

Highlights
-
•
We performed a high-throughput screen of 60,000 compounds against A. fumigatus chitinase A1.
-
•
Novel low micromolar competitive inhibitors were identified.
-
•
These represent the most potent selective plant-type A. fumigatus chitinase inhibitors to date.
-
•
We provide new tools for probing chitinase inhibition in A. fumigatus and other fungi.
Abbreviations: HTS, high-throughput screen/screening; GlcNAc, N-acetylglucosamine; AfChiA1, Aspergillus fumigatus chitinase A1; ScCTS1, Saccharomyces cerevisiae chitinase 1; AfChiB, Aspergillus fumigatus chitinase B1; AMCase, acidic mammalian chitinase; HsCHT, Homo sapiens chitotriosidase (chitinase 1); PI, percentage inhibition
Keywords: Chitinases, Inhibitors, High-throughput screen (HTS), Aspergillus fumigatus
Abstract
A limited therapeutic arsenal against increasing clinical disease due to Aspergillus spp. necessitates urgent characterisation of new antifungal targets. Here we describe the discovery of novel, low micromolar chemical inhibitors of Aspergillus fumigatus family 18 plant-type chitinase A1 (AfChiA1) by high-throughput screening (HTS). Analysis of the binding mode by X-ray crystallography confirmed competitive inhibition and kinetic studies revealed two compounds with selectivity towards fungal plant-type chitinases. These inhibitors provide new chemical tools to probe the effects of chitinase inhibition on A. fumigatus growth and virulence, presenting attractive starting points for the development of further potent drug-like molecules.
1. Introduction
Aspergillus fumigatus is a filamentous opportunistic fungus and regarded as one of the most pernicious pathogens in immunosuppressed individuals. The spectrum and burden of clinical disease due to A. fumigatus is becoming increasingly recognised [1], [2]. Neutropenia in patients with haematological malignancies remains an important risk factor for invasive pulmonary aspergillosis (IPA). Despite therapeutic management, overall mortality remains around 50% for IPA [1], increasing up to 90% in disseminated disease [3]. Evidence is emerging that chronic pulmonary aspergillosis, affecting approximately 3–5 million patients globally [1], [4], may be alleviated by adjunct long-term oral antifungal therapy [5]. These contrasting infections rely on a limited repertoire of antifungal classes (polyenes, azoles and echinocandins), none of which are without significant drawbacks in terms of toxicity, drug–drug interactions and/or efficacy [6], [7], [8]. Azole resistance through one of several lanosterol 14 α-demethylase (cyp51A) mutations or environmental sources is an increasing concern [9]. With no new antifungal classes in the immediate pipeline, what is urgently needed is the characterisation of targets possessing novel modes of action [10].
The fungal cell wall is a complex polysaccharide composed predominantly of chitin, β-(1,3) glucan and galactomannan that provides structural stability and is essential for survival [11]. Chitin, a linear polymer of β-(1,4) linked N-acetylglucosamine (GlcNAc) forms the rigid inner layer of the cell wall and is also partially hydrolysed during morphogenesis [11]. This is performed by glycoside hydrolase family 18 (GH18) chitinases that catalyse the cleavage of β-(1,4) glycosidic bonds between GlcNAc residues. While GH18 chitinases are found in all kingdoms of life [12], chitin is absent in mammalian cells and these enzymes have been considered potential drug targets [13].
In A. fumigatus there are 17 chitinase genes phylogenetically divided into three subgroups forming two broad families [14], [15], [16]: subgroup B “plant-type” (chiA1–5, class III) and subgroup A/C “bacterial-type” (chiB1–12, class V). The latter, found most frequently in bacteria and also humans, are almost exclusively soluble secreted enzymes with exochitinase activity and unclear physiological function [11]. In contrast, “plant-type” enzymes are predominantly cell wall associated endochitinases. These endochitinases are potentially attractive antifungal targets, as they (i) provide selectivity since the distant human orthologues are non-essential and (ii) are extracellular. Nevertheless, chitinases pose a formidable challenge as a family containing presumably redundant genes. Comprehensive genetic validation involving targeted disruption of all 17 genes in a single strain is not feasible. Neither a quintuple mutant deleting all five plant-type chitinase genes [16] nor a single deletion of chiB1 in A. fumigatus yielded a significant phenotype under standard growth conditions [17]. Interestingly, recent work suggests chitinases may be involved in biofilm maturation [18]. A fungal biofilm is defined as a surface associated, highly structured community of hyphae encased by a polysaccharide extracellular matrix [19], [20]. Most clinical infections are biofilm related and, taken together, this finding sparked further investigation into chitinases as potential antifungal targets. A crucial first step is the generation of potent chemical tools to probe concurrent inhibition of all A. fumigatus chitinases, and thus allow for investigation of chemical validation as an alternative to genetic validation.
The natural product allosamidin was the first chitinase inhibitor reported [21]. This pseudotrisaccharide structurally mimics chitin and competitively inhibits all characterised GH18 family chitinases, albeit in the mid-micromolar range for the plant-type chitinase class. Limited availability and unfavourable chemical characteristics preclude use as a tool for chemical validation. Crucially, within the separate two families, A. fumigatus chitinases possess highly conserved active sites [22], [23], suggesting that it may be possible to generate separate pan-AfChiAx and pan-AfChiBx inhibitors that could be combined to investigate the phenotype of inhibiting all 17 chitinases concurrently. While inhibitors originally designed to inhibit AfChiB1 also potently inhibit other “bacterial-type” chitinases [22], [24], a potent nanomolar AfChiAx inhibitor to allow chemical validation of this attractive class of targets remains elusive. Natural product derivatives based on fragments of the bacterial-type chitinase inhibitor argifin yielded micromolar inhibitors of A. fumigatus chitinase A1 (AfChiA1) [23]. Another study screened a model fungal plant-type chitinase, Saccharomyces cerevisiae CTS1 (ScCTS1) against a library containing 880 off-patent drugs [25]. Despite extensive optimisation through structure-based activity relationships, derivatives failed to improve the potency of the parent compound, acetazolamide (AfChiA1 IC50 164 μM) [26].
Having exhausted all previous tractable chemical starting points we performed a high-throughput screen (HTS) of approximately 60,000 compounds against AfChiA1. Here we describe the discovery of the most potent, novel, low micromolar scaffolds reported to date together with the crystal structures of the enzyme in complex with hits selective for plant-type chitinases.
2. Materials and methods
2.1. AfChiA1 cloning, expression and purification
AfChiA1 (Arg28-His337) was expressed and purified as described previously [23]. In brief, the enzyme was expressed in Pichia pastoris as a secreted protein, the culture supernatant was dialysed, concentrated and AfChiA1 purified using anion exchange followed by size exclusion chromatography. This protein sample was then used for the HTS, kinetics and structural biology described below.
2.2. AfChiA1 enzyme assay and high-throughput screen
Information according to published guidelines on the standardised reporting of HTS [27] is provided in Table S1. The Dundee Drug Discovery Unit (DDU) diversity set of 59,904 compounds was solubilised in DMSO (final maximum assay concentration of 1% (v/v) in all samples including controls) and a HTS performed in singlet at a concentration of 30 μM. Library compounds and DMSO controls were transferred to 384-well black polystyrene plates (Matrix) using a Hummingbird (Genomic Solutions). Columns 1–22 received library compounds, columns 23 and 24 were reserved for high/low controls (DMSO) and an 8-point standard inhibitory curve with acetazolamide (Sigma), a micromolar AfChiA1 inhibitor [26]. Quality control (QC) plates, two per six assay plates, consisted of low control (columns 1–10), high control (columns 11–20) and a standard curve (columns 21–24). AfChiA1 catalyses the hydrolysis of chitin and an existing assay liberating 4-methylumbelliferyl (4-MU) from the fluorogenic substrate 4-methylumbelliferyl β-d-N,N′,N″-triacetylchitotrioside (4-MU-GlcNAc3, Sigma) was optimised for microtitre plate format compatible with HTS [26]. AfChiA1 activity was assayed in McIlvaine’s buffer (100 mM citric acid, 200 mM sodium phosphate [pH 5.5]) and 0.05 mg/ml BSA (Pierce) in a final reaction volume of 42 μl. Each reaction contained 10 nM AfChiA1 except eight wells in column 24 (screening plates) and columns 1–10 (QC plates), to which only buffer was added to determine background signal. The reaction was initiated by 100 μM 4MU-GlcNAc3 and both additions were executed using a FlexDrop reagent dispenser (PerkinElmer). Assay plates were incubated on a microtitre plate shaker (Heidolph) at room temperature for 70 min. Fluorescence generated from the release of 4-MU was quantified using an EnVision 2102 multilabel reader (Perkin Elmer).
2.3. Data analysis
The Activity Base Suite (Abase) version 5.4 from IDBS was used for all data processing and analyses. For all compounds, the raw relative fluorescent units (RFUs) were corrected and normalised to percentage inhibition (PI) according to the equations in Table S1. Calculation of the Z-factor to determine the quality of the screening assay was as follows [28]:
for which SDhigh control and SDlow control represent the standard deviations of the response from 8 to 12 control wells (uninhibited signal) and 8 to 12 background signal wells, respectively. Subsequent curve fitting to determine the IC50 was with XLFit version 4.2 (IDBS) using a four-parameter dose response curve.
2.4. Verification of primary hits and potency determination
Initial hits were (a) re-assayed at 30 μM for verification and (b) potency determined to generate an approximate IC50. Compounds were transferred into columns 1 and 13 of a 384-well polypropylene plate (Matrix) and serially diluted in 100% DMSO through 10 half-log increments in row orientation using a JANUS 8-channel Varispan automated workstation (PerkinElmer). This produced a source plate containing thirty test and two standard inhibitor curves (100× final assay concentration) in columns 1–10 and 13–22. An additional QC plate containing acetazolamide was also generated. From these source plates 0.5 μl, corresponding to a final concentration range of 30 μM to 1.5 nM, were transferred into replicate black 384-well polystyrene assay plates in duplicate. AfChiA1 and substrate were added as in the original screen.
Confirmed hits were re-purchased from commercial sources (Table 1 shows the corresponding structure of each compound denoted in bold). Compounds 1, 2, 6 and 7 (ChemBridge Corporation, San Diego, CA, USA); compounds 3, 4 and 8 (ChemDiv, San Diego, CA, USA); compounds 9 and 11 (Asinex Europe, Rijswijk, NED); compound 5 (Maybridge, Fisher Scientific, Loughborough, UK); compound 10 (InterBioScreen, Moscow, RUS) and compound 12 (Enamine, Monmouth, NJ, USA). For accurate duplicate potency determinations, the starting concentration was increased from 30 μM to 120 μM and diluted as described.
Table 1.
Chemical structure of HTS compound hits identified against AfChiA1 and corresponding inhibitory constants across other GH18 family 18 chitinases. Compounds were assigned to chemical series based on structural similarity.
| Compound (series) | Structure | GH18 family chitinase IC50 (μM) |
||||
|---|---|---|---|---|---|---|
| Plant-type |
Bacterial-type |
|||||
| AfChiA1a | ScCTS1 | AfChiB1 | AMCase | HsCHT | ||
| 1 (1) | 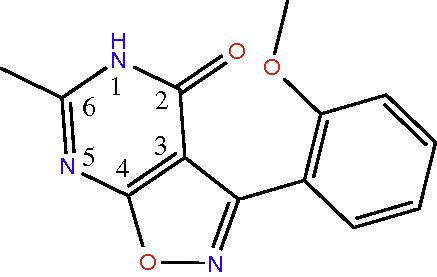 |
2.6 | 0.5 | >100 | 9.8 | >100 |
| 2 (2) | 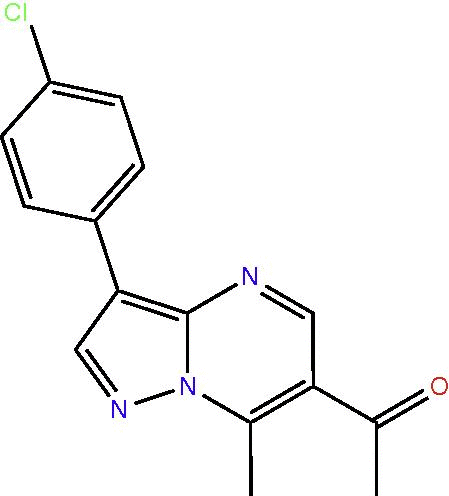 |
34.9 | N/D | N/D | N/D | N/D |
| 3 (3) | 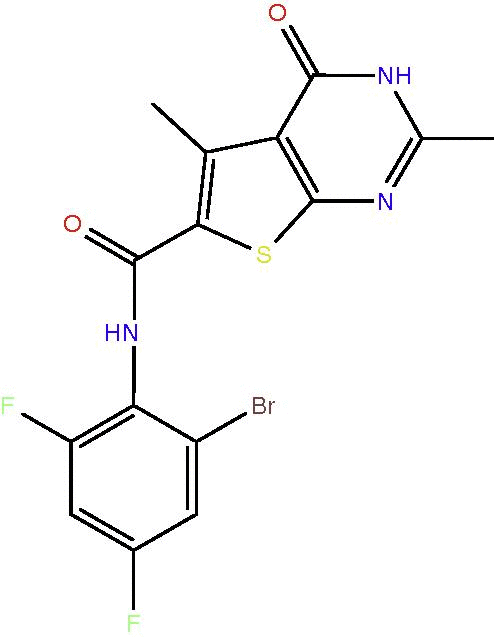 |
6.6 | N/D | N/D | N/D | N/D |
| 4 (3) | 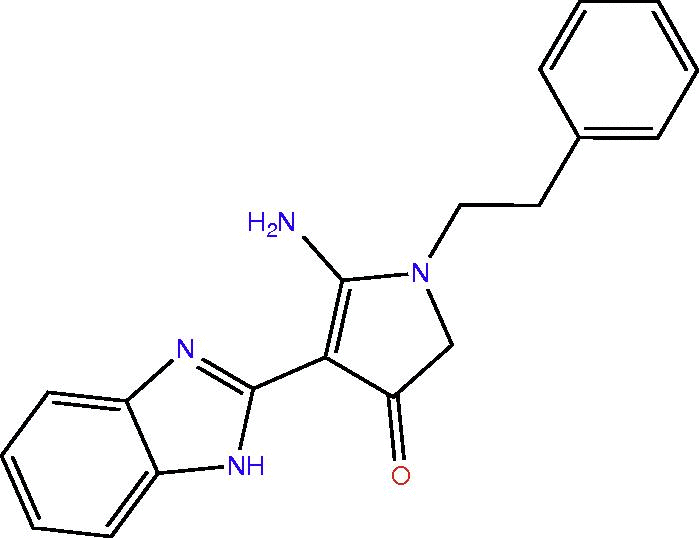 |
19.4 | >100 | N/A | N/A | N/A |
| 5 (4) | 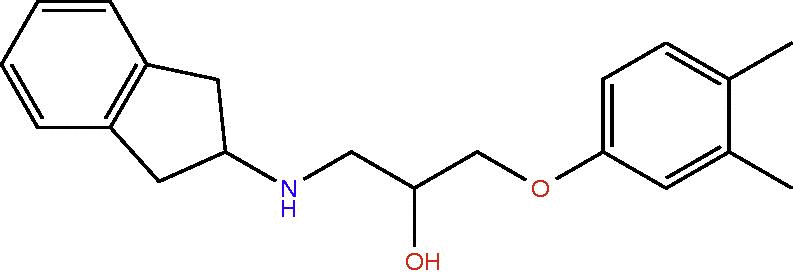 |
9.2 | 2.4 | >100 | >100 | >100 |
| 6 (3) | 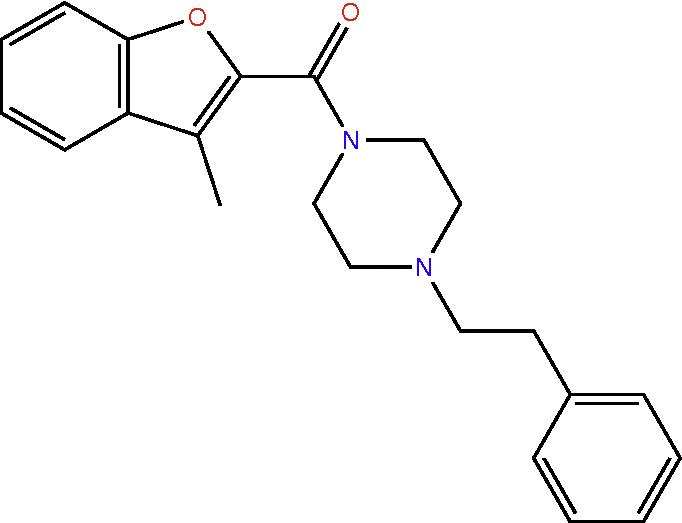 |
11.4 | 63.0 | >100 | >100 | >100 |
| 7 (1) | 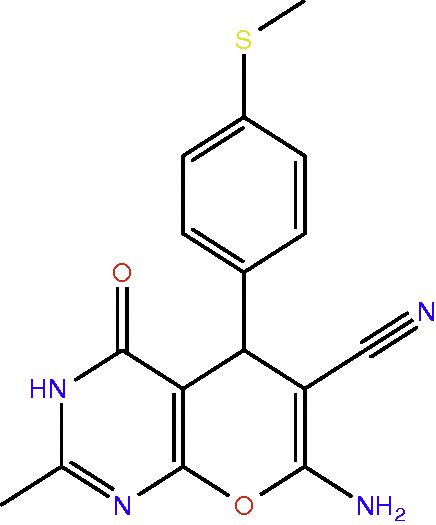 |
14.3 | >100 | N/A | N/A | N/A |
| 8 (1) | 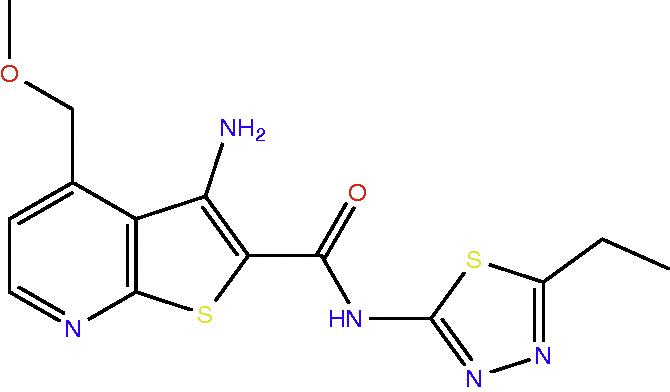 |
16.2 | >100 | N/A | N/A | N/A |
| 9 (1) | 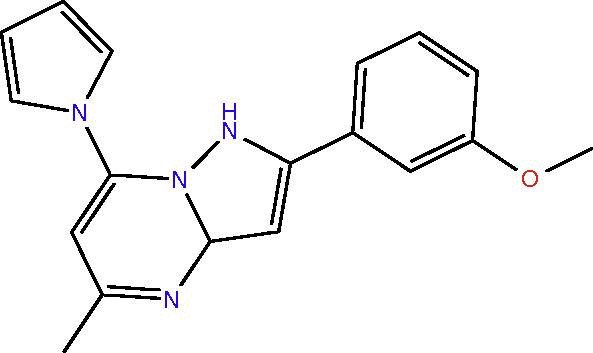 |
11.8 | >100 | N/A | N/A | N/A |
| 10 (1) | 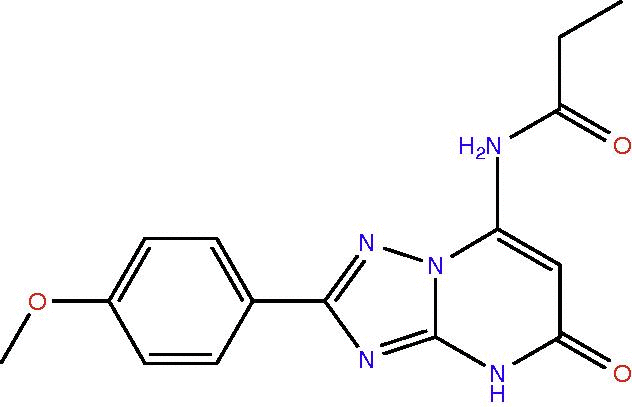 |
45.3 | >100 | N/A | N/A | N/A |
| 11 (2) | 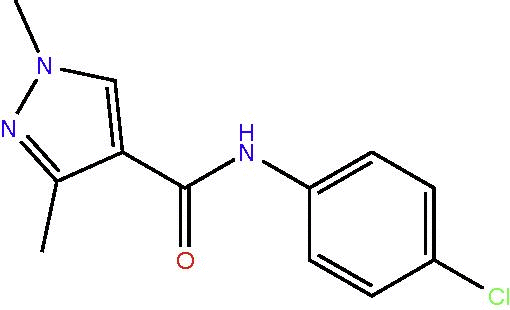 |
58.8 | >100 | N/A | N/A | N/A |
| 12 (1) | 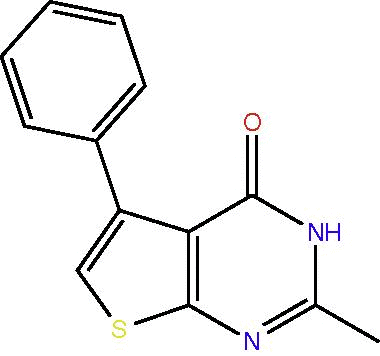 |
23.7 | 16.1 | 65.5 | 13.6 | >100 |
N/D, not determined. N/A, not applicable.
The IC50 values quoted for AfChiA1 are representative of the repeat potency determinations (performed in duplicate) and with the exception of compound 2 were within 5-fold of the original IC50.
2.5. Inhibition profiles across the GH18 family chitinases
Counter screening assays to determine an IC50 against other GH18 family chitinases included S. cerevisiae CTS1 (ScCTS1, model plant-type chitinase) and three bacterial-type chitinases (A. fumigatus chitinase B1 [AfChiB], acidic mammalian chitinase [AMCase] and Homo sapiens chitinase 1/chitotriosidase [HsCHT]). Assays were performed as per AfChiA1 with the following modifications [25], [29]. Bacterial-type chitinases (AfChiB, AMCase and HsCHT) utilised 4-methylumbelliferyl β-d-N-N-diacetylchitobioside (4MU-NAG2) as a substrate [29] and for these pentoxifylline (Sigma) was used as a control inhibitor [30]. Substrate concentrations for IC50 determinations were selected according to the published Km values for each enzyme [25], [29], [31], [32]. The mode of inhibition was established by Lineweaver–Burk plots of steady-state kinetics with 50 μM to 1 mM of 4MU-GlcNAc3 in the presence of different concentrations of selected compounds [23].
2.6. Crystallography of AfChiA1 and ScCTS1 in complex with selected HTS hits
Native crystals were obtained using the hanging (AfChiA1) and sitting drop (ScCTS1) vapour diffusion methods using standard protocols [23], [25]. Crystals were soaked by the addition of 4 mM of the respective ligand (AfChiA:compound 1 and ScCTS1:compound 5; prepared from a 0.2 M stock in 100% DMSO) to a crystallization drop. After incubation at 20 °C for 30 min, crystals were transferred to cryoprotectant (2.5 M Li2SO4 for AfChiA1 and 0.1 M HEPES, 40% PEG 600 pH 7.5 for ScCTS1) using a nylon loop for approximately 10 s before being flash frozen in liquid nitrogen. Diffraction data were collected at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. Diffraction data were processed and scaled with the HKL suite [33] to resolutions of 1.9 Å (AfChiA:compound 1) and 1.8 Å (ScCTS1:compound 5), respectively. Each complex was solved by manual molecular replacement with the apo structure of AfChiA1 (PBD ID 2XVP [23]) or ScCTS1 (PDB ID 2UY2, [25]). Programs from the CCP4 suite [34] were used throughout the refinement process, which was initiated by rigid-body refinement and proceeded through iterative cycles of minimisation using REFMAC5 [35] and model building with Coot [36]. Ligand coordinates and topologies were produced with PRODRG [37]; ligands were not included until their conformations were completely defined by unbiased σA-weighted |Fo| − |Fc|, φcalc electron density maps. Further refinement yielded the final models described in Table S2. Figures were generated using PyMol [38].
3. Results and discussion
3.1. High-throughput screening identifies novel AfChiA1 inhibitors
Further exploration of chitinases as potential antifungal targets requires new potent chemical tools for the plant-type subclass to complement the nanomolar inhibitor, bisdionin C, discovered for AfChiB [39]. To identify new AfChiA1 inhibitor scaffolds, a HTS was performed using a fluorescent assay. The Dundee Drug Discovery Unit (DDU) diversity set containing 59,904 compounds was screened against AfChiA1 at 30 μM. This purpose-designed library adheres to lead-like properties of drug-like compounds [40]. The 172 assay screening plates generated a robust mean Z factor (±SD) of 0.79 (±0.05) indicative of an excellent assay with wide separation between the high and low controls [28]. The hit distribution profile (Fig. 1) showed nearly half of the library compounds (28,094/59,904) clustered around an AfChiA1 inhibition of 1–10%. Primary screening identified 48 compounds with ⩾35% inhibition (0.08% hit rate) using greater than two times the standard deviation of the mean of the uninhibited control signal across all screening plates as a threshold. All 48 compounds were selected for re-confirmation and potency determination in duplicate (R2 = 0.9991, Fig. 1) using 10-point dose–response curves. From this, 23 compounds were verified hits with inhibition rates ⩾35% in two separate experiments giving an overall confirmed hit rate of 0.04%. Hits were ranked according to IC50 and 12 compounds produced values ⩽20 μM with the most potent hit (compound 1, Table 1) returning an initial IC50 of 1.7 μM. Compounds were grouped into four chemical series according to their structural similarity (Table 1). Series 1 included the top hit and six additional compounds with a common heterocyclic core: a six-membered substructure fused to either a five- or six-membered ring. On the other hand, compound (5) was the sole member of series 4. After re-purchasing from the original vendor, hits were further evaluated for AfChiA1 inhibition using a 10-point dilution series starting at 120 μM. The results were in accordance (<5-fold difference) with the original potency determinations with one exception (compound 2, 15-fold increase).
Fig. 1.

HTS of AfChiA1 against the DDU diversity set. (A) Hit distribution profile representing percentage inhibition (PI) values for the HTS. Hits were designated as compounds that displayed a PI equal to or greater than two standard deviation units above the mean (35% inhibition). (B) Comparison of replicate potency determinations for the 48 primary screen hits. A R2 value of 1.0 indicates the regression line perfectly fits the data.
3.2. Two compounds selectively inhibit plant-type chitinases in the low micromolar range
Of the confirmed hits identified by HTS, we wished to identify those selective for plant-type fungal chitinases in general. Given apparent redundancy in this enzyme class we postulated that scaffolds possessing pan plant-type chitinase inhibition provide more favourable tools for probing biological function than those with selectivity towards an individual enzyme within the family. The amino acid residues lining active site of the five plant-type chitinases in A. fumigatus are completely conserved apart from a single tyrosine residue (Y125) in AfChiA1 [26]. Based on this, AfChiA1 is considered a suitable model for the active sites of AfChiA2–5. Additionally, we chose the well-characterised plant-type chitinase from S. cerevisiae (ScCTS1), which has close homologs in Candida albicans (CHT1–3) [41] as well as, putatively, in Aspergillus flavus, Aspergillus niger, Aspergillus clavatus and Aspergillus oryzae, as a second ‘reference’ plant-type chitinase. To investigate our hypothesis, ten hits representative of each chemical series were evaluated for inhibition against two model plant-type chitinases. Of these, six compounds (4, 7, 8, 9, 10 and 11) did not show significant inhibition of ScCTS1 with IC50 values exceeding 100 μM suggestive of discrepancies towards substrate binding and/or specificity between the two plant-type chitinases. Given our selection criteria these hits were not further evaluated. Previous work [25] revealed the active site pocket in A. fumigatus is shallower than in S. cerevisiae due to a large methionine (Met310) side chain and this may account for these hits having greater affinity for AfChiA1 than ScCTS1. On the other hand, four compounds (1, 5, 6 and 12) inhibited both AfChiA1 and ScCTS1 within half an order of magnitude suggestive of a common binding mode (Table 1) and pan plant-type chitinase activity. Compounds 1 and 5 are of particular interest with IC50 values <10 μM for both AfChiA1 and ScCTS1.
Next, we assessed the selectivity of compounds 1, 5, 6 and 12 across GH18 family chitinases. In particular we sought to identify compounds only with selectivity towards plant-type chitinases to complement existing nanomolar inhibitors of the bacterial-type chitinases. Counter screening was performed against three bacterial-type chitinases: A. fumigatus chitinase B1 [AfChiB], acidic mammalian chitinase [AMCase] and H. sapiens chitinase 1/chitotriosidase [HsCHT]). The most potent hit, compound 1, had no activity against AfChiB or HsCHT, but inhibited AMCase with a fourfold drop-off in potency compared to AfChiA1 (Table 1). Compounds 5 and 6 were selective for plant-type chitinases with IC50 values above 100 μM for all of the bacterial-type chitinases tested. Finally, compound 12 displayed comparable activity between both classes (Table 1) and was discarded from subsequent analysis. Although chitin is absent from humans, the human genome includes two active bacterial-type chitinases HsCHT and AMCase. Despite extensive work the precise role of these enzymes is unresolved although they may offer a protective role [12]. Hence, we wished to focus on potent scaffolds that did not display selectivity towards these enzymes.
Additional kinetic experiments were performed on compounds 1, 5 and 6 against AfChiA1. Steady state kinetics were measured to determine the mode of inhibition and the corresponding inhibition constant, Ki. Inhibitor concentrations were chosen according to the previously determined AfChiA1 IC50 (Table 1). Lineweaver–Burk analysis indicates that each compound competes with the pseudo-substrate 4MU-GlcNAc3 for binding to the AfChiA1 active site (Fig. 2). Inhibition constants were 1.2 μM, 9.5 μM and 10.6 μM for compounds 1, 5 and 6 respectively.
Fig. 2.

Lineweaver–Burk plots of compounds 1 (A) and 5 (B) measured against AfChiA1 using different concentrations of each inhibitor. The data were fitted against a competitive inhibition model and resulted in a Ki of 1.2 μM (compound 1) and 9.5 μM (compound 5). Km 300 ± 27 μM [23].
3.3. Crystal structures of compound 1 and 5 reveal a competitive binding mode
As compounds 1 and 5 were thought to represent the most promising inhibitor scaffolds, we determined the molecular basis of binding to allow for the future design of derivatives. AfChiA1 crystals were soaked with compound 1, diffraction data were collected and scaled to 1.9 Å resolution, the AfChiA1-compound 1 complex structure solved by molecular replacement and refined to an Rfree of 0.22 (Table S2). Electron density corresponding to the ligand (compound 1) was observed in the active site for both molecules in the asymmetric unit. As previously observed [26], the active site was partially obstructed in chain A by a symmetry-related protein molecule and further discussion focuses on chain B. Comparison of the AfChiA-compound 1 complex with a published AfChiA-acetazolamide complex (PBD 2XTK [26]) revealed similar overall conformations with an RMSD of 0.15 Å for 309 Cα atoms. Compound 1 consists of two ring systems, an isoxazolopyrimidine stacking on Trp312 and an attached methoxybenzene. The pyrimidine moiety inserts deeply into the AfChiA active site and thus is likely most important for binding (Fig. 3A). The 6-methyl of the pyrimidine moiety inserts into a pocket at the bottom of the binding cleft that is formed by Tyr34, Met310, Ala205 and Gln230. Residues lining the AfChiA1 active site form a number of hydrogen bonds with the ligand (Fig. 3A): the N1 ring atom donates a hydrogen bond to the side chain of Asp172, while the pyrimidine carbonyl group originating from the C2 position accepts a hydrogen bond from the backbone amine of Ala124 and the N5 ring atom accepts a third hydrogen bond from the side chain hydroxyl of Tyr232. The isoxazolo moiety points out of the binding cleft (Fig. 3A). It forms no direct hydrogen bonds with the protein, but engages in a (weak) water-mediated hydrogen bonding interaction with the side chain amide of Asn233 and the backbone carbonyl of Ala279. The methoxyphenyl moiety makes limited van der Waals contacts with the side chains of Trp312, Gln37, Phe60 and Ala124, but otherwise points towards the bulk solvent suggesting that the ring could be replaced. Crucially, compound 1 only interacts with side chains that are identical in all five AfChiA enzymes. Strikingly the non-conserved Tyr125 side chain has flipped out to accommodate the methoxy group of the isoxazolo moiety and this may account for the improved inhibition of compound 1 over acetazolamide in AfChiA1 by facilitating tight interactions of the neighbouring pyrimidine moiety within the depths of binding pocket. In ScCTS1 a serine residue replaces the bulky flexible tyrosine unique to AfChiA1 at the opening of the active site pocket. Interestingly both acetazolamide [26] and compound 1 displayed greater potency (8 and 5-fold respectively) against ScCTS1 compared to AfChiA1 providing contributory evidence that Tyr125 is not required for ligand specificity in plant-type chitinases.
Fig. 3.

Stereo view of HTS hit compounds (purple) binding to the active site of plant-type chitinases. (A) Compound 1 bound to AfChiA1. (B) Compound 5 bound to ScCTS1. Surface representation of the protein active site is shown as a grey cartoon. Side chains of key amino acid residues interacting with the ligand (purple) are depicted as grey sticks and labelled. Potential hydrogen bonds are indicated by black dotted lines, a water molecule participating in indirect hydrogen bonding between compound 1 and AfChiA1 is represented as a red sphere. The unbiased (calculated before the addition of the ligand to the model) |Fo| − |Fc|, φcalc electron density map covering each ligand (purple) is contoured at 3.0 σ.
Exhaustive attempts to obtain a complex of compound 5 with AfChiA1 were unsuccessful. Due to comparable IC50 values between AfChiA1 and ScCTS1 (9.2 μM versus 2.4 μM, Table 1), as an alternative approach, we selected ScCTS1 as a model to study this interaction. The beta-barrel containing the active site is highly conserved between ScCTS1 (PDB 2UY2 [25]) and AfChiA1 (PDB 2XVP [23]) with an RMSD of 1.2 Å for 270 Cα atoms providing further rationale for our choice of surrogate. Compound 5 was soaked into ScCTS1 crystals and diffraction data were collected to 1.8 Å. The structure was solved by molecular replacement and refined to a final Rfree of 0.23 (Table S2). Clear electron density for compound 5 was observed in the active site (Fig. 3B), corroborating the kinetics data showing that it, like compound 1, is a competitive inhibitor. The ScCTS1–compound 5 complex (Fig. 3B) shows the ligand filling a similar space to that occupied by compound 1 despite the lack of obvious chemical similarities. The dihydroindene moiety inserts into the same pocket as the pyrimidine methyl of compound 1. The hydrogen bonding requirements of the Asp172 side chain (Asp155 in ScCTS1) is satisfied by the linker amine of compound 5, which also donates a potential hydrogen bond to the carboxylate of Glu157 (Glu174 in AfChiA1). Continuing the ‘hydrogen bond zipper’, the linker hydroxyl of compound 5 also donates a hydrogen bond to the Glu157 side chain. Finally the ether oxygen of the ligand accepts a hydrogen bond from the Ala110 backbone (equivalent to Ala124 in AfChiA1). An aromatic group stacking with the active site tryptophan side chain (Trp312 in AfChiA1, Trp285 in ScCTS1) is a feature common to both compound 1 and acetazolamide as well as numerous other family 18 chitinase inhibitors [26]. Consequently, the absence of stacking moieties in compound 5 is unusual, though it may help explain the specificity of this molecule for plant-type over bacterial-type family 18 chitinases, as the latter group generally possesses a second tryptophan that forms a ‘lid’ for the active site [39]. Bacterial-type chitinase inhibitors generally bind sandwiched between the two Trp side chains, suggesting that these enzymes may have a stronger preference for flat/aromatic ligands. Binding of the bulky dihydroindene deep in the ScCTS1 active site induces a conformational change in the protein, with the side chain of Tyr214 (Tyr232 in AfChiA1) and the following loop being pushed out by ∼2 Å compared to the superimposed ScCTS1–acetazolamide complex structure. It is likely that binding of compound 5 to AfChiA1 in an equivalent pose will require more dramatic conformational changes to the binding site, as the dihydroindene-binding pocket is smaller in AfChiA1 due to the presence of Met310 (equivalent to Ala283 in ScCTS1) It is possible that this explains the slight decrease in affinity of compound 5 for AfChiA1 compared to ScCTS1.
4. Concluding remarks
Chitinases represent a fascinating group of enzymes from a chemical perspective although their precise physiological roles remain elusive. In particular, the bacterial-type family have been extensively studied in terms of competitive inhibitors that mimic the carbohydrate substrate interaction [39], [42], [43]. Although both a natural product cyclopentapeptide inhibitor (argifin [43]) and a synthesised derivative based on linking a two-xanthine ring system (bisdionin C [39]) have high nanomolar affinities for AfChiB they provide limited starting-points for plant-type chitinase inhibitors [23]. This is highlighted by fundamental differences in substrate specificity and active site architecture between the different families. Comparison of the crystal structures (AfChiA1 PDB 2XVP [23] and AfChiB1 PDB 1W9P [29]) of both chitinase subclasses in A. fumigatus reveals a deep pocket unique to plant-type chitinases in the base of the substrate binding groove. On the other hand, bacterial-type chitinases posses a more shallow and accessible groove with the tight binding of bisdionin C (AfChiB IC50 200 nM) attributed to tryptophan stacking beyond the single one conserved between AfChiA1 and AfChiB1 [39].
In contrast plant-type chitinases are underexplored in terms of chemical inhibitors. Prior to this study the only reported plant-type chitinase inhibitors were allosamidin [21], acetazolamide [26] and guanylurea derivatives [23], all of which are remarkably poor in terms of their inhibition against AfChiA1. Structurally they are unfavorable: allosamidin extends beyond the conserved binding pocket while all attempts to obtain acetazolamide/guanylurea based derivatives failed to improve potency.
Our HTS against AfChiA1 has provided novel, low micromolar plant-type chitinase competitive inhibitors (Table 1) that allow for tailored ligand specificity. Of the two hits for which we obtained structural information to illustrate the binding mode (Fig. 3), compound 1 (IC50 2.6 μM), a novel pyrimidinone scaffold, could be a platform for chemical modification to inhibit a broad range of fungal plant-type chitinases and increase potency towards nanomolar inhibition. Specifically, as the 6-methyl moiety of the pyrimidine ring engages the depth of the AfChiA1 binding pocket there may be scope for larger substituents in this position to fully occupy this space. This is supported by the known binding mode of acetazolamide, which interacts in the same pocket with an ethyl group. Taken together with information derived from the ScCTS1-compound 5 complex, an ideal substitution could involve anything from a small methyl group to a more bulky ring in terms of size. Due to the tight affinity of the pyrimidine ring for the AfChiA binding pocket and three key hydrogen bond interactions (Fig. 3A) no further extensions are possible. Currently the methoxyphenyl moiety provides little contribution to binding and therefore could be replaced. We hope these modifications will concurrently drive selectivity further towards the plant-type chitinases and create chemical tools to probe their biological role.
This work represents a significant advance in generating chemical starting points and provides a platform for the development of nanomolar AfChiA1 inhibitors that are required to fully dissect the biological importance of these fungal plant-type chitinases. Evidence of an upregulation in plant-type chitinase activity in mature A. fumigatus biofilms is emerging suggestive of a role in the composition of the extracellular matrix potentially through the liberation of extracellular DNA [18]. Intriguingly acetazolamide, a weak plant-type chitinase inhibitor (AfChiA1 IC50 > 150 μM) [25], [26], was shown to reduce A. fumigatus biofilm biomass [18]. If further work elaborating our novel pyrimidinone scaffold succeeds and the role of plant-type chitinases in A. fumigatus biofilm maturation is conclusive, this would open up translational prospects perhaps one day leading to the clinical use of chitinase-inhibitors as anti-biofilm agents.
Acknowledgements
We wish to thank the Dundee Drug Discovery Unit for access to the diversity set library and the European Synchrotron Radiation Facility, Grenoble, for time at the beamline. This work was supported by a MRC Programme Grant (G0900138) and a Wellcome Trust Senior Research Fellowship (WT087590MA) to D.M.F.v.A. D.E.A.L. is the recipient of a MRC Clinical Research Training Fellowship (G1100430). The structures have been deposited in the Protein Data Bank with accession codes 4TX6 and 4TXE.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.febslet.2014.07.015.
Appendix A. Supplementary data
References
- 1.Brown G.D., Denning D.W., Gow N.A., Levitz S.M., Netea M.G., White T.C. Hidden killers: human fungal infections. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3004404. 165rv113. [DOI] [PubMed] [Google Scholar]
- 2.Lass-Florl C. The changing face of epidemiology of invasive fungal disease in Europe. Mycoses. 2009;52:197–205. doi: 10.1111/j.1439-0507.2009.01691.x. [DOI] [PubMed] [Google Scholar]
- 3.Lin S.J., Schranz J., Teutsch S.M. Aspergillosis case-fatality rate: systematic review of the literature. Clin. Infect. Dis. 2001;32:358–366. doi: 10.1086/318483. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal R., Chakrabarti A., Shah A. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin. Exp. Allergy. 2013;43:850–873. doi: 10.1111/cea.12141. [DOI] [PubMed] [Google Scholar]
- 5.Al-Shair K., Atherton G.T., Harris C., Ratcliffe L., Newton P.J., Denning D.W. Long-term antifungal treatment improves health status in patients with chronic pulmonary aspergillosis: a longitudinal analysis. Clin. Infect. Dis. 2013;57:828–835. doi: 10.1093/cid/cit411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Committee J.F. BMJ Group and Pharmaceutical Press; 2013. British National Formulary (online) [Google Scholar]
- 7.Andes D. Optimizing antifungal choice and administration. Curr. Med. Res. Opin. 2013;29(Suppl. 4):13–18. doi: 10.1185/03007995.2012.761135. [DOI] [PubMed] [Google Scholar]
- 8.Epaulard O., Villier C., Ravaud P. A multistep voriconazole-related phototoxic pathway may lead to skin carcinoma: results from a French nationwide study. Clin. Infect. Dis. 2013;57:e182–e188. doi: 10.1093/cid/cit600. [DOI] [PubMed] [Google Scholar]
- 9.Vermeulen E., Lagrou K., Verweij P.E. Azole resistance in Aspergillus fumigatus: a growing public health concern. Curr. Opin. Infect. Dis. 2013;26:493–500. doi: 10.1097/QCO.0000000000000005. [DOI] [PubMed] [Google Scholar]
- 10.Steinbach W.J. Are we there yet? Recent progress in the molecular diagnosis and novel antifungal targeting of Aspergillus fumigatus and invasive aspergillosis. PLoS Pathog. 2013;9:e1003642. doi: 10.1371/journal.ppat.1003642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gastebois A., Clavaud C., Aimanianda V., Latge J.P. Aspergillus fumigatus: cell wall polysaccharides, their biosynthesis and organization. Future Microbiol. 2009;4:583–595. doi: 10.2217/fmb.09.29. [DOI] [PubMed] [Google Scholar]
- 12.Adrangi S., Faramarzi M.A. Biotechnol. Adv.; 2013. From bacteria to human: a journey into the world of chitinases. [DOI] [PubMed] [Google Scholar]
- 13.Ruiz-Herrera J., San-Blas G. Chitin synthesis as target for antifungal drugs. Curr. Drug Targets Infect. Disord. 2003;3:77–91. doi: 10.2174/1568005033342064. [DOI] [PubMed] [Google Scholar]
- 14.Henrissat B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991;280(Pt 2):309–316. doi: 10.1042/bj2800309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seidl V. Chitinases of filamentous fungi: a large group of diverse proteins with multiple physiological functions. Fungal Biol. Rev. 2008;22:36–42. [Google Scholar]
- 16.Alcazar-Fuoli L., Clavaud C., Lamarre C. Functional analysis of the fungal/plant class chitinase family in Aspergillus fumigatus. Fungal Genet. Biol. 2011;48:418–429. doi: 10.1016/j.fgb.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Jaques A.K., Fukamizo T., Hall D. Disruption of the gene encoding the ChiB1 chitinase of Aspergillus fumigatus and characterization of a recombinant gene product. Microbiology. 2003;149:2931–2939. doi: 10.1099/mic.0.26476-0. [DOI] [PubMed] [Google Scholar]
- 18.Rajendran R., Williams C., Lappin D.F., Millington O., Martins M., Ramage G. Extracellular DNA release acts as an antifungal resistance mechanism in mature Aspergillus fumigatus biofilms. Eukaryot. Cell. 2013;12:420–429. doi: 10.1128/EC.00287-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loussert C., Schmitt C., Prevost M.C. In vivo biofilm composition of Aspergillus fumigatus. Cell. Microbiol. 2010;12:405–410. doi: 10.1111/j.1462-5822.2009.01409.x. [DOI] [PubMed] [Google Scholar]
- 20.Beauvais A., Schmidt C., Guadagnini S. An extracellular matrix glues together the aerial-grown hyphae of Aspergillus fumigatus. Cell. Microbiol. 2007;9:1588–1600. doi: 10.1111/j.1462-5822.2007.00895.x. [DOI] [PubMed] [Google Scholar]
- 21.Sakuda S., Isogai A., Matsumoto S., Suzuki A. Search for microbial insect growth regulators. II. Allosamidin, a novel insect chitinase inhibitor. J. Antibiot. (Tokyo) 1987;40:296–300. doi: 10.7164/antibiotics.40.296. [DOI] [PubMed] [Google Scholar]
- 22.Schuttelkopf A.W., Andersen O.A., Rao F.V. Screening-based discovery and structural dissection of a novel family 18 chitinase inhibitor. J. Biol. Chem. 2006;281:27278–27285. doi: 10.1074/jbc.M604048200. [DOI] [PubMed] [Google Scholar]
- 23.Rush C.L., Schuttelkopf A.W., Hurtado-Guerrero R. Natural product-guided discovery of a fungal chitinase inhibitor. Chem. Biol. 2010;17:1275–1281. doi: 10.1016/j.chembiol.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sutherland T.E., Andersen O.A., Betou M. Analyzing airway inflammation with chemical biology: dissection of acidic mammalian chitinase function with a selective drug-like inhibitor. Chem. Biol. 2011;18:569–579. doi: 10.1016/j.chembiol.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurtado-Guerrero R., van Aalten D.M. Structure of Saccharomyces cerevisiae chitinase 1 and screening-based discovery of potent inhibitors. Chem. Biol. 2007;14:589–599. doi: 10.1016/j.chembiol.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Schuttelkopf A.W., Gros L., Blair D.E., Frearson J.A., van Aalten D.M., Gilbert I.H. Acetazolamide-based fungal chitinase inhibitors. Bioorg. Med. Chem. 2010;18:8334–8340. doi: 10.1016/j.bmc.2010.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inglese J., Shamu C.E., Guy R.K. Reporting data from high-throughput screening of small-molecule libraries. Nat. Chem. Biol. 2007;3:438–441. doi: 10.1038/nchembio0807-438. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 29.Rao F.V., Houston D.R., Boot R.G. Specificity and affinity of natural product cyclopentapeptide inhibitors against A. fumigatus, human, and bacterial chitinases. Chem. Biol. 2005;12:65–76. doi: 10.1016/j.chembiol.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 30.Rao F.V., Andersen O.A., Vora K.A., Demartino J.A., van Aalten D.M. Methylxanthine drugs are chitinase inhibitors: investigation of inhibition and binding modes. Chem. Biol. 2005;12:973–980. doi: 10.1016/j.chembiol.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Renkema G.H., Boot R.G., Muijsers A.O., Donker-Koopman W.E., Aerts J.M. Purification and characterization of human chitotriosidase, a novel member of the chitinase family of proteins. J. Biol. Chem. 1995;270:2198–2202. doi: 10.1074/jbc.270.5.2198. [DOI] [PubMed] [Google Scholar]
- 32.Chou Y.T., Yao S., Czerwinski R. Kinetic characterization of recombinant human acidic mammalian chitinase. Biochemistry. 2006;45:4444–4454. doi: 10.1021/bi0525977. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z., Minor W. Kluwer Academic Publishers; London: 2001. Denzo & Scalepack. [Google Scholar]
- 34.Winn M.D., Ballard C.C., Cowtan K.D. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vagin A.A., Steiner R.A., Lebedev A.A. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuttelkopf A.W., van Aalten D.M. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 38.L. Schrodinger, The PyMOL Molecular Graphics System. Version 1.5.0.4., http://www.pymol.org/citing.
- 39.Schuettelkopf A.W.A.O., Rao F.V., Allwood M., Rush C.L., Eggleston I.M., van Aalten D.M. Bisdionin C – a rationally designed, submicromolar inhibitor of family 18 chitinases. ACS Med. Chem. Lett. 2011;2:428–432. doi: 10.1021/ml200008b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brenk R., Schipani A., James D. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem. 2008;3:435–444. doi: 10.1002/cmdc.200700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCreath K.J., Specht C.A., Liu Y., Robbins P.W. Molecular cloning of a third chitinase gene (CHT1) from Candida albicans. Yeast. 1996;12:501–504. doi: 10.1002/(SICI)1097-0061(199604)12:5%3C501::AID-YEA931%3E3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 42.Rao F.V., Houston D.R., Boot R.G., Aerts J.M., Sakuda S., van Aalten D.M. Crystal structures of allosamidin derivatives in complex with human macrophage chitinase. J. Biol. Chem. 2003;278:20110–20116. doi: 10.1074/jbc.M300362200. [DOI] [PubMed] [Google Scholar]
- 43.Houston D.R., Shiomi K., Arai N. High-resolution structures of a chitinase complexed with natural product cyclopentapeptide inhibitors: mimicry of carbohydrate substrate. Proc. Natl. Acad. Sci. USA. 2002;99:9127–9132. doi: 10.1073/pnas.132060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further reading
- 44.Brand S., Cleghorn L.A., McElroy S.P. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 2012;55:140–152. doi: 10.1021/jm201091t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.