

Abstract
Cystinosis is caused by mutations in the CTNS gene (17p13.2), which encodes for a lysosomal cystine/proton symporter termed cystinosin. It is the most common cause of inherited renal Fanconi syndrome in young children. Because of its rarity, the diagnosis and specific treatment of cystinosis are frequently delayed, which has a significant impact on the overall prognosis. In this document, we have summarized expert opinions on several aspects of the disease to improve knowledge and provide guidance for diagnosis and treatment.
Keywords: CTNS gene, cysteamine treatment, cystinosis, extra-renal complications, renal Fanconi syndrome
INTRODUCTION
Cystinosis is a rare autosomal-recessive lysosomal storage disease with an incidence of ∼0.5–1.0 per 100 000 live births. Although phenotypes may overlap, three clinical forms are recognized. The most severe and frequent form, affecting ∼95% of patients, is termed infantile nephropathic cystinosis (OMIM 219800); patients generally develop clinical symptoms related to renal Fanconi syndrome during the first year of life. Juvenile or late-onset nephropathic cystinosis (OMIM 219900) is usually diagnosed later in childhood or during adolescence; patients present with milder forms of renal Fanconi syndrome or with isolated proteinuria. Finally, the ocular or adult form (OMIM 219750) is characterized by isolated symptoms related to corneal cystine crystal depositions and is rarely diagnosed before adulthood [1].
Owing to the rarity of nephropathic cystinosis, the diagnosis is often delayed; some patients are diagnosed only when they present with end-stage renal disease (ESRD). This text summarizes shared opinions among experts to increase disease awareness and to guide diagnosis and treatment. Unless specified, this document focuses on infantile nephropathic cystinosis.
MOLECULAR BASIS AND PATHOGENESIS OF CYSTINOSIS
What is the molecular basis of cystinosis?
The three clinical forms of cystinosis are caused by bi-allelic mutations in the CTNS gene (17p13.2) that encodes the lysosomal cystine transporter cystinosin. Current evidences indicate that cystinosis is a monogenic-recessive disease with complete penetrance. Severe truncating mutations cause infantile cystinosis, while milder mutations in at least one allele are usually observed in late-onset and ocular forms [2, 3]. More than 100 mutations have been reported, including deletions, small insertions, duplications, missense, nonsense and splice site mutations, mutations in the promotor sequence and genomic rearrangements (uniparental heterodisomy of chromosome 17) [4]. The most frequent mutation, affecting ∼76% of northern European alleles, is a large deletion of 57 257 base pairs involving the first 9 CTNS exons and part of exon 10 [5]. The deletion also encompasses the CARKL gene that encodes the enzyme sedoheptulokinase and the first two non-coding exons of the TRPV1 gene that encodes the transient receptor potential vanilloid 1. Patients with homozygous 57 kb deletions have increased sedoheptulose levels in tissues, serum and urine [6]. No clinical disorders related to sedoheptulose deficiency or to TRPV1 dysfunction have been reported.
Is a prenatal diagnosis possible?
Prenatal diagnosis can be reliably performed on DNA samples isolated from chorionic villi or from amniotic fluid cells [4].
How does lack of functional cystinosin translate into clinical symptoms?
Cystinosin is ubiquitously expressed on lysosomal membranes and functions as a proton/cystine symporter (1 : 1 molar ratio); cystine efflux is driven by the lysosomal proton gradient [7, 8]. In the absence of a functional cystinosin protein, cystine accumulates and crystallizes in the lysosomal lumen. The exact mechanisms that cause clinical symptoms remain incompletely understood and may differ in different tissues.
Recent studies in Ctns −/− mice have shown a decreased expression of the multi-ligand receptors megalin and cubilin in the proximal tubule apical surfaces, associated with cell dedifferentiation [9, 10]. The S1 segments adjacent to the glomeruli are particularly vulnerable and undergo enhanced apoptosis, resulting in segmental atrophy of proximal tubules, also termed ‘swan neck deformities’ [10, 11]. Enhanced cell death due to activation of pro-apoptotic signals probably also occurs in other tissues where it contributes to organ damage [12, 13]. In addition, cystinosin may play key roles in lysosomal kinetics and exocytosis [14], autophagy [15] and in sensing cell oxidative state [16]. The mechanisms by which cystinosin regulates lysosomal trafficking are unknown; involvement of specific small GTPases has recently been suggested. Specifically, expression of a constitutively active form of Rab-GTPase 27a in cystinosin-deficient cells has been shown to rescue intracellular lysosomal transport, to enhance lysosomal exocytosis and to partially decrease lysosomal cystine accumulation [14]. In this respect, a second cystinosin isoform, termed cystinosin-LKG, that is not exclusively expressed in lysosomes, may be of particular relevance; however, its exact role in the pathogenesis of the disease remains unclear [17, 18].
Cysteamine, a cystine-depleting agent, efficiently decreases apoptotic cell death and cell oxidation in vitro, but has no significant effect on the renal Fanconi syndrome [12, 13, 19].
CLINICAL PRESENTATION AND DIAGNOSIS OF NEPHROPATHIC CYSTINOSIS
What is the natural history of nephropathic cystinosis not treated with cysteamine?
Cystine accumulation begins during fetal life and affects all tissues. Cell damage and organ dysfunction, however, are heterogeneous and vary in severity and progression.
At birth, renal tubular function appears well preserved. The renal Fanconi syndrome usually manifests by 4–6 months of age with polyuria, polydipsia, failure to thrive, vomiting, constipation, dehydration, growth retardation and/or rickets, in association with biochemical evidence of proximal tubular dysfunction [1]. This includes substantial losses of electrolytes, low-molecular weight proteinuria and severe acidosis; hypophosphatemia and impaired calcitriol metabolism often cause severe rickets. Without treatment with cysteamine, renal function declines with time, causing ESRD at ∼10–12 years of age [20, 21].
Corneal cystine crystals are usually visible by a slit lamp exam after the first year of life. Cystine accumulation in thyroid follicular cells causes fibrosis, atrophy and dysfunction of the thyroid gland in 75% of patients by the age of 10 years [22]. Males develop hypergonadotropic hypogonadism [23]; testosterone replacement therapy allows pubertal development in males, but does not prevent infertility, which is invariably observed [24]. Conversely, female patients usually have pubertal retardation, but their gonadal function is not as severely compromised and successful pregnancies have been reported [25].
Pancreatic dysfunction can cause insulin-dependent diabetes mellitus in the second and third decade of life [26]. Lysosomal cystine accumulation also causes hepatomegaly and splenomegaly in approximately one-third of subjects by the age of 15 years. Hepatomegaly is related to enlargement of Kupffer's cells and foam cells containing cystine crystals [27]. Likewise, foam cells also accumulate in the red pulp of the spleen [27]. In intestinal biopsies, cystine crystals are visible in histiocytes and are enumerable in the mucosal tissue [28].
In young adults, cystine deposition in muscles causes generalized muscle atrophy and muscle weakness, initially involving the distal extremities [29].
Pulmonary dysfunction parallels the severity of the myopathy and causes severe exercise intolerance and respiratory failure.
Generally, patients have normal intelligence. However, mild neurocognitive abnormalities have been reported even in very young children and could be related to abnormalities in the cerebral white matter microstructure. Reduced white matter integrity in the parietal lobes has been demonstrated using magnetic resonance diffusion tensor imaging [30]. Relative weaknesses are often found in visual motor, visual spatial and visual memory skills, and may be associated with academic difficulties, primarily in arithmetic. Subtle motor deficits, primarily fine motor incoordination, are common in children with cystinosis and may persist even into adult life [31]. A different pattern of major central nervous system involvement represents a ‘late’ complication that rarely occurs before the age of 20 years. The main symptoms include speech impairment, memory losses, reduced intellectual functions and dementia. Despite successful renal transplantation, many patients do not survive past the age of 30 years if they have not been treated with cysteamine (see below).
When should the diagnosis of cystinosis be suspected?
Diagnosis should be made as soon as possible, because early initiation of cysteamine treatment has a considerable impact on the long-term prognosis [1, 29, 32]. It is estimated that at least 50% of children worldwide are diagnosed beyond 1 year of age, unless other affected siblings are present in the family.
Since cystinosis is the most common hereditary cause of renal Fanconi syndrome in young children, cystinosis should always be suspected in children presenting with the above listed symptoms [1, 29, 32]. Urine dipstick usually shows low specific gravity, overt glucosuria and mild albuminuria. Serum creatinine is generally normal in young children, unless patients are dehydrated [1].
How should the diagnosis of nephropathic cystinosis be confirmed?
The diagnosis can be confirmed by performing the following tests:
measurement of leukocyte cystine levels (LCL);
demonstration of corneal cystine crystals by the slit lamp exam and
genetic analysis of the CTNS gene.
Cystine crystals in the cornea may not be apparent in the first months of life, but are always present by 18 months of age [1]. Measuring LCL requires dedicated laboratories; local reference values should be used. In general, levels are >2 nmol ½ cystine/mg protein in affected patients, whereas normal subjects have LCL <0.2 nmol ½ cystine/mg protein [1].
TREATMENT OF CYSTINOSIS
Non-specific, symptomatic treatment of the renal Fanconi syndrome includes providing appropriate nutrition and substituting renal losses; these are crucial to allow satisfactory growth. After kidney transplantation patients require immunosuppressive therapies. Additional treatments may be necessary to treat extra-renal manifestations such as hypothyroidism or diabetes.
Specific cystine-depleting treatment with cysteamine currently represents the mainstay of therapy, allowing depletion of lysosomal cystine in most tissues. It should be initiated as early as possible and continued lifelong. Although cysteamine does not cure the disease, it dramatically improves the overall prognosis.
What dietary recommendations should be given to parents of infants and children with nephropathic cystinosis?
Most young children with cystinosis develop severe progressive failure to thrive, unless they receive high caloric diets [33]. Early gastric tube placement should be considered in all children who have poor appetite or frequent vomiting; gastric tubes also facilitate administration of oral medications. Jejunal tubes should be considered in some children. Patients with frequent vomiting and gastroesophageal reflux may respond to proton pump inhibitor therapy, but those with severe symptoms could also be considered for fundoplication [34].
Overall, children should receive at least 100% of the recommended diet allowance (RDA) for their age; no evidence indicates that caloric intake in excess to 150% of the RDA is useful. The composition of the diet should be balanced and should include salt and fluid supplementation, as needed.
Should indomethacin be prescribed to cystinotic patients with renal Fanconi syndrome?
Fluid and electrolyte supplementation in young patients with renal Fanconi syndrome is often challenging due to extremely high urinary losses and frequent vomiting. In 1982, Haycock et al. reported three children with cystinosis, aged 9–18 months, in whom indomethacin significantly improved polyuria and clinical symptoms [35]. Later, several other centers, mostly in Europe, reported decreased polyuria ranging between 30–70% and improved weight gain with indomethacin therapy. Indomethacin is used orally at a dose of 1–3 mg/kg/day, divided into two or three doses. It inhibits prostaglandin (PG) synthesis in the renal parenchyma. PGs, mainly PGE 2, increase the medullary blood flow, inhibit sodium chloride reabsorption in the medullary thick ascending limb and collecting duct, and decrease the expression of aquaporin 2. Thus, indomethacin enhances salt reabsorption in the loop of Henle and in the collecting duct, which helps compensate for proximal tubule losses and decreases renal salt wasting [35–37]. In normal subjects, indomethacin has limited effects on glomerular filtration rate (GFR) because synthesis of PGs is not stimulated in euvolemic states [37]. However, in patients with decreased renal perfusion, such as in renal Fanconi syndrome, indomethacin may have a detrimental effect, because local production of PGs is upregulated as part of the renal compensatory mechanisms aimed at preserving GFR. Chronic indomethacin toxicity on the renal interstitium is of particular concern, since renal lesions in cystinosis involve predominantly the tubulo interstitial compartment. Other side-effects, such as thrombocyte dysfunction or gastro intestinal complaints, are usually mild and do not preclude the use of indomethacin in cystinosis patients. For these reasons, there is currently no consensus on the use of this drug in nephropathic cystinosis. Overall, indomethacin appears to be relatively safe and well tolerated in young children in whom control of the renal Fanconi syndrome is particularly challenging. Indomethacin should be stopped if patients are dehydrated, hypotensive or if renal function deteriorates. It should not be prescribed in association with angiotensin-converting enzyme inhibitors (ACEi), which also reduce renal perfusion, although no clinical evidence is currently available. Most centers that use indomethacin in infants with nephropathic cystinosis discontinue treatment when patients grow older.
Should cystinosis patients receive ACEis or angiotensin receptor blockers (ARBs)?
ACEis and angiotensin receptor blockers (ARBs) are well-established therapies for chronic renal failure. In most cases, these drugs reduce the rate of decline in GFR, especially in glomerular diseases that are associated with proteinuria. Limited data suggest that ACEis may also decrease albuminuria and delay progression of renal failure in nephropathic cystinosis [21, 38]. However, no firm evidence currently exists supporting the systematic use of these agents in cystinosis patients. Moreover, ACEis and ARBs can reduce renal perfusion, which is usually already decreased in children with severe water and salt losses secondary to renal Fanconi syndrome. Therefore, ACEis and ARBs should be used with caution in nephropathic cystinosis. High doses should always be avoided, and doses should be reduced or stopped during hot weather.
Growth retardation in cystinosis: when should growth hormone be prescribed?
Severe growth retardation is a clinical hallmark in children with nephropathic cystinosis [27, 39–42]. In historical cohorts of patients not treated with cysteamine, only very few patients attained an adult height >150 cm [27].
Cysteamine treatment may prevent growth retardation when administered from early infancy [40, 43, 44], but does not usually induce catch-up growth in children who are already growth-retarded [21, 44]. When cysteamine, adequate nutrition and other symptomatic therapies fail to prevent growth retardation, treatment with recombinant human growth hormone (GH) should be considered, even in the absence of renal insufficiency.
GH treatment in cystinosis is safe and effective [45–47], and should be initiated early. Young children with GH treatment prior to starting renal replacement therapy show the best response [46]. The highest improvement in height is obtained within the first 3 years of treatment; thereafter GH treatment mostly allows the preservation of growth velocity [46]. Long-term GH treatment improves final height, which ranges on average from −2.6 to −2.0 SDS [21, 47]. In peri- or post-pubertal cystinosis children on renal replacement therapy, GH treatment is less likely to improve height [46].
What is the optimal dose of cysteamine?
Cysteamine was approved for clinical use in the USA and Europe in the 1990s, based on its demonstrated efficacy in reducing LCL and GFR decline [20]. Current evidence obtained in patients born between 1970 and 2000 indicates that early initiation of cysteamine delays the age of end-stage renal failure by approximately 6–10 years [1, 42, 48]. The best effects of cysteamine on renal function are obtained if the drug is started at very young ages [48].
The most widely used cysteamine preparation is cysteamine bitartrate (Cystagon®), which needs to be given every 6 h. Cys-teamine should be started at a low dose (e.g. 1 of 6 of the target dose), due to poor gastrointestinal tolerance, and increased progressively, over 4–6 weeks, to reach a target dose of 1.30 g/m2/day of free base (for patients up to 12 years) and 2 g/day for patients older than 12 years or weighting > 50 kg. LCL is traditionally used as a biomarker to monitor the effectiveness of cystine depletion, but no definitive demonstration of this approach is available at the tissue level. Depending on local reference values, the dose of cysteamine is prescribed to keep trough levels below the upper limit for heterozygous carriers, who are asymptomatic (<1 nmol ½ cystine/mg protein) [20]. The maximum dose should not exceed 1.95 g/m2/day. Non-compliance is frequent and is secondary to the acrid sulfur body and breath smell, gastrointestinal upset and tight administration schedules (every 6 h) [49]. Strategies to reduce foul odor include oral supplements of vitamin B2 (riboflavin) or the use of chlorophyll tablets to mask the odor produced by dimethyl sulfide, the culprit metabolite of cysteamine that causes bad smell. However, the benefits of these compounds have not been proved and are particularly difficult to monitor in the clinical practice; therefore, their use is purely based on subjective assessment from patients. Very rarely patients develop symmetric angioendotheliomatosis lesions on their elbows, and less frequently long-bone deformations, striae rubrae and muscular or neurological symptoms [50]. Such lesions appear in part related to excessive doses of cysteamine, so it is recommended not to exceed the maximum dose.
What is the effect of cysteamine on extra-renal manifestations of cystinosis?
Serious, non-renal, manifestations of nephropathic cystinosis develop mostly in the second decade of life, due to the continuous accumulation of cystine in tissues [1, 51]. Powerful evidence indicates that oral cysteamine lowers cystine content in muscle and other tissues, and prevents or delays late, non-renal, complications of cystinosis [29, 52, 53]. Patients aged 21–30 years that have been well treated with cysteamine have significantly decreased frequencies of myopathy, pulmonary dysfunction, diabetes and death (Figure 1) [53]. Cysteamine treatment decreases the risk of developing swallowing impairment associated with myopathy, which can be life-threatening [54], and preserves thyroid and pancreatic functions, eliminating the need for hormone replacement [55, 56]. Vascular calcifications also occur less frequently in adult patients who receive cysteamine [53].
FIGURE 1:

Percent of the patients with diabetes, myopathy, pulmonary dysfunction, death and hypothyroidism between 21 and 30 years old not treated with cysteamine (off cysteamine, blue) and well-treated with cysteamine (on cysteamine, red). There is a significance decrease in all above complications in the group of the patients well treated with cysteamine (original data provided by Dr W. Gahl and Dr G. Nesterova).
Cysteamine promotes growth and bone maturation, allowing several patients to reach normal adult height without GH therapy [1, 51, 56].
Unlike the effects on renal function, benefits of cysteamine therapy are apparent even if the drug is started later, although existing complications are generally not reversible [48]. Because this therapy was approved in the 1990s, the number of patients treated since infancy that are older than 30 years of age is still limited.
The transition of patients from pediatric to adult care providers requires special attention to meet all challenges presented by adult patients with cystinosis.
Prevention and treatment of eye complications
The pathognomonic and most frequently described ocular manifestation of cystinosis is deposition of cystine crystals in the conjunctiva and cornea [57]. Corneal crystals typically appear as needle-shaped and highly reflective crystals by a slit lamp biomicroscopy (Figure 2). They increase with age, leading to photophobia, blepharospasm, superficial punctate keratopathy and recurrent corneal erosions. In older patients, filamentous keratopathy, band keratopathy and peripheral corneal neovascularization can occur [58]. Other less frequently reported eye abnormalities include glaucoma, pigmentary retinopathy, retinal degeneration and optic nerve elevation [59].
FIGURE 2:
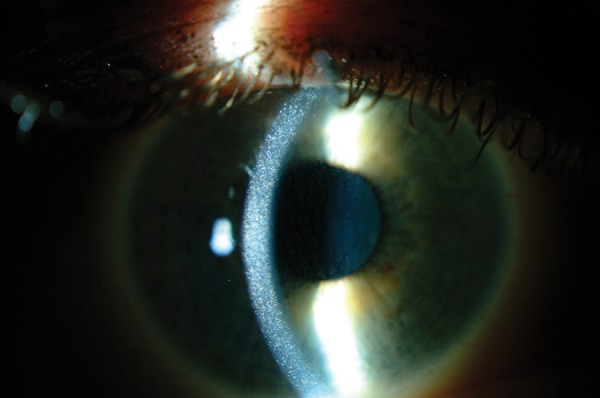
Slit-lamp photography of corneal cystine crystals. Corneal crystals appear as needle-shaped and highly reflective crystals within the cornea (original photograph provided by Dr. A. Labbé).
Oral cysteamine has no effect on corneal cystine crystals [57–59]. Patients need to be treated topically with cysteamine hydrochloride eye drops that dissolve crystals and alleviate symptoms at all ages [57, 58]. These results have been obtained with a 0.55% collyrium taken 6–10 times per day. Most patients, however, apply cysteamine eye drops less frequently (four to six times per day), in part because they cause eye burning, due to the low pH of the solution. In addition, the free thiol can oxidize to cystamine at room temperature, so it is recommended to store frozen aliquots. A commercial 0.44% cysteamine ophthalmic solution (Cystaran®) has recently been approved for clinical use in the USA. A 0.55% gel formulation (Cystadrops®) has also been developed [60]. Very rarely, patients not treated with cysteamine eye drops develop corneal lesions severe enough to require a corneal transplant. Cystine crystals can reappear in the transplanted cornea due to invading host cells, if topical cysteamine treatment is not used.
What is the renal outcome after kidney transplantation?
Renal transplantation is the treatment of choice of ESRD in cystinosis. Both living donor kidneys and cadaveric kidneys perform well. Proximal tubular disease does not recur in the transplanted kidney since the metabolic disorder is not present in the grafted organ, but native kidneys can cause renal Fanconi syndrome if they still produce urine. Protocol biopsies have shown that cystine crystals may be deposited within the interstitial tissue and less frequently within the glomeruli of the transplanted kidney, without clinical or biological manifestation. These crystals develop in the recipient mononuclear cells that infiltrate the transplant [61]. Overall, the results of kidney transplantation in cystinosis patients are better than in other patients undergoing transplantation (Figure 3) [42, 43].
FIGURE 3:

Superior 5-year renal graft survival in patients with cystinosis (NC) compared with patients with end-stage renal disease due to other causes (non-NC); adapted from Van Stralen et al. with permission [42].
Do patients with cystinosis require different immunosuppressive regimens?
Although less frequently, patients with cystinosis can experience rejection episodes if they are not appropriately treated with immunosuppressive medications.
Renal transplantation does not correct the basic metabolic defect in other tissues, and cystine continues to accumulate, causing organ damage. Immunosuppressive therapies (mainly corticosteroids and tacrolimus) may increase the risk of developing diabetes mellitus; however, the benefits of these medications exceed their risks [26].
EMERGING THERAPIES IN CYSTINOSIS
Delayed release cysteamine formulation
The pharmacokinetics (PK) of cysteamine released immediately in the stomach requires administration every 6 h around the clock, which limits compliance [49]. Using a micro-spheronized enteric-coated formulation of cysteamine bitartrate, the PK characteristics have been modified to allow 12 h dosing, with delayed-release cysteamine bitartrate being approved in the USA and Europe in 2013 (Procysbi®). In a prospective, controlled clinical trial, short-term non-inferiority for pharmacodynamics (PD) (control of LCL) of Procysbi® has been demonstrated, with reduced need for concomitant gastro-protective agent usage, and at a dose of 82% of the total daily dose of immediately released cysteamine [62]. Recent prospective, controlled data demonstrate continued PK and PD efficacy after 24 months of treatment [63]).
Gene therapy
The two major challenges for gene therapy in nephropathic cystinosis are that CTNS is expressed in all tissues and that cystinosin is a lysosomal transmembrane protein. To this end, a mouse model of cystinosis [64] has been used to test adult bone marrow stem cells as vehicles to bring wild-type CTNS to tissues. Hematopoietic stem cell (HSC) transplantation leads to abundant integration of bone marrow-derived cells in tissues and produces a significant decrease in tissue cystine content (94%—in the liver, 70%—in the kidney and 57%—in the brain) [65]. This treatment also results in long-term preservation of kidney function in mice [66]. These exciting findings usher in a new concept that HSCs might prevent serious consequences of systemic diseases such as cystinosis and probably involve paracrine effects of healthy HSC-derived cells on adjacent host cells [67]. Because allogeneic transplantation is associated with significant morbidity and mortality, research is currently focused in developing an autologous HSC transplantation strategy whereby the patient's own stem cells are gene-modified ex vivo using a lentiviral vector and re-injected into the patient to introduce a functional CTNS copy in tissues. Harrison et al. have established proof-of-concept for this approach in the Ctns −/− mice, showing that gene-modified HSCs can induce significant decrease in tissue cystine content and improvement in renal function [68]. If successful, autologous transplantation of gene-modified HSCs may represent definitive therapy. However, many studies have reported discrepancies between mice and man in the efficacy of treatments [69]. Enthusiasms should be therefore tempered until proof is established that this strategy can be safely and efficiently applied to humans.
SUMMARY
Cystinosis is a partially treatable genetic disorder that should be recognized as early as possible, because most of its clinical manifestations can be prevented or retarded with cysteamine. In awaiting novel therapies, cysteamine should be used in all patients, regardless of age and renal transplant status.
CONFLICT OF INTEREST STATEMENT
None declared.
ACKNOWLEDGEMENTS
E.L. is supported by the fund for Scientific Research, Flanders (F.W.O. Vlaanderen) grant 1801110N. W.G. and G.N. were supported in part by the Intramural Research Program of the National Human Genome Research Institute. C.A., P.G., F.E., S.C. and E.L. are supported by the Cystinosis Research Foundation. This manuscript is developed as a part of collaboration between Working Groups on Inherited Kidney Disorders of European Society of Pediatric Nephrology (ESPN) and European Renal Association – European Dialysis and Transplantation Association (ERA-EDTA).
REFERENCES
- 1.Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347:111–121. doi: 10.1056/NEJMra020552. [DOI] [PubMed] [Google Scholar]
- 2.Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- 3.Kalatzis V, Nevo N, Cherqui S, et al. Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum Mol Genet. 2004;13:1361–1371. doi: 10.1093/hmg/ddh152. [DOI] [PubMed] [Google Scholar]
- 4.Levtchenko E, van den Heuvel L, Emma F, et al. Clinical utility gene card for: cystinosis. Eur J Hum Genet. 2013 doi: 10.1038/ejhg.2013.204. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forestier L, Jean G, Attard M, et al. Molecular characterization of CTNS deletions in nephropathic cystinosis: development of a PCR-based detection assay. Am J Hum Genet. 1999;65:353–359. doi: 10.1086/302509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wamelink MM, Struys EA, Jansen EE. Sedoheptulokinase deficiency due to a 57-kb deletion in cystinosis patients causes urinary accumulation of sedoheptulose: elucidation of the CARKL gene. Hum Mutat. 2008;29:532–536. doi: 10.1002/humu.20685. [DOI] [PubMed] [Google Scholar]
- 7.Kalatzis V, Cherqui S, Antignac C, et al. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20:5940–5949. doi: 10.1093/emboj/20.21.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruivo R, Bellenchi GC, Chen X, et al. Mechanism of proton/substrate coupling in the heptahelical lysosomal transporter cystinosin. Proc Natl Acad Sci U S A. 2012;109:E210–E217. doi: 10.1073/pnas.1115581109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raggi C, Luciani A, Nevo N, et al. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddt617. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 10.Chevronnay HP, Janssens V, Van Der Smissen P, et al. Time-course of pathogenic and adaptive mechanisms in cystinotic 1 mice kidneys. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2013060598. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahoney CP, Striker GE. Early development of the renal lesions in infantile cystinosis. Pediatr Nephrol. 2000;15:50–56. doi: 10.1007/pl00013448. [DOI] [PubMed] [Google Scholar]
- 12.Park M, Helip-Wooley A, Thoene J. Lysosomal cystine storage augments apoptosis in cultured human fibroblasts and renal tubular epithelial cells. J Am Soc Nephrol. 2002;13:2878–2887. doi: 10.1097/01.asn.0000036867.49866.59. [DOI] [PubMed] [Google Scholar]
- 13.Park MA, Pejovic V, Kerisit KG, et al. Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase Cdelta. J Am Soc Nephrol. 2006;17:3167–3175. doi: 10.1681/ASN.2006050474. [DOI] [PubMed] [Google Scholar]
- 14.Johnson JL, Napolitano G, Monfregola J, et al. Upregulation of the Rab27a-dependent trafficking and secretory mechanisms improves lysosomal transport, alleviates endoplasmic reticulum stress, and reduces lysosome overload in cystinosis. Mol Cell Biol. 2013;33:2950–2962. doi: 10.1128/MCB.00417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sansanwal P, Yen B, Gahl WA, et al. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol. 2010;21:272–283. doi: 10.1681/ASN.2009040383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellomo F, Corallini S, Pastore A, et al. Modulation of CTNS gene expression by intracellular thiols. Free Radic Biol Med. 2010;48:865–872. doi: 10.1016/j.freeradbiomed.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Taranta A, Petrini S, Palma A, et al. Identification and subcellular localization of a new cystinosin isoform. Am J Physiol Renal Physiol. 2008;294:1101–1108. doi: 10.1152/ajprenal.00413.2007. [DOI] [PubMed] [Google Scholar]
- 18.Taranta A, Petrini S, Citti A, et al. Distribution of cystinosin-LKG in human tissues. Histochem Cell Biol. 2012;138:351–363. doi: 10.1007/s00418-012-0958-8. [DOI] [PubMed] [Google Scholar]
- 19.Wilmer MJ, Kluijtmans LA, van der Velden TJ, et al. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim Biophys Acta. 2011;1812:643–651. doi: 10.1016/j.bbadis.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Markello TC, Bernardini IM, Gahl WA. Improved renal function in children with cystinosis treated with cysteamine. New Engl J Med. 1993;328:1157–1162. doi: 10.1056/NEJM199304223281604. [DOI] [PubMed] [Google Scholar]
- 21.Greco M, Brugnara M, Zaffanello M, et al. Long-term outcome of nephropathic cystinosis: a 20-year single-center experience. Pediatr Nephrol. 2010;25:2459–2467. doi: 10.1007/s00467-010-1641-8. [DOI] [PubMed] [Google Scholar]
- 22.Lucky AW, Howley PM, Megyesi K, et al. Endocrine studies in cystinosis: compensated primary hypothyroidism. J Pediatr. 1977;91:204–210. doi: 10.1016/s0022-3476(77)80813-5. [DOI] [PubMed] [Google Scholar]
- 23.Chik CL, Friedman A, Merriam GR, et al. Pituitary-testicular function in nephropathic cystinosis. Ann Intern Med. 1993;119:568–575. doi: 10.7326/0003-4819-119-7_part_1-199310010-00004. [DOI] [PubMed] [Google Scholar]
- 24.Besouw MT, Kremer JA, Janssen MC, et al. Fertility status in male cystinosis patients treated with cysteamine. Fertil Steril. 2010;93:1880–1883. doi: 10.1016/j.fertnstert.2008.12.113. [DOI] [PubMed] [Google Scholar]
- 25.Reiss RE, Kuwabara T, Smith ML, et al. Successful pregnancy despite placental cystine crystals in a woman with nephropathic cystinosis. N Engl J Med. 1988;319:223–226. doi: 10.1056/NEJM198807283190406. [DOI] [PubMed] [Google Scholar]
- 26.Robert JJ, Tête MJ, Guest G, et al. Diabetes mellitus in patients with infantile cystinosis after renal transplantation. Pediatr Nephrol. 1999;13:524–529. doi: 10.1007/s004670050651. [DOI] [PubMed] [Google Scholar]
- 27.Broyer M, Tete MJ, Gubler MC. Late symptoms in infantile cystinosis. Pediatr Nephrol. 1987;1:519–524. doi: 10.1007/BF00849263. [DOI] [PubMed] [Google Scholar]
- 28.Dohil R, Carrigg A, Newbury R. A potential new method to estimate tissue cystine content in nephropathic cystinosis. J Pediatr. 2012;161:531–535.e1. doi: 10.1016/j.jpeds.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Nesterova G, Gahl W. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol. 2013;28:51–59. doi: 10.1007/s00467-012-2242-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bava S, Theilmann RJ, Sach M, et al. Developmental changes in cerebral white matter microstructure in a disorder of lysosomal storage. Cortex. 2010;46:206–216. doi: 10.1016/j.cortex.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trauner DA, Spilkin AM, Williams J, et al. Specific cognitive deficits in young children with cystinosis: evidence for an early effect of the cystinosin gene on neural function. J Pediatr. 2007;151:192–196. doi: 10.1016/j.jpeds.2007.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilmer MJ, Schoeber JP, van den Heuvel LP, et al. Cystinosis: practical tools for diagnosis and treatment. Pediatr Nephrol. 2011;26:205–215. doi: 10.1007/s00467-010-1627-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elenberg E, Norling LL, Kleinman RE, et al. Feeding problems in cystinosis. Pediatr Nephrol. 1998;5:365–370. doi: 10.1007/s004670050467. [DOI] [PubMed] [Google Scholar]
- 34.Dohil R, Fidler M, Barshop B, et al. Esomeprazole therapy for gastric acid hypersecretion in children with cystinosis receiving cysteamine. Pediatr Nephrol. 2005;20:1786–1793. doi: 10.1007/s00467-005-2027-1. [DOI] [PubMed] [Google Scholar]
- 35.Haycock GB, Al-Dahhan J, Mak RH, et al. Effect of indomethacin on clinical progress and renal function in cystinosis. Arch Dis Child. 1982;57:934–939. doi: 10.1136/adc.57.12.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Usberti M, Pecoraro C, Federico S, et al. Mechanism of action of indomethacin in tubular defects. Pediatrics. 1985;75:501–507. [PubMed] [Google Scholar]
- 37.Bennett WM, Henrich WL, Stoff JS. The renal effects of nonsteroidal anti-inflammatory drugs: summary and recommendations. Am J Kidney Dis. 1996;28:S56–S62. doi: 10.1016/s0272-6386(96)90570-3. [DOI] [PubMed] [Google Scholar]
- 38.Levtchenko E, Blom H, Wilmer M, et al. ACE inhibitor enalapril diminishes albuminuria in patients with cystinosis. Clin Nephrol. 2003;60:386–389. doi: 10.5414/cnp60386. [DOI] [PubMed] [Google Scholar]
- 39.Broyer M, Guillot M, Gubler MC, et al. Infantile cystinosis: a reappraisal of early and late symptoms. Adv Nephrol Necker Hosp. 1981;10:137–166. [PubMed] [Google Scholar]
- 40.Gahl WA, Reed GF, Thoene JG, et al. Cysteamine therapy for children with nephropathic cystinosis. New Engl J Med. 1987;316:971–977. doi: 10.1056/NEJM198704163161602. [DOI] [PubMed] [Google Scholar]
- 41.Winkler L, Offner G, Krull F, et al. Growth and pubertal development in nephropathic cystinosis. Eur J Pediar. 1993;152:244–249. doi: 10.1007/BF01956154. [DOI] [PubMed] [Google Scholar]
- 42.Van Stralen KJ, Emma F, Jager KJ, et al. Improvement in the renal prognosis in nephropathic cystinosis. Clin J Am Soc Nephrol. 2011;6:2485–2491. doi: 10.2215/CJN.02000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ehrich JH, Brodehl J, Byrd DI, et al. Renal transplantation in 22 children with nephropathic cystinosis. Pediatr Nephrol. 1991;5:708–714. doi: 10.1007/BF00857880. [DOI] [PubMed] [Google Scholar]
- 44.van't Hoff WG, Gretz N. The treatment of cystinosis with cysteamine and phosphocysteamine in the United Kingdom and Eire. Pediatr Nephrol. 1995;9:685–689. doi: 10.1007/BF00868711. [DOI] [PubMed] [Google Scholar]
- 45.Wuhl E, Haffner D, Gretz N, et al. Treatment with recombinant human growth hormone in short children with nephropathic cystinosis: no evidence for increased deterioration rate of renal function. The European Study Group on Growth Hormone Treatment in Short Children with Nephropathic Cystinosis. Pediatr Res. 1998;43:484–488. doi: 10.1203/00006450-199804000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Wuhl E, Haffner D, Offner G, et al. Long-term treatment with growth hormone in short children with nephropathic cystinosis. J Pediatr. 2001;138:880–887. doi: 10.1067/mpd.2001.113263. [DOI] [PubMed] [Google Scholar]
- 47.Besouw MT, Van Dyck M, Francois I, et al. Detailed studies of growth hormone secretion in cystinosis patients. Pediatr Nephrol. 2012;27:2123–2127. doi: 10.1007/s00467-012-2213-x. [DOI] [PubMed] [Google Scholar]
- 48.Brodin-Sartorius A, Tête MJ, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012;81:179–189. doi: 10.1038/ki.2011.277. [DOI] [PubMed] [Google Scholar]
- 49.Levtchenko EN, van Dael CM, de Graaf-Hess AC, et al. Strict cysteamine dose regimen is required to prevent nocturnal cystine accumulation in cystinosis. Pediatr Nephrol. 2006;21:110–113. doi: 10.1007/s00467-005-2052-0. [DOI] [PubMed] [Google Scholar]
- 50.Besouw MT, Bowker R, Dutertre JP, et al. Cysteamine toxicity in patients with cystinosis. J Pediatr. 2011;159:1004–1011. doi: 10.1016/j.jpeds.2011.05.057. [DOI] [PubMed] [Google Scholar]
- 51.Gahl WA, Thoene J, Schneider JA. Cystinosis: a disorder of lysosomal membrane transport. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill, c.; 2001. pp. 5085–5108. 199. [Google Scholar]
- 52.Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008;23:863–878. doi: 10.1007/s00467-007-0650-8. [DOI] [PubMed] [Google Scholar]
- 53.Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. 2007;147:242–250. doi: 10.7326/0003-4819-147-4-200708210-00006. [DOI] [PubMed] [Google Scholar]
- 54.Sonies BC, Almajid P, Kleta R, et al. Swallowing dysfunction in 101 patients with nephropathic cystinosis: benefit of long-term cysteamine therapy. Medicine (Baltimore) 2005;84:137–146. doi: 10.1097/01.md.0000164204.00159.d4. [DOI] [PubMed] [Google Scholar]
- 55.Kimonis VE, Troendle J, Rose SR, et al. Effects of early cysteamine therapy on thyroid function and growth in nephropathic cystinosis. J Clin Endocrinol Metab. 1995;80:3257–3261. doi: 10.1210/jcem.80.11.7593434. [DOI] [PubMed] [Google Scholar]
- 56.Kleta R, Bernardini I, Ueda M, et al. Long-term follow-up of well-treated nephropathic cystinosis patients. J Pediatr. 2004;145:555–560. doi: 10.1016/j.jpeds.2004.03.056. [DOI] [PubMed] [Google Scholar]
- 57.Gahl WA, Kuehl EM, Iwata F, et al. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol Genet Metab. 2000;71:100–120. doi: 10.1006/mgme.2000.3062. [DOI] [PubMed] [Google Scholar]
- 58.Tsilou E, Zhou M, Gahl W, et al. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: report of a case and review of the literature. Surv Ophthalmol. 2007;52:97–105. doi: 10.1016/j.survophthal.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Labbé A, Niaudet P, Loirat C, et al. In vivo confocal microscopy and anterior segment optical coherence tomography analysis of the cornea in nephropathic cystinosis. Ophthalmology. 2009;116:870–876. doi: 10.1016/j.ophtha.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 60.Labbé A, Baudouin C, Deschênes G, et al. A new gel formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis: the Cystadrops OCT-1 study. Mol Genet Metab. 2014;111:314–320. doi: 10.1016/j.ymgme.2013.12.298. [DOI] [PubMed] [Google Scholar]
- 61.Spear GS, Gubler MC, Habib R, et al. Renal allografts in cystinosis and mesangial demography. Clin Nephrol. 1989;32:256–261. [PubMed] [Google Scholar]
- 62.Langman CB, Greenbaum LA, Sarwal M, et al. A randomized controlled crossover trial with delayed-release cysteamine bitartrate in nephropathic cystinosis: effectiveness on white blood cell cystine levels and comparison of safety. Clin J Am Soc Nephrol. 2012;7:1112–1120. doi: 10.2215/CJN.12321211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langman C, Rioux P, Greenbaum L, et al. Extended treatment of patients with cystinosis and CKD with RP103 demonstrates efficacy and safety. Pediatr Nephrol. 2013;28:1363. [Google Scholar]
- 64.Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 2010;25:1059–1066. doi: 10.1093/ndt/gfp553. [DOI] [PubMed] [Google Scholar]
- 65.Syres K, Harrison F, Tadlock M, et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood. 2009;114:2542–2552. doi: 10.1182/blood-2009-03-213934. [DOI] [PubMed] [Google Scholar]
- 66.Yeagy BA, Harrison F, Gubler MC, et al. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 2011;79:1198–1206. doi: 10.1038/ki.2010.537. [DOI] [PubMed] [Google Scholar]
- 67.Iglesias DM, El-Kares R, Taranta A, et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS One. 2012;7:e42840. doi: 10.1371/journal.pone.0042840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harrison F, Yeagy BA, Rocca CJ, et al. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol Ther. 2013;21:433–444. doi: 10.1038/mt.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van der Worp HB, Howells DW, Sena ES, et al. Can animal models of disease reliably inform human studies? PLoS Med. 2010;7:e1000245. doi: 10.1371/journal.pmed.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]