-domain activity in the context of EbVP40 (Fig. 1) and when transferred to an MLV Gag context (Fig. 2 and 3).
FIG.4.

Failure of PPXY-type L domains to support HIV-1 budding. (A) 293T cells were transfected with wild-type (WT) or mutant forms of full-length HIV-1 proviral plasmids containing the natural PTAP motif, a variant EbVP40 PTAPPEY motif, or a PPPY motif from MLV. (B) Western blot analysis with an anti-HIV-1 CA antibody of cell lysates and extracellular virions harvested by ultracentrifugation is shown; the scale on the left shows the migration of molecular size markers (in kilodaltons). (C) 293T cell were transfected with a p6-deleted HIV-1 provirus along with trans-complementing Gagδp6 protein fused to either HIV-1 p6, equine infectious anemia virus p9, Rous sarcoma virus p2, or wild-type or mutant forms of MLV p12, as indicated. Cell lysates and virion particles were analyzed by Western blotting as in panel B.
To determine whether lack of activity is a general feature of PPXY L domains when placed in the context of HIV-1, we next replaced the HIV-1 PTAP motif with a short PPPY-containing sequence derived from the MLV p12 L domain (Fig. 4A). The resulting construct (HIV-1/mlvPY) was defective for viral particle release (Fig. 4B) and infectious virus production (data not shown). This result suggested that this PPPY-containing peptide is not able to substitute for a PTAP motif to mediate HIV-1 budding.
To determine whether other PPXY-type L domains could be active in the context of HIV-1, we next used a previously described trans-complementation assay in which an HIV-1 Gag protein lacking p6 is fused to various candidate L domains and coexpressed with an L-domain-defective proviral plasmid. Gag multimerization leads to complementation of the defective provirus by a Gag protein carrying a functional L domain and the release of particles that contain processed HIV-1 Gag proteins and are infectious. This assay has the advantage that it is not limited to the analysis of short peptide sequences; larger heterologous L domains such as equine infectious anemia virus p9 or MLV p12 can be analyzed in the context of infectious HIV-1 virion production.
With this assay, we observed that either HIV-1 p6 or equine infectious anemia virus p9 L domains or PTAP-containing peptides can restore virus release by an L-domain-defective HIV-1 proviral plasmid (23, 25, 26). However, as shown in Fig. 4C, PPXY-type L domains that are active in the MLV context, Rous sarcoma virus p2b (TSAPPPPYVG) and MLV p12, were unable to stimulate particle release in the context of HIV-1. Importantly, however, a chimeric MLV p12 in which the PPPY motif was replaced with the HIV-1 PTAP motif (MLVp12/hPTAP; Fig. 1A) exhibited L-domain activity similar to that of HIV-1 p6 (Fig. 4C). Thus, while both MLV p12 and MLV p12/hPTAP can serve as L domains in the context of MLV Gag, only the latter is active in the context of HIV-1. Overall, the experiments in Fig. 4 demonstrate that PTAP motifs are active L domains in the context of full-length HIV-1, but PPXY motifs from EbVP40, Rous sarcoma virus, and MLV are not. Therefore, retroviral L domains are not universally transferable, as was previously thought.
Opposing influences of PTAP and PPXY motifs on HIV-1 Gag ubiquitination.
Previously, it has been reported that stimulation of Gag ubiquitination is a general feature of retroviral L domains (28, 38, 40). Given that PTAP motifs are active L domains in the context of HIV-1 and PPXY motifs are not (Fig. 4), we next compared the ability of each to influence the ubiquitination status of HIV-1 Gag. Ubiquitination was determined with an assay in which chimeric MLV or HIV-1 Gag proteins were coexpressed with an epitope-tagged ubiquitin expression plasmid. Thereafter, Gag was immunoprecipitated or purified from cell lysates. Conjugation of epitope-tagged ubiquitin to Gag could therefore be assessed by Western blot analysis of the immunoprecipitated or purified Gag protein.
We first applied this approach with an MLV Gag-GST fusion protein. As shown in Fig. 5A, the fusion protein formed extracellular virus-like particles efficiently (as efficiently as unfused MLV Gag; data not shown), and this activity was highly dependent on the PPXY motif that is contained therein. Intracellular MLV Gag-GST was indeed found to be ubiquitinated. As expected, this ubiquitination was at least partly dependent on the presence of the PPXY motif in p12, since MLV Gag(δPY) carried fewer ubiquitin epitopes than did the wild-type protein (Fig. 5A). A similar analysis with HIV-1 Gag revealed that a truncated protein lacking p6 (Gagδp6) carried a low but easily detectable level of ubiquitin, in the form of mono- or diubiquitinated as well as a low level of oligo- and polyubiquitinated forms (Fig. 5B) This was somewhat surprising, given that two lysine residues within p6 were thought to be the major sites of HIV-1 Gag ubiquitination (38). Even more remarkable, the addition of an HIV-1 p6-derived PTAP-containing peptide (PEPTAPPEES) to the C terminus of Gagδp6 actually reduced the level of Gag ubiquitination. In contrast, peptide sequences derived from EbVP40 (ILPTAPPEYMEA), Rous sarcoma virus p2 (TSAPPPPYVG), and the cellular sodium transporter ENaC (TAPPPAYATLG), a binding site for the Nedd4 ubiquitin ligase, each promoted HIV-1 Gag ubiquitination (Fig. 5B). Thus, PTAP- and PPXY-containing sequences apparently had opposite effects on HIV-1 Gag ubiquitination that did not necessarily correlate with their ability to stimulate virus release.
FIG. 5.

Opposite effects of L domains on MLV and HIV-1 Gag ubiquitination. (A) Wild-type (WT) and mutant (PPPY-deleted [δPY]) forms of MLV Gag fused to GST were expressed in 293T cells along with an HA-tagged form of ubiquitin. The left panels show Western blot analyses of extracellular virus-like particles and cell lysates with an anti-MLV CA antibody. The right two panels show anti-HA and anti-GST Western blot analyses of the MLV Gag-GST fusion proteins purified from cell lysates with glutathione-agarose beads; the positions of Gag GST- and HA-ubiquitin conjugates are indicated. (B) HIV-1 Gagδp6 proteins fused to the indicated viral L domains or PTAP- or PPXY-containing peptide motifs were coexpressed in 293T cells along with an HA-tagged form of ubiquitin. In this case, HIV-1 Gag was immunoprecipitated with serum from an HIV-positive patient, and the immunoprecipitates were analyzed by Western blotting with anti-HIV-1 CA and anti-HA monoclonal antibodies. The band marked with an asterisk in the blots probed with anti-HA is the immunoglobulin G heavy chain. In both A and B, the scales to the side of the blots indicate the positions of molecular size markers (in kilodaltons).
Like the HIV-1 p6-derived PTAP-containing peptide, an intact HIV-1 p6 domain clearly reduced the level of ubiquitin conjugated to HIV-1 Gag (Fig. 5B). This effect was at least partly due to the PTAP motif contained therein, because a protein carrying two substitutions in the PTAP motif (LTAL) was more heavily ubiquitinated than that containing an intact PTAP sequence. Similarly, equine infectious anemia virus p9, which is a functional L domain in the context of HIV-1, reduced the level of ubiquitin conjugated to HIV-1 Gag (Fig. 5B). Conversely, MLV p12, which was unable to stimulate virus release (Fig. 4C), potently induced HIV-1 Gag ubiquitination (Fig. 5B), and this effect was due largely to the presence of the PPXY motif, as was the case in the natural MLV Gag context (Fig. 5A). In contrast, the chimeric MLV p12/hPTAP protein did not stimulate HIV-1 Gag ubiquitination (Fig. 5B), but did stimulate virus release (Fig. 4C). In fact, overall there was no correlation between the ability of L domains to induce HIV-1 Gag ubiquitination and to support virion release. Rather, there appeared to be a better correlation between the ability of L domains to reduce the level of Gag ubiquitination (Fig. 5B) and to stimulate virus production (Fig. 4C).
Dissociation of HIV-1 release and Gag ubiquitination functions in an L domain that exhibits both activities.
The only L domain that stimulated both HIV-1 Gag ubiquitination and particle release was the EbVP40-derived sequence ILPTAPPEYMEA. However, as shown in Fig. 6, these two properties could be functionally separated, at least in the context of HIV-1 Gag. Specifically, Gagδp6/ePTAP was moderately ubiquitinated upon expression in mammalian cells (Fig. 6A) and stimulated infectious HIV-1 release by an L-domain-defective provirus with the trans-complementation assay (Fig. 6B). However, the introduction of a single-amino-acid mutation into the PTAP motif (P7L) increased the level of Gagδp6/ePTAP ubiquitination (Fig. 6A) but completely eliminated the ability of Gagδp6/ePTAP to stimulate infectious virus production (Fig. 6B). Conversely, selective mutation of the PPXY motif (P11L or Y13A) dramatically inhibited Gagδp6/ePTAP ubiquitination (Fig. 6A) but had no effect on its ability to function as an L domain (Fig. 6B). This result indicates that the virion release function of the EbVP40-derived peptide in the context of HIV-1 Gag appears to be entirely due to a PTAP-Tsg101 interaction and that the ubiquitination induced by the overlapping PPEY motif has no effect on HIV-1 release. Consistent with this, and in contrast to results obtained in the context of MLV Gag, HIV-1 virion release mediated by the EbVp40 PTAP sequence (Fig. 6C) was equivalently sensitive to siRNA-mediated Tsg101 depletion to that mediated by HIV-1 PTAP (Fig. 6D). Overall, the results in Fig. 5 and 6 indicate that HIV-1 Gag ubiquitination and L-domain activity do not correlate and that L domains that are functional in the context of HIV-1 appear to decrease rather than increase the amount of ubiquitin carried by the HIV-1 Gag protein.
FIG. 6.

Opposite effects of PTAP and PPEY motifs in the EbVP40 L domain on ubiquitination and lack of contribution of the PPEY motif to HIV-1 budding. (A) HIV-1 Gagδp6 proteins fused to the indicated wild-type (WT) and mutant forms of the EbVP40 L domain peptide (ILPTAPPEYMEA; the P7, P11, and Y13 mutated residues are in italics) were coexpressed with HA-tagged ubiquitin in 293T cells. Subsequently, HIV-1 Gag was immunoprecipitated with serum from an HIV-positive patient, and the immunoprecipitates were analyzed by Western blotting with anti-HIV-1 CA and anti-HA monoclonal antibodies, as in Fig. 5B. (B) Ability of the same Gagδp6-EbVP40 L domain fusion proteins to complement PTAP-defective HIV-1 budding. Infectious virion formation by 293T cells cotransfected with plasmids expressing a PTAP-defective HIV-1 provirus and Gagδp6 fusion proteins containing the indicated wild-type and mutant L domain motifs was measured by inoculation of HeLa P4/R5 cells. Gagδp6-p6 and Gagδp6-hPTAP were included as positive controls. (C and D) Equivalent effects of Tsg101 depletion on infectious HIV-1 virion formation mediated by HIV-1 (C) and EbVP40 (D) L-domain motifs.
DISCUSSION
In this study, we demonstrate that PPXY motifs exhibit clear context dependence in their ability to promote the release of enveloped viral particles. In EbVP40, a PPEY motif was essential for efficient virus release; in MLV, the same element was able to promote virus release but was not essential; and in HIV-1, this and several other PPXY motifs were unable to replace the L-domain function that is normally provided by a PTAP motif. Nonetheless, in the last context, all PPXY motifs examined were able to stimulate Gag ubiquitination. This finding indicates that ubiquitination of HIV-1 Gag is clearly not sufficient to promote viral egress.
In contrast to previous findings (38, 40), the ability of viral L domains to promote HIV-1 particle release appeared to correlate with their ability to reduce rather than increase the amount of ubiquitin carried by the Gag protein. This was true of both the PTAP- and YPDL-type L domains that stimulated HIV-1 particle release and reduced ubiquitination. Each of these L domains has been demonstrated to recruit class E VPS factors via direct interactions with Tsg101 and AIP1/ALIX, respectively. Although mammalian class E VPS factors have not been unequivocally demonstrated to recruit deubiquitinating enzymes, this clearly occurs in S. cerevisiae (1), and one mammalian class E VPS factor, HPB/STAM, has been reported to bind to the deubiquitinating enzyme UBPY (15). Thus, we propose that the propensity of PTAP- and YPDL-type L domains to reduce the burden of ubiquitin carried by HIV-1 Gag is due to the recruitment of deubiquitinating enzymes that occurs as a secondary consequence of class E VPS factor recruitment. However, we cannot formally exclude the possibility that the greater burden of ubiquitin carried by Gag in the absence of the functional L domain is due to occlusion of ubiquitin ligase recognition sites by class E VPS factor recruitment. Alternatively, it is possible that prolonged exposure to cytoplasmic ubiquitin ligases, due to failure to bud into the extracellular medium, might increase the levels of ubiquitin conjugated by HIV-1 Gag.
The inability of PPXY motifs to stimulate HIV-1 budding even though they do stimulate Gag ubiquitination was surprising, given that current dogma suggests that viral L domains are generally interchangeable and that stimulation of Gag ubiquitination has previously been correlated with L-domain function (38, 40). Because we and others measured only the steady-state levels of Gag ubiquitination, it remains possible that all L-domain types recruit both ubiquitin ligases and deubiquitinating enzymes and that levels of Gag ubiquitination observed are a reflection of the relative efficiency with which each is recruited by the different L domains.
Intruigingly, several of the PPXY motifs that we demonstrate to be inactive in the context of a full-length HIV-1 Gag protein have previously been shown to strongly facilitate the release of minimal HIV-1 or “mini-Gag” particles (38, 40). In fact, in the context of a mini-Gag protein, PTAP motifs were found to be inactive (40). Thus, with respect to the ability of PTAP and PPXY motifs to stimulate particle budding, mini-Gag and full-length HIV-1 Gag proteins have precisely opposite properties. This is not easily explained. However, it may be that different morphologies of budding structures during virion or virus-like particle formation might impose different requirements for host factors that are recruited by L domains that could, for example, mediate membrane curvature and/or membrane-membrane contacts as well as the membrane fusion event that ultimately separates the nascent particle from the cell.
Consistent with this idea, mutation of the PPXY motifs found in HTLV-I or Mason-Pfizer monkey virus induces a budding defect that differs from a classical L-domain defect in that budding is arrested at an earlier stage than is typical of other retroviruses harboring mutant L domains (9, 20). In addition, mutations in the MLV L domain or inhibition of MLV budding by dominant negative ESCRT-III components can lead to the formation of tubes rather than the simple membrane-tethered immature but virtually complete virions that characterize other retroviral L-domain mutants (24, 51). Taken together, these findings suggest that L domains can indeed recruit factors that play a more central role in virion morphogenesis and that the requirement for such factors may vary depending on the innate contribution of the viral structural proteins to particle morphogenesis. In their cellular context, it appears that class E VPS factors are responsible for at least three conceptually distinct activities: (i) sorting of vesicular cargo, (ii) formation of a budding vesicular structure, and (iii) fission of the vesicular membrane from the limiting membrane of the endosome. Thus, it seems plausible that different viruses might depend on these activities to different extents and could recruit different partial or complete subsets of class E VPSs via different L-domain motifs.
An additional consideration that could impact the apparent context dependence of different viral L-domain activities is the notion that multiple, as yet undiscovered, but required L domains exist within some viral proteins and that the measured activity of a particular motif is impacted by the presence or absence of others of the same or of a different class. For example, the apparent inability of PPXY motifs to mediate budding in the context of HIV-1 might mean that the Gag protein already contains an unrecognized motif that performs the same function. In such a context, a PTAP or YPDL motif might strongly stimulate budding, but an additional PPXY-type motif might be redundant. Conversely, in the context of a mini-Gag protein, which lacks a large proportion of HIV-1 Gag sequences, a PTAP motif might be inactive due to the absence of other required L-domain motifs. Strack et al. recently proposed a related notion and suggested that the NC-p1 domain of Gag might be necessary for the induction of ubiquitination and particle release function of PTAP motifs in the context of HIV-1 Gag (40). However, such considerations do not readily account for the observations that PPXY motifs strongly promote mini-Gag but not full-length HIV-1 Gag budding.
The notion that L domains can for the most part be transplanted both within and between viral proteins (31) while retaining function has proved to be a highly useful and informative concept. Nonetheless, recent evidence that both PTAP (40) and (as described herein) PPXY motifs exhibit a high degree of context dependence and have opposing effects on Gag ubiquitination suggests additional and previously unappreciated complexity in the relationship between viral and host proteins during the morphogenesis of enveloped virus particles.
Acknowledgments
We thank Stephen Goff and Dirk Bohmann for reagents and Trinty Zang and Anton Yarovoy for technical assistance.
This work was supported by the NIH (RO1AI52774, RO1AI50111) and AmFAR (02865-31). J.M.-S is the recipient of a postdoctoral fellowship from the Spanish Ministerio de Educacion, Cultura y Deporte. P.D.B. is an Elizabeth Glaser Scientist of the Elizabeth Glaser Pediatric AIDS Foundation.
REFERENCES
- 1.Amerik, A. Y., J. Nowak, S. Swaminathan, and M. Hochstrasser. 2000. The Doa4 deubiquitinating enzyme is functionally linked to the vacuolar protein-sorting and endocytic pathways. Mol. Biol. Cell 11:3365-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babst, M., D. J. Katzmann, E. J. Estepa-Sabal, T. Meerloo, and S. D. Emr. 2002. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell 3:271-282. [DOI] [PubMed] [Google Scholar]
- 3.Babst, M., D. J. Katzmann, W. B. Snyder, B. Wendland, and S. D. Emr. 2002. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev. Cell 3:283-289. [DOI] [PubMed] [Google Scholar]
- 4.Bogerd, H. P., R. A. Fridell, W. S. Blair, and B. R. Cullen. 1993. Genetic evidence that the Tat proteins of human immunodeficiency virus types 1 and 2 can multimerize in the eukaryotic cell nucleus. J. Virol. 67:5030-5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouamr, F., J. A. Melillo, M. Q. Wang, K. Nagashima, M. de Los Santos, A. Rein, and S. P. Goff. 2003. PPPYEPTAP motif is the late domain of human T-cell leukemia virus type 1 Gag and mediates its functional interaction with cellular proteins Nedd4 and Tsg101. J. Virol. 77:11882-11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demirov, D. G., A. Ono, J. M. Orenstein, and E. O. Freed. 2002. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 99:955-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garrus, J. E., U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka, and W. I. Sundquist. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for hiv-1 budding. Cell 107:55-65. [DOI] [PubMed] [Google Scholar]
- 8.Gottlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 88:3195-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gottwein, E., J. Bodem, B. Muller, A. Schmechel, H. Zentgraf, and H. G. Krausslich. 2003. The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J. Virol. 77:9474-9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harty, R. N., M. E. Brown, J. P. McGettigan, G. Wang, H. R. Jayakar, J. M. Huibregtse, M. A. Whitt, and M. J. Schnell. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75:10623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harty, R. N., M. E. Brown, G. Wang, J. Huibregtse, and F. P. Hayes. 2000. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. USA 97:13871-13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harty, R. N., J. Paragas, M. Sudol, and P. Palese. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73:2921-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hui, E. K., S. Barman, T. Y. Yang, and D. P. Nayak. 2003. Basic residues of the helix six domain of influenza virus M1 involved in nuclear translocation of M1 can be replaced by PTAP and YPDL late assembly domain motifs. J. Virol. 77:7078-7092. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Jasenosky, L. D., G. Neumann, I. Lukashevich, and Y. Kawaoka. 2001. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 75:5205-5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato, M., K. Miyazawa, and N. Kitamura. 2000. A deubiquitinating enzyme UBPY interacts with the Src homology 3 domain of Hrs-binding protein via a novel binding motif PX(V/I)(D/N)RXXKP. J. Biol. Chem. 275:37481-37487. [DOI] [PubMed] [Google Scholar]
- 16.Katoh, K., H. Shibata, H. Suzuki, A. Nara, K. Ishidoh, E. Kominami, T. Yoshimori, and M. Maki. 2003. The ALG-2-interacting protein Alix associates with CHMP4b, a human homologue of yeast Snf7 that is involved in multivesicular body sorting. J. Biol. Chem. 278:39104-39113. [DOI] [PubMed] [Google Scholar]
- 17.Katzmann, D. J., M. Babst, and S. D. Emr. 2001. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106:145-155. [DOI] [PubMed] [Google Scholar]
- 18.Katzmann, D. J., G. Odorizzi, and S. D. Emr. 2002. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3:893-905. [DOI] [PubMed] [Google Scholar]
- 19.Kikonyogo, A., F. Bouamr, M. L. Vana, Y. Xiang, A. Aiyar, C. Carter, and J. Leis. 2001. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA 98:11199-11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Blanc, I., M. C. Prevost, M. C. Dokhelar, and A. R. Rosenberg. 2002. The PPPY motif of human T-cell leukemia virus type 1 Gag protein is required early in the budding process. J. Virol. 76:10024-10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li, F., C. Chen, B. A. Puffer, and R. C. Montelaro. 2002. Functional replacement and positional dependence of homologous and heterologous L domains in equine infectious anemia virus replication. J. Virol. 76:1569-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Licata, J. M., M. Simpson-Holley, N. T. Wright, Z. Han, J. Paragas, and R. N. Harty. 2003. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77:1812-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Serrano, J., and P. D. Bieniasz. 2003. A bipartite late-budding domain in human immunodeficiency virus type 1. J. Virol. 77:12373-12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin-Serrano, J., A. Yaravoy, D. Perez-Caballero, and P. D. Bieniasz. 2003. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. USA 100:12414-12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313-1319. [DOI] [PubMed] [Google Scholar]
- 26.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2003. Role of ESCRT-I in retroviral budding. J. Virol. 77:4794-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noda, T., H. Sagara, E. Suzuki, A. Takada, H. Kida, and Y. Kawaoka. 2002. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J. Virol. 76:4855-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ott, D. E., L. V. Coren, E. N. Chertova, T. D. Gagliardi, and U. Schubert. 2000. Ubiquitination of HIV-1 and MuLV Gag. Virology 278:111-121. [DOI] [PubMed] [Google Scholar]
- 29.Ott, D. E., L. V. Coren, R. C. Sowder, 2nd, J. Adams, K. Nagashima, and U. Schubert. 2002. Equine infectious anemia virus and the ubiquitin-proteasome system. J. Virol. 76:3038-3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ott, D. E., L. V. Coren, R. C. Sowder, 2nd, J. Adams, and U. Schubert. 2003. Retroviruses have differing requirements for proteasome function in the budding process. J. Virol. 77:3384-3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parent, L. J., R. P. Bennett, R. C. Craven, T. D. Nelle, N. K. Krishna, J. B. Bowzard, C. B. Wilson, B. A. Puffer, R. C. Montelaro, and J. W. Wills. 1995. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 69:5455-5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patnaik, A., V. Chau, F. Li, R. C. Montelaro, and J. W. Wills. 2002. Budding of equine infectious anemia virus is insensitive to proteasome inhibitors. J. Virol. 76:2641-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patnaik, A., V. Chau, and J. W. Wills. 2000. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 97:13069-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peck, J. W., E. T. Bowden, and P. D. Burbelo. 2003. Structure and function of human Vps20 and Snf7 proteins. Biochem. J. 377:693-700. [DOI] [PMC free article] [PubMed]
- 35.Piper, R. C., and J. P. Luzio. 2001. Late endosomes: sorting and partitioning in multivesicular bodies. Traffic 2:612-621. [DOI] [PubMed] [Google Scholar]
- 36.Puffer, B. A., L. J. Parent, J. W. Wills, and R. C. Montelaro. 1997. Equine infectious anemia virus utilizes a YXXL motif within the late assembly domain of the Gag p9 protein. J. Virol. 71:6541-6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schubert, U., D. E. Ott, E. N. Chertova, R. Welker, U. Tessmer, M. F. Princiotta, J. R. Bennink, H. G. Krausslich, and J. W. Yewdell. 2000. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 97:13057-13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strack, B., A. Calistri, M. A. Accola, G. Palu, and H. G. Gottlinger. 2000. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 97:13063-13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strack, B., A. Calistri, S. Craig, E. Popova, and H. G. Gottlinger. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689-699. [DOI] [PubMed] [Google Scholar]
- 40.Strack, B., A. Calistri, and H. G. Gottlinger. 2002. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J. Virol. 76:5472-5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strecker, T., R. Eichler, J. Meulen, W. Weissenhorn, H. Dieter Klenk, W. Garten, and O. Lenz. 2003. Lassa virus Z protein is a matrix protein sufficient for the release of virus-like particles. J. Virol. 77:10700-10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanzi, G. O., A. J. Piefer, and P. Bates. 2003. Equine infectious anemia virus utilizes host vesicular protein sorting machinery during particle release. J. Virol. 77:8440-8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Timmins, J., S. Scianimanico, G. Schoehn, and W. Weissenhorn. 2001. Vesicular release of Ebola virus matrix protein VP40. Virology 283:1-6. [DOI] [PubMed] [Google Scholar]
- 44.Treier, M., L. M. Staszewski, and D. Bohmann. 1994. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78:787-798. [DOI] [PubMed] [Google Scholar]
- 45.VerPlank, L., F. Bouamr, T. J. LaGrassa, B. Agresta, A. Kikonyogo, J. Leis, and C. A. Carter. 2001. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 98:7724-7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vincent, O., L. Rainbow, J. Tilburn, H. N. Arst, Jr., and M. A. Penalva. 2003. YPXL/I is a protein interaction motif recognized by Aspergillus PalA and its human homologue, AIP1/Alix. Mol. Cell. Biol. 23:1647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.von Schwedler, U. K., M. Stuchell, B. Muller, D. M. Ward, H. Y. Chung, E. Morita, H. E. Wang, T. Davis, G. P. He, D. M. Cimbora, A. Scott, H. G. Krausslich, J. Kaplan, S. G. Morham, and W. I. Sundquist. 2003. The protein network of HIV budding. Cell 114:701-713. [DOI] [PubMed] [Google Scholar]
- 48.Wang, H., K. M. Norris, and L. M. Mansky. 2002. Analysis of bovine leukemia virus gag membrane targeting and late domain function. J. Virol. 76:8485-8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wills, J. W., C. E. Cameron, C. B. Wilson, Y. Xiang, R. P. Bennett, and J. Leis. 1994. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J. Virol. 68:6605-6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yasuda, J., and E. Hunter. 1998. A proline-rich motif (PPPY) in the Gag polyprotein of Mason-Pfizer monkey virus plays a maturation-independent role in virion release. J. Virol. 72:4095-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan, B., S. Campbell, E. Bacharach, A. Rein, and S. P. Goff. 2000. Infectivity of Moloney murine leukemia virus defective in late assembly events is restored by late assembly domains of other retroviruses. J. Virol. 74:7250-7260. [DOI] [PMC free article] [PubMed] [Google Scholar]