

Abstract
Background
Mirtazapine has a unique mechanism of antidepressive action and is one of the commonly used antidepressants in clinical practice.
Objectives
The aim of the present review was to assess the evidence on the efficacy and acceptability of mirtazapine compared with other antidepressive agents in the acute-phase treatment of major depression in adults.
Search methods
We searched the Cochrane Collaboration Depression, Anxiety and Neurosis review group’s specialised register (CCDANCTR), which includes relevant randomised controlled trials from the following bibliographic databases: The Cochrane Library (all years to April 2011), EMBASE, (1980 to July 2011) MEDLINE (1950 to July 2011) and PsycINFO (1974 to July 2011). Reference lists of the reports of relevant studies were checked and experts in the field contacted. The review was not limited to English-language articles.
Selection criteria
Randomised controlled trials (RCTs) allocating participants with major depression to mirtazapine versus any other antidepressive agent.
Data collection and analysis
Two authors independently checked eligibility and extracted data on an intention-to-treat basis. The primary outcome was response to treatment. The secondary outcomes included dropouts and individual adverse events.
Meta-analyses were conducted using the random-effects model.
Main results
A total of 29 RCTs (n = 4974), mostly following up the participants for six weeks in outpatient clinics and inadequately reporting the risk of bias, were included. In comparison with tricyclic antidepressants (10 trials, n = 1553) there was no robust evidence to detect a difference between mirtazapine and tricyclics in terms of response at two weeks (odds ratio (OR) 0.85, 95% confidence interval (CI) 0.64 to 1.13) or at the end of acute-phase treatment (at 6 to 12 weeks) (OR 0.89, 95% CI 0.72 to 1.10). In comparison with selective serotonin reuptake inhibitors (SSRIs) (12 trials, n = 2626) mirtazapine was significantly more effective at two weeks (OR 1.57, 95% CI 1.30 to 1.88) and at the end of acute-phase treatment (OR 1.19, 95% CI 1.01 to 1.39). Mirtazapine was significantly more effective than a serotonin-noradrenaline reuptake inhibitor (venlafaxine only, two trials, n = 415) at two weeks (OR 2.29, 95% CI 1.45 to 3.59) and at the end of acute-phase treatment (OR 1.53, 95% CI 1.03 to 2.25).
In terms of dropouts, there was no robust evidence to detect a difference between mirtazapine and other antidepressants. Mirtazapine was more likely to cause weight gain or increased appetite and somnolence than SSRIs but less likely to cause nausea or vomiting and sexual dysfunction.
Authors’ conclusions
Some statistically significant and possibly clinically meaningful differences between mirtazapine and other antidepressive agents were found for the acute-phase treatment of major depression. Mirtazapine is likely to have a faster onset of action than SSRIs during the acute-phase treatment. Dropouts occur similarly in participants treated with mirtazapine and those treated with other antidepressants, although the adverse event profile of mirtazapine is unique.
Medical Subject Headings (MeSH): Antidepressive Agents [therapeutic use]; Antidepressive Agents, Tricyclic [* therapeutic use]; Cyclohexanols [therapeutic use]; Depression [* drug therapy]; Mianserin [* analogs & derivatives; therapeutic use]; Randomized Controlled Trials as Topic
MeSH check words: Adult, Humans
BACKGROUND
Description of the condition
Major depression is generally diagnosed in people with a persistent and unreactive low mood and loss of all interest and pleasure accompanied by a range of symptoms including appetite loss, insomnia, fatigue, loss of energy, poor concentration, psychomotor symptoms, inappropriate guilt and morbid thoughts of death (APA 1994). It was the third leading cause of burden among all diseases of humankind, after lower respiratory infections and HIV/AIDS, in the year 2002 and accounted for 4.5% of total human suffering (WHO 2006a). Moreover, it is expected to show a rising trend during the coming 20 years (WHO 2006b). This condition is associated with marked personal, social and economic morbidity, loss of functioning and productivity, and creates significant demands on service providers in terms of workload (NICE 2004).
Description of the intervention
Although both pharmacological and psychological interventions are effective for major depression, in primary and secondary care settings antidepressant drugs remain the mainstay of treatment (APA 2000; Ellis 2004; NICE 2004). Amongst antidepressants many different agents are available, including tricyclics (TCAs) (for example amitriptyline, amoxapine, clomipramine, desipramine, dosulepin, doxepin, imipramine, nortriptyline), monoamine oxidase inhibitors (MAOIs) (for example moclobemide, selegiline, tranylcypromine), selective serotonin reuptake inhibitors (SSRIs) (for example citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline), serotonin-noradrenaline reuptake inhibitors (SNRIs) (for example venlafaxine, duloxetine, milnacipran) and other newer agents (mirtazapine, reboxetine, bupropion).
In many western countries, antidepressant consumption has risen dramatically during the last 20 years, mainly because of the increasing consumption of SSRIs and newer antidepressants that have progressively become the most commonly prescribed antidepressants (Ciuna 2004; Guaiana 2005). SSRIs are generally better tolerated than TCAs (Barbui 2000; Hotopf 1997; Steffens 1997), and there is some evidence of similar efficacy (Anderson 2000; Williams 2000). However, head-to-head comparisons provide contrasting findings. Amitriptyline, for example, may have the edge over SSRIs in terms of efficacy (Guaiana 2003) and individual SSRIs and SNRIs may differ in terms of efficacy and tolerability (Cipriani 2005; Puech 1997; Smith 2002).
Given that the most recently available evidence refers to the SSRIs as a homogeneous group (Arroll 2005; Hansen 2005), it is still unclear how each of the SSRIs or newer agents compare with other antidepressants in terms of effects and side effects.
How the intervention might work
Mirtazapine is a prescription only antidepressant introduced in 1996. It has a unique pharmacological profile, including potent antagonism of central alpha 2-adrenergic autoreceptors and heteroreceptors and antagonism of both serotonin 5-hydroxytryptamine-2 (5-HT2) and 5-HT3 receptors (Kent 2000). Unlike venlafaxine and nefazodone, mirtazapine has minimum effects on monoamine reuptake and is classified as a NaSSA (noradrenergic and specific serotonergic antidepressant). Antagonism of alpha 2-adrenergic receptors leads to blockade of presynaptic autoreceptors and thus enhances norepinephrine release, while blockade of heteroreceptors on serotonergic neurons increases serotonin release. With 5-HT2 and 5-HT3 receptor blockade, enhanced serotonin release results in a net increase in 5-HT1 mediated neurotransmission (de Boer 1995), which is considered to be related to the antidepressant effect of mirtazapine (de Boer 1996).
Mirtazapine is usually prescribed to patients at 15 mg/day as the starting dose and the maintenance dose is generally not more than 45 mg/day. Because of the unique pharmacology of mirtazapine, antihistaminergic effects have been thought to predominate at lower doses (causing drowsiness, sedation), whilst noradrenergic neurotransmission increases with increasing doses to counteract some of the antihistaminergic effects. Dry mouth, sedation, and increases in appetite and body weight have been reported as the most common adverse effects (Anttila 2001).
Why it is important to do this review
The efficacy of mirtazapine has been investigated through several meta-analyses, especially compared with a placebo or amitriptyline. A meta-analysis of eight randomised controlled clinical trials (RCTs) showed that mirtazapine is superior to placebo and is comparable to amitriptyline for the treatment of patients with major depression (Fawcett 1998). However, that review is outdated because 11 years have elapsed since its publication.
Regarding a comparison with SSRIs, only limited evidence has been established to date. A Cochrane review for fluoxetine showed that mirtazapine was more effective than fluoxetine (Cipriani 2005). Several RCTs have examined the efficacy of mirtazapine in comparison with other newer antidepressants, including paroxetine (Benkert 2000; Schatzberg 2002), sertraline (Behnke 2003), citalopram (Leinonen 1999) and trazodone (van Moffaert 1995). In those RCTs, mirtazapine was consistently reported to have a faster onset of action than the other agents on core symptoms of depression (Thase 2005; Thase 2006) but, to our knowledge, no overall systematic quantitative review has been published for these comparisons.
In addition, the profile of adverse events related to mirtazapine has been controversial. Mirtazapine was thought to have no association with sexual side effects (Anttila 2001; Kent 2000). However, a cross-sectional survey has shown that 41% of patients taking mirtazapine in primary care clinics experienced sexual dysfunction, similar to paroxetine (Clayton 2002). Therefore, the comparative efficacy and adverse effects of mirtazapine against antidepressants other than fluoxetine remain uncertain.
A group of researchers agreed to join forces under the rubric of the Multiple Meta-Analysis of New Generation Antidepressants Study (MANGA study) to systematically review all available evidence for each specific, newer antidepressant. We have, to date, completed or planned head-to-head meta-analyses for individual newer antidepressants in comparison with all the other antidepressive agents. The newer antidepressants included bupropion, citalopram (Imperadore 2007), duloxetine (Nose 2007), escitalopram (Cipriani 2009b), fluoxetine (Cipriani 2005), fluvoxamine (Omori 2010), milnacipran (Nakagawa 2009), paroxetine (Cipriani 2007a), sertraline (Cipriani 2009), venlafaxine (Cipriani 2007b), reboxetine (Churchill 2009) and mirtazapine.
Although the overall efficacy, tolerability and information about the adverse event profile of mirtazapine in treatment for major depression, in comparison with other antidepressants, has been published elsewhere (Watanabe 2008; Watanabe 2010) the presented data have been outdated.
This review aims to provide a comprehensive summary of the information about the overall efficacy, tolerability and adverse events for mirtazapine compared with other antidepressants used in the treatment of major depression, and will update previously published findings (Watanabe 2008; Watanabe 2010).
OBJECTIVES
To determine the efficacy of mirtazapine in comparison with other antidepressive agents in acute-phase treatment for major depression
To review the acceptability of mirtazapine in comparison with that of other antidepressive agents in acute-phase treatment for major depression
To investigate the adverse effects of mirtazapine in comparison with other antidepressive agents in acute-phase treatment for major depression
METHODS
Criteria for considering studies for this review
Types of studies
Randomised controlled trials, with individual participant or cluster randomisation, were included. Quasi-randomised trials, such as those allocating participants by using alternate days of the week, were excluded. For trials which had a crossover design only results from the first randomisation period were considered.
Types of participants
Inclusion criteria
participants aged 18 years or older, of both sexes and with a primary diagnosis of unipolar major depression diagnosed according to any of the standardised criteria: Feighner criteria, Research Diagnostic Criteria, DSM-III (APA 1980), DSM-III-R (APA 1987), DSM-IV (APA 1994) or ICD-10 (WHO 1992) were included. We included the following subtypes: chronic, with catatonic features, with melancholic features, with atypical features, postpartum onset, seasonal pattern. participants with co-morbid mental disorders that were not their primary diagnosis were included. Studies in which less than 20% of the participants suffered from bipolar depression were included.
Exclusion criteria
A concurrent primary diagnosis of Axis I or II disorders was an exclusion criterion. We excluded participants with the subtype: with psychotic features. Participants with a serious concomitant medical illness were also excluded.
Types of interventions
Experimental intervention
The experimental intervention was mirtazapine used for acutephase treatment of major depression. No restrictions on dose, frequency, intensity or duration of treatment were applied.
Comparator intervention
All other antidepressive agents in the treatment of acute depression, including:
conventional tricyclic ADs (TCAs)
SSRIs (e.g. fluoxetine, fluvoxamine, citalopram, escitalopram, paroxetine, sertraline)
SNRIs (e.g. duloxetine, milnacipran, venlafaxine)
heterocyclic ADs (e.g. maprotiline)
newer antidepressants (MAOIs or newer agents such as bupropion and reboxetine) and non-conventional ADs, such as herbal products (e.g. hypericum).
No restrictions on dose, frequency, intensity and duration were applied.
Trials in which mirtazapine was compared to another type of psychopharmacological agent (e.g. anxiolytics, anti-convulsants, antipsychotics or mood-stabilisers) were excluded. Trials in which mirtazapine or the comparator agent was used as an augmentation strategy were also excluded.
Types of outcome measures
We decided, a priori, to subdivide the treatment outcome indices into:
at two weeks after commencement of treatment;
after the conclusion of the acute-phase treatment (between 6 and 12 weeks)
after the conclusion of continuation treatment (between 4 and 6 months).
After the conclusion of the acute-phase treatment was defined as the primary time point. For each outcome, a risk ratio (RR) of mirtazapine in comparison with each type of antidepressant class was examined in the primary analyses (see Data synthesis).
Primary outcomes
Response
The primary outcome in our systematic review was defined as a response, after the conclusion of acute-phase treatment, represented by a reduction of at least 50% in the score on the Hamilton Rating Scale for Depression (HAM-D) (Hamilton 1960), Montgomery-Asberg Depression Rating Scale (MADRS) (Montgomery 1979), or ‘much or very much improved’ (score 1 or 2) on the CGI-Improvement measure (Guy 1970). We did not employ the original authors’ definitions of the primary outcomes per se because investigators or journal editors might selectively withhold some of the measured outcomes because of the poor strength of the result (outcome reporting bias) (Furukawa 2007). Among the three response criteria, we used the HAM-D for the primary outcome whenever possible, even when we needed to impute response rates (see Data synthesis) because the HAM-D has been the gold standard measure of depression severity for clinical trials of antidepressants (Williams 2001).
Secondary outcomes
Efficacy outcomes
Remission
We used remission as the secondary outcome, represented by a score of 7 or less on the 17-item HAM-D, of 8 or less in all the other longer versions of HAM-D, and of 11 or less on the MADRS.
Depression severity
Group mean scores at the end of the trial on the HAM-D, or MADRS, or any other depression scale. We applied a looser form of ITT analyses, whereby all the participants with at least one post-baseline measurement were represented by their last observations carried forward.
Social adjustment
Social adjustment, social functioning including the Global Assessment of Function (Luborsky 1962) scores.
Health-related quality of life (HRQoL)
The HRQoL outcomes were included in the analysis when they were reported in a validated scale such as SF-12 or SF-36 (Ware 1993), HoNOS (Wing 1994) and WHO-QOL (WHOQOL Group 1998).
With regard to continuous outcomes, when data were provided in a trial both as endpoint scores and as change scores, change scores were included in the analysis because change scores were preferable in meta-analyses (Norman 1989; Wiebe 2003).
Tolerability and acceptability outcomes
Number of participants who dropped out during the trial due to any reason.
Number of participants who dropped out during the trial due to the development of adverse event.
Total number of participants experiencing at least some adverse events during the trial.
Number of participants experiencing the following specific individual adverse events: hypertension or tachycardia; hypotension or bradycardia; sweating; constipation; diarrhoea; dry mouth or decreased salivation; nausea, vomiting or gastric distress; weight gain or increased appetite; weight loss or anorexia; sexual dysfunction; anxiety or agitation; dizziness, vertigo, faintness; fatigue, tiredness, asthenia; headache; tremor; sleep disturbance; sleepiness, drowsiness, somnolence; completed suicide, and suicide attempt.
To avoid missing any relatively rare or unexpected side effects in the data extraction phase, we collected all adverse events data reported in the literature and discussed ways to summarise them post hoc. Descriptive data regarding adverse event profiles were extracted from all available studies. Only studies reporting the number of participants experiencing individual adverse events were retained. Due to the variety in reporting of adverse events as presented from the study authors’ descriptions, terms describing similar adverse events were combined.
Search methods for identification of studies
The Cochrane, Depression, Anxiety and Neurosis Review Group’s Specialised Register (CCDANCTR)
CCDAN maintain two clinical trials registers at their editorial base in Bristol, UK, a references register and a studies based register. The CCDANCTR-References Register contains over 27,500 reports of trials in depression, anxiety and neurosis. Approximately 60% of these reports have been tagged to individual trials. Coded trial records are held in the CCDANCTR-Studies Register and records are linked between the two registers through the use of unique Study ID tags. Reports of trials for inclusion in the Group’s registers are collated from routine (weekly), generic searches of MEDLINE (1950-date), EMBASE (1980 - date) and PsycINFO (1967 - date); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review specific searches of additional databases. Reports of trials are also sourced from international trials registers c/o the World Health Organisation’s trials portal (ICTRP), drug companies, the hand-searching of key journals, conference proceedings and other (non-Cochrane) systematic reviews and meta-analyses. Details of CCDAN’s generic search strategies used to identify RCTs can be found on the CCDAN website.
Electronic searches
The CCDANCTR-Studies Register was searched using the following terms:
DIAGNOSIS = Depress* or Dysthymi* or “Adjustment Disorder*” or “Mood Disorder*” or “Affective Disorder” or “Affective Symptoms”
And
INTERVENTION = Mirtazapine
The CCDANCTR-References Register was searched using free-text terms to identify any additional untagged references: ((Depress* or Dysthymi* or “Adjustment Disorder*” or “Mood Disorder*” or “Affective Disorder” or “Affective Symptoms”) and Mirtazapine)
Both registers were searched up to July 2011.
Searching other resources
Trial databases and trial results registers were searched for additional unpublished or ongoing studies. These included: the World Health Organisation’s trials portal (ICTRP); ClinicalTrials.gov and ClinicalStudyResults.org. The FDA database drugs@fda.gov was also searched in June 2010.
Personal communication
Experts in the field were asked if they knew of any study which met the inclusion criteria for this review. The pharmaceutical company Organon who market (and developed) Mirtazapine was also contacted and asked to provide unpublished data (December 8, 2006).
Reference checking
The reference lists of reports of all included studies, previous systematic reviews and major textbooks of affective disorder, written in English, were checked for published reports and citations of unpublished research. A citation search was conducted to identify articles citing any of the included studies.
Data collection and analysis
Selection of studies
Studies relating to mirtazapine were identified by the electronic search of CCDANCTR-Studies and CCDANCTR-References and other complementary searches by the Trials Search Coordinator of CCDAN. They were scanned by one author (NW) as over-inconclusively as possible, firstly based on the title and abstracts. Those studies which met the following inclusion criteria constituted the preliminary list and their full texts were retrieved.
Randomised trial.
Comparing mirtazapine against any other antidepressant.
participants with depression, regardless of the diagnostic criteria used.
All the full text articles were then assessed independently by two review authors (NW and IMO) to see if they meet the strict inclusion criteria. When the raters disagreed the final rating was made by consensus, with the involvement of another author (TAF). Considerable care was taken to exclude duplicate publications.
Data extraction and management
Two authors (NW and IMO) independently extracted data from the included studies concerning participant characteristics (age, sex, depression diagnosis, comorbidity, depression severity, antide-pressant treatment history for the index episode, study setting), intervention details (intended dosage range, mean daily dosage actually prescribed, co-intervention if any, mirtazapine as investigational drug or as comparator drug, sponsorship) and outcome measures of interest. When any discrepancies between the data extracted by each author occurred, the final decision was made by consensus through discussion between the authors.
When the trial was a three (or more) armed trial that involved a placebo arm, the data were extracted from the placebo arm as well. Data were entered by a review author (NW) with double data entry to avoid input errors.
Assessment of risk of bias in included studies
The methodological quality of the selected trials was assessed by using criteria based on the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008). Each potential risk of bias was assessed by two review authors (NW, TAF) independently. All the judgements were made as low risk of bias, high risk of bias or unclear risk of bias (when insufficient information provided to permit judgement) and described for each trial in a ‘Methodological quality summary’ table (Figure 1).
Figure 1. Methodological quality summary: review authors’ judgements about each methodological quality item for each included study.

Sequence generation
The method used to generate the allocation sequence was assessed to ascertain whether the sequence was adequately generated. Judgements were made as low risk of bias when the investigators described a random component in the sequence generation process, such as referring to a random number table, using a computerised random number generator, coin tossing, shuffling cards or envelopes, throwing dice, drawing of lots and minimization.
Allocation concealment
Quality of allocation concealment in the randomisation to treatment conditions was assessed to ascertain whether allocation was adequately concealed. The judgements were made as low risk of bias when participants and investigators enrolling participants could not foresee assignment because one of the following, or an equivalent method, was used to conceal allocation: central allocation (including telephone, web-based and pharmacy-controlled randomisation); sequentially numbered drug containers of identical appearance; and sequentially numbered, opaque sealed envelopes.
Blinding
Quality of blinding was rated to ascertain whether the outcome measures were assessed by an independent assessor who was blind to treatment allocation. A self-report questionnaire can be a ‘blind’ measurement if the participants were adequately blinded to their allocated treatment. The judgements were made as low risk of bias when any one of the following was done: no blinding, but the review authors judged that the outcome and the outcome measurement were not likely to be influenced by lack of blinding; blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken; either participants or some key study personnel were not blinded but outcome assessment was blinded and the non-blinding of others was unlikely to introduce bias.
Incomplete outcome data
Adequate addressing of incomplete outcome data was regarded as low risk of bias both when less than 20% of the allocated participants to each group were assessed and results of the assessment were reported at the primary time point of the present review in both intervention groups regardless of the number of participants who dropped out from the allocated intervention, and when reasons for missing outcome data were unlikely to be related to the true outcome.
Selective outcome reporting
The completeness of outcome data was assessed to ascertain whether the primary outcome ‘response’ was adequately provided in the original report and no imputing (see Dealing with missing data) was required for our analysis at the primary time point, which was the end of acute-phase treatment.
Other sources of bias
Sponsorship bias was regarded as high risk of bias when the original study was funded by a pharmaceutical company or when other sources of potential bias were detected. Other sources of bias referred to any bias in certain circumstances, for example, in relation to trial design or setting.
Measures of treatment effect
See also Data synthesis.
Dichotomous data
Odds ratios (ORs) and their 95% confidence intervals (CIs) were calculated.
Continuous data
Mean differences or standardised mean differences (SMDs) and their 95% CIs were calculated (See Data synthesis).
Unit of analysis issues
Cluster-randomised trials
The effect size in cluster-randomised trials was estimated using the intracluster correlation coefficient (ICC), where provided, to adjust for cluster effects.
Crossover trials
It was planned to use the first active treatment phase in analyses.
Multi-intervention trials
For studies with two active or control arms, the results of both comparisons were pooled by dividing the opposite arm into two equal numbers for the purpose of avoiding unit of analysis errors (Ramsay 2005).
Dealing with missing data
Dichotomous data
The analysis was calculated on an intention-to-treat (ITT) basis, where dropouts were always included in the analysis. Where participants withdrew from the trial before the endpoint, it was assumed that they had not experienced the outcome by the end of the trial.
When the efficacy dichotomous outcomes of interest were not reported but baseline and endpoint means and standard deviations (SD) of the HAM-D (or any other depression scale) were provided, we imputed the number of participants with responses and remission by using a validated statistical method, for example according to the following formula for the response outcome (Furukawa 2005):
number of responders at endpoint = number of participants at endpoint * normal standard distribution corresponding to (50% of the baseline score - endpoint score)/SD.
When the SD was not reported, its value was imputed by pooling the SDs reported in the other included trials (Altman 1996; Furukawa 2006), although using both of these imputing methods at the same time has not yet been empirically supported.
Continuous data
When there were missing data and the method of ‘last observation carried forward’ (LOCF) had been used to do an ITT analysis, then the LOCF data were used with due consideration of the potential bias and uncertainty introduced.
Only data reported with a point estimate, SDs and number of participants at the time point were included in the analysis. The above imputation method for SDs was not used because we could not assess the skewness of the data without SDs (See Data synthesis).
Assessment of heterogeneity
Heterogeneity between the studies was assessed by using the Chi2 test, the I2 statistic and by visual inspection of the results. A P value of less than 0.1 for the Chi2 test or an I2 value of greater than 50% were considered to be suggestive of heterogeneity, although these definitions as recommended in the Cochrane Handbook might be arbitrary because they depends on the number of studies included in the analysis, the direction and magnitude of the treatment effects and the strength of evidence against the null hypothesis of homogeneity. Where significant heterogeneity was detected and was unexplained by subgroup analysis, other potential sources of the heterogeneity were investigated.
Assessment of reporting biases
Funnel plot analysis was performed to check for existence of small study effects including publication bias.
Data synthesis
All the analyses were conducted by NW using RevMan 5.0.
A random-effects model was used to pool the results of single studies because we anticipated this model was likely to provide the best fit to the data given the heterogeneity (Furukawa 2002). In addition, the model is more conservative than a fixed-effect model and incorporates both within-study and between-study variance.
Dichotomous data
A random-effects model using the odds ratio (OR) was used for the primary analysis rather than a random-effects model using risk ratio (RR) because it has been shown to have the highest generalisability in our empirical examination of summary effect measures for meta-analyses (Furukawa 2002). The 95% confidence interval (CI) was presented along with its precise P value. The robustness of this summary measure was routinely examined by checking the fixed-effect model using ORs, and the random-effects model using RRs. Fixed-effect analyses were done routinely for the continuous outcomes as well, to investigate the effect of the choice of method on the estimates. Material differences between the models were reported.
Continuous data
Continuous data were analysed using mean differences or standardised mean differences (where different measurement scales were used) and the random-effects model. The 95% CI was presented along with its precise P value. Skewed data were presented descriptively and were not included in the meta-analyses. Outcomes were considered skewed when the mean was smaller than twice the SD.
P values and statistical significance
We did not set any alpha level for ‘statistical significance’ in the outcomes. Instead, the effect estimate, its 95% CI and the precise P value were always presented because the conventional significance threshold at P value of 0.05 is an arbitrary one, as P values are smaller in a larger study than in a smaller study (Higgins 2008).
Subgroup analysis and investigation of heterogeneity
Subgroup analyses should be performed and interpreted with caution because multiple analyses could lead to false positive conclusions (Oxman 1992). We planned to perform the following subgroup analyses for the primary outcome.
For individual comparator drugs.
For the treatment settings (e.g. psychiatric inpatients or outpatients in primary care), because the treatment setting is thought to reflect the severity of depression.
Elderly participants (aged 65 years or older) separately from other adult participants.
The subgroup analyses have been amended from the published protocol, in which there are five subgroup analyses (Mirtazapine dosing, Comparator dosing, Depression severity, Treatment settings, Elderly participants), because adequate numbers of studies were not available for these analyses.
Where significant heterogeneity was unexplained by subgroup analysis other potential sources, such as depression severity at baseline, focusing on refractory depression and inclusion of bipolar participants, were investigated.
Sensitivity analysis
Sensitivity analyses were also planned. By limiting the studies to those with higher quality we examined if the results changed and we meant to check for the robustness of the observed findings for the primary outcome by:
excluding trials for which the response rate at the end of the acute-phase treatment had to be calculated based on the above imputation method;
excluding trials funded by or with at least one author affiliated to a pharmaceutical company marketing mirtazapine. This latter sensitivity analysis is particularly important in view of the recent repeated findings that funding strongly affects outcomes of research studies (Als-Nielsen 2003; Bhandari 2004; Lexchin 2003; Montgomery 2004; Perlis 2005; Procyshyn 2004) and because industry sponsorship and authorship of clinical trials have been increasing over the last 20 years (Buchkowsky 2004).
The sensitivity analyses have been amended from the published protocol, in which there are six sensitivity analyses (excluding trials with unclear concealment of random allocation and/or unclear double blinding, excluding trials whose drop out rate is greater than 20%, performing the worst case/best case scenario ITT, excluding trials for which the response rates had to be calculated based on the imputation method/borrowed from other trials, examination of “wish bias” by comparing mirtazapine as investigational drug vs mirtazapine as comparator, excluding studies funded by the pharmaceutical company marketing mirtazapine), because adequate numbers of studies were not available for these analyses.
RESULTS
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies.
Results of the search
Our initial search strategy yielded 94 trials including 135 references (Figure 2). After examining their titles and abstracts, 82 trials appeared to meet the inclusion criteria and their full texts were obtained. Among these, 45 studies were written in Chinese. We commissioned a professional translator for the full translation of these papers. The translation process is still ongoing, so in the present review we considered all Chinese studies as awaiting assessment. Through a contact with authors of the trials, experts in the area and the manufacturer of mirtazapine, we also obtained information on unpublished data not yet available in the published information (Hoyberg 1996; Schoemaker 2002; Thase 2000) and unpublished data from the manufacturer (Organon 85146). Eight studies were finally excluded. Twenty-nine studies with a total enrolment of 4974 participants were identified as satisfying our inclusion criteria and were included in our final analyses.
Figure 2.

Included studies
Design
All the included trials employed randomisation for individual participants. Neither cluster nor crossover trials were identified. The participants were followed up for six weeks (range: 2 to 24 weeks) in a majority of the trials (15 trials). Only one trial reported data after the end of the continuation treatment (at 24 weeks) (Wade 2003).
Setting
Seven trials enrolled psychiatric inpatients only (Brunnauer 2008; Guelfi 2000; Organon 85146; Richou 1995; Schule 2006; van Moffaert 1995; Zivkov 1995), the focus was placed on participants in primary care in one study (Wade 2003) and both psychiatric inpatients and outpatients were included in the other trials.
Participants
In all but one trial (Marttila 1995) the participants were diagnosed to have depression based on the DSM. Elderly participants (over 65 years of age) were included in 16 trials, two trials focused only on older adults or elderly participants. One trial limited the participants to those aged 60 years or older (Hoyberg 1996) and the other limited participants to those aged 65 years or older (Schatzberg 2002). The participants with psychiatric or physical disorders as co-morbidities with depression were excluded in a great majority of the trials (25 trials). Female participants who were potentially or actually pregnant or breastfeeding on entry into the trials were also excluded in 20 trials and the other trials did not state whether such participants were included or excluded. Two trials focused on refractory or treatment-resistant depression (Fava 2006; Thase 2000).
Interventions
All but four trials (Amini 2005; Benkert 2006; Schule 2006; Winokur 2003) employed flexible dosing regimens for both the mirtazapine and comparator arms. No trials examined different doses or schedules of the same therapy.
In terms of the comparator drug, in ten trials a TCA (amitriptyline in seven; clomipramine, doxepin and nortriptyline each in one) was used as the comparator drug; in 13 trials an SSRI (citalopram in one, fluoxetine in five, fluvoxamine in one, paroxetine in four and sertraline in two) was used; in two an SNRI (venlafaxine) was used; in two an heterocyclic AD (trazodone) was used; and in two a newer AD (reboxetine) was used. No trials using any other SNRIs as the comparator, such as duloxetine or milnacipran, were identified.
A placebo pill was used as a comparator intervention in three trials (Bremner 1995; Halikas 1995; Smith 1990).
Outcomes
In all but two trials (Fava 2006; Leinonen 1999) the 17- or 21-item HAM-D was used for reporting the response. With regard to the acceptability outcomes, 23 trials provided the number of dropouts due to any reason and 23 trials reported the number of dropouts due to the development of adverse event during the trials; these trial results did not always overlap.
Excluded studies
See: Characteristics of excluded studies. Eight studies were excluded for the following reasons: not a relevant diagnostic status (Kremer 1995; Peyron 1996; Tulen 1996), a review of other studies (Bruijin 1996; Kasper 1997a; Kasper 1997b), not employing random allocation (Zourkova 2001), and combined therapy of mirtazapine and another antidepressant (Blier 2004).
Ongoing studies
No ongoing studies were identified.
Studies awaiting classification
Forty-six studies written in Chinese are awaiting classification. One study (Catterson 1996a) needs further data for checking if the study meets the strict eligibility criteria. Seven studies (Blier 2009; Fang 2010; Kang 2009; Kim 2011; Paslakis 2010; Sarginson 2010; Scharnholz 2010) identified after the completion of the review are classified in this category.
Risk of bias in included studies
Two of the three review authors (NW, IMO, TAF) independently assessed study quality. Any discrepancy between two authors was dissolved upon discussion (see: Figure 1; Figure 3).
Figure 3. Methodological quality graph: review authors’ judgements about each methodological quality item presented as percentages across all included studies.

Sequence generation
None of the trials described the sequence generation process.
Allocation
None of the trials reported whether allocation concealment was adequately performed.
Blinding
All trials but one (Fava 2006) were undertaken on a double-blind basis, but none of the trials reported information to judge whether it was likely or unlikely that the blinding had been broken.
Incomplete outcome data
Only three trials (Amini 2005; Bremner 1995; Leinonen 1999) were rated as adequate in terms of addressing incomplete outcome data.
Selective reporting
Outcomes in terms of the primary outcome (response) at the primary time point (at the end of acute-phase treatment) were obtained in all but two trials (Hoyberg 1996; Winokur 2003) without using the imputation method.
Other potential sources of bias
One study (Fava 2006) was funded by the National Institute of Mental Health (NIMH), and one study (Amini 2005) was unclear in terms of sponsorship bias. The other studies were sponsored by, or had at least one author affiliated to, a pharmaceutical company.
No other potential sources of bias were identified.
Effects of interventions
ORs for the efficacy data that were larger than one (falling to the right of the midline in a graph) and those for the acceptability and tolerability data that were smaller than one indicate a difference in favour of mirtazapine.
1. Mirtazapine versus tricyclic antidepressants (TCAs)
Primary analysis
1.1 Primary outcome: response
There was no robust evidence to detect a difference between mirtazapine and TCAs in terms of the response outcome at two weeks (8 studies; OR 0.85, 95% CI 0.64 to 1.13, P = 0.27) (Analysis 1.1; Figure 4) and at end of the acute-phase treatment (9 studies; OR 0.89, 95% CI 0.72 to 1.10, P = 0.29) (Analysis 1.2; Figure 5). No substantial heterogeneity was observed.
Figure 4. Forest plot of comparison: 1 MIRTAZAPINE vs TCAs, outcome: 1.1 Primary outcome (response) at 2 weeks.

Figure 5. Forest plot of comparison: 1 MIRTAZAPINE vs TCAs, outcome: 1.2 Primary outcome (response) at end of the acute-phase treatment.

1.2 Secondary outcomes
1.2.1 Remission
There was robust evidence to detect any difference between mirtazapine and TCAs in terms of the remission outcome at two weeks (8 studies; OR 0.85, 95% CI 0.55 to 1.32, P = 0.47) (Analysis 1.3) and at end of the acute-phase treatment (9 studies; OR 0.86, 95% CI 0.69 to 1.08, P = 0.19) (Analysis 1.4).
1.2.2 Depression severity
Two studies (Mullin 1996; Zivkov 1995) contributed to the meta-analysis at two weeks and one study (Organon 85146) did so at the end of acute-phase treatment. Information from studies with skewed data were not included in the meta-analysis but were included in a descriptive table (Analysis 1.32; Analysis 1.33). From the results of the meta-analysis, there was no robust evidence to detect a difference between mirtazapine and TCAs in terms of depression severity on a continuous scale at two weeks (2 studies; Analysis 1.5) and at the end of the acute-phase treatment (1 study; Analysis 1.6).
1.2.3 Social adjustment
One study (Mullin 1996) contributed to the meta-analysis at two weeks and three studies (Marttila 1995; Mullin 1996; Richou 1995) did so at the end of the acute-phase treatment. There was no robust evidence to detect a difference between mirtazapine and TCAs in terms of social adjustment at two weeks (1 study; Analysis 1.7) and at end of the acute-phase treatment (3 studies; Analysis 1.8).
1.2.4 Health-related quality of life (HRQoL)
No data were available.
1.2.5 Withdrawal due to any reason
There was no robust evidence to detect a difference between mirtazapine and TCAs in terms of withdrawal due to any reason during the acute-phase treatment (7 studies; Analysis 1.9).
1.2.6 Withdrawal due to the development of an adverse event
There was no robust evidence to detect a difference between mirtazapine and TCAs in terms of withdrawal due to the development of an adverse event during the acute-phase treatment (8 studies; Analysis 1.10).
1.2.7 Having some adverse events
There was no robust evidence to detect a difference between mirtazapine and TCAs in terms of developing adverse events during the acute-phase treatment (2 studies; Analysis 1.11).
1.2.8 Individual adverse events
No data were available with regard to diarrhoea, weight loss and completed suicide. Mirtazapine was less likely than TCAs to bring on hypertension or tachycardia (4 studies; OR 0.44, 95% CI 0.24 to 0.81, P = 0.008) (Analysis 1.12) and tremor (7 studies; OR 0.36, 95% CI 0.22 to 0.57, P < 0.0001) (Analysis 1.24). There was no robust evidence to detect a difference between mirtazapine and TCAs in terms of developing other types of individual adverse events.
Secondary analysis
1.3 Subgroup analysis and investigation of heterogeneity
1.3.1 Individual comparator drugs
There was no robust evidence to detect a difference between mirtazapine and any specific type of TCA comparator in terms of the response outcome during the acute-phase treatment (8 studies at 2 weeks: Analysis 1.1, 9 studies at end of acute-phase treatment:Analysis 1.2).
1.3.2 Treatment settings
No studies focused on participants in primary care clinics only. Limiting findings to studies focusing on psychiatric inpatients, there was no robust evidence to detect a difference between mirtazapine and TCAs in terms of the response to acute-phase treatment (2 studies at 2 weeks: Analysis 1.28, 3 studies at end of the acute-phase treatment: Analysis 1.29).
1.3.3 Elderly participants
No studies focused on elderly participants only.
1.4 Sensitivity analysis
1.4.1 Studies without imputation
All but one study (Hoyberg 1996) did not need any imputation method for the primary outcome at the end of the acute-phase treatment. Limiting findings to these studies, there was no robust evidence to detect a difference between mirtazapine and TCAs in terms of the response to acute-phase treatment (7 studies at 2 weeks: Analysis 1.30, 8 studies at end of the acute-phase treatment:Analysis 1.31).
1.4.2 Studies without sponsorship bias
From one study (Fava 2006), there was no robust evidence to detect a difference between mirtazapine and TCAs in the response to acute-phase treatment.
2. Mirtazapine versus selective serotonin reuptake inhibitors (SSRIs)
Primary analysis
2.1 Primary outcome: response
In comparison with SSRIs, mirtazapine was shown to be significantly more effective in terms of response at two weeks (12 studies; OR 1.57, 95% CI 1.30 to 1.88, P < 0.00001) (Analysis 2.1; Figure 6) and at the end of acute-phase treatment (12 studies; OR 1.19, 95% CI 1.01 to 1.39, P = 0.04) (Analysis 2.2; Figure 7). There was no robust evidence to detect a difference between mirtazapine and SSRIs at the end of the continuation treatment (at 24 weeks) based on one study (Wade 2003) (1 study; OR 1.60, 95% CI 0.91 to 2.81, P = 0.10) (Analysis 2.3). No substantial heterogeneity was observed at any of the three time points.
Figure 6. Forest plot of comparison: 2 MIRTAZAPINE vs SSRIs, outcome: 2.1 Primary outcome (response) at 2 weeks.

Figure 7. Forest plot of comparison: 2 MIRTAZAPINE vs SSRIs, outcome: 2.2 Primary outcome (response) at end of the acute-phase treatment.

2.2 Secondary outcomes
2.2.1 Remission
Mirtazapine was shown to be significantly more effective than SSRIs in terms of the remission outcome at two weeks (12 studies; OR 1.82, 95% CI 1.36 to 2.44, P < 0.0001) (Analysis 2.4). At the end of the acute-phase treatment, there was no robust evidence to detect a difference between mirtazapine and SSRIs (12 studies; OR 1.17, 95% CI 0.98 to 1.40, P = 0.08) (Analysis 2.5). There was no robust evidence to detect a difference between mirtazapine and SSRIs at the end of the continuation treatment (1 study; OR 1.89, 95% CI 1.01 to 3.54, P = 0.05) (Analysis 2.6).
2.2.2 Depression severity
No data were available for the meta-analysis. Information from studies with skewed data were not included in the meta-analysis but in a descriptive table (2 studies at 2 weeks: Analysis 2.31, 3 studies at end of the acute-phase treatment: Analysis 2.32).
2.2.3 Social adjustment
No data were available.
2.2.4 Health-related quality of life (HRQoL)
No data were available.
2.2.5 Withdrawal due to any reason
There was no robust evidence to detect a difference between mirtazapine and SSRIs in terms of withdrawal due to any reason during the acute-phase treatment (11 studies; Analysis 2.7).
2.2.6 Withdrawal due to the development of an adverse event
There was no robust evidence to detect a difference between mirtazapine and SSRIs in terms of withdrawal due to the development of an adverse event during the acute-phase treatment (11 studies; Analysis 2.8).
2.2.7 Having some adverse events
There was no robust evidence to detect a difference between mirtazapine and SSRIs in terms of developing any adverse events during the acute-phase treatment (7 studies; Analysis 2.9).
2.2.8 Individual adverse events
No data were available with regard to hypertension or tachycardia and completed suicide. In comparison with SSRIs, mirtazapine was more likely to cause dry mouth (10 studies; OR 1.80, 95% CI 1.37 to 2.36, P < 0.0001) (Analysis 2.14), weight gain or increased appetite (11 studies; OR 4.23, 95% CI 2.93 to 6.11, P < 0.00001) (Analysis 2.16), fatigue (8 studies; OR 1.53, 95% CI 1.08 to 2.15, P = 0.02) (Analysis 2.21) and somnolence (11 studies; OR 1.81, 95% CI 1.39 to 2.37, P < 0.0001) (Analysis 2.25) but less likely to cause sweating (5 studies; OR 0.25, 95% CI 0.15 to 0.44, P < 0.00001) (Analysis 2.11), diarrhoea (8 studies; OR 0.57, 95% CI 0.41 to 0.80, P = 0.001) (Analysis 2.13), nausea or vomiting (11 studies; OR 0.33, 95% CI 0.26 to 0.43, P < 0.00001) (Analysis 2.15), sexual dysfunction (4 studies; OR 0.31, 95% CI 0.13 to 0.74, P = 0.009) (Analysis 2.18), headache (11 studies; OR 0.69, 95% CI 0.56 to 0.86, P = 0.0008) (Analysis 2.22), tremor (5 studies; OR 0.34, 95% CI 0.18 to 0.66, P = 0.001) (Analysis 2.23) or sleep disturbance (5 studies; OR 0.52, 95% CI 0.31 to 0.86, P = 0.01) (Analysis 2.24). There was no robust evidence to detect a difference between mirtazapine and SSRIs in terms of developing other types of individual adverse events.
Secondary analysis
2.3 Subgroup analysis and investigation of heterogeneity
2.3.1 Individual comparator drugs
In terms of response at two weeks, mirtazapine was shown to be significantly more effective than paroxetine (3 studies; OR 2.39, 95% CI 1.42 to 4.02, P = 0.001) and sertraline (2 studies; OR 1.45, 95% CI 1.04 to 2.02, P = 0.03) (Analysis 2.1). Mirtazapine was shown to be significantly more effective than fluoxetine at the end of the acute-phase treatment (5 studies; OR 1.55, 95% CI 1.07 to 2.23, P = 0.02) (Analysis 2.2). There was no robust evidence to detect a difference between mirtazapine and the other individual comparator drugs.
2.3.2 Treatment settings
No studies focused on psychiatric inpatients or participants in primary care clinics only.
2.3.3 Elderly participants
Based on one study (Schatzberg 2002), mirtazapine was shown to be significantly more effective than paroxetine at two weeks (OR 2.59, 95% CI 1.35 to 4.97, P = 0.004) (Analysis 2.1). There was no robust evidence to detect a difference between mirtazapine and paroxetine at the end of the acute-phase treatment (OR 1.41, 95% CI 0.86 to 2.32, P = 0.17) (Analysis 2.2).
2.4 Sensitivity analysis
2.4.1 Studies without imputation
All but one study (Winokur 2003) did not need any imputation method for the primary outcome at the end of the acutephase treatment. Limiting findings to these studies, mirtazapine was shown to be significantly more effective than SSRIs in terms of response at two weeks (11 studies; OR 1.58, 95% CI 1.31 to 1.90, P < 0.00001) (Analysis 2.29). There was no robust evidence to detect a difference between mirtazapine and SSRIs at the end of the acute-phase treatment (11 studies; OR 1.17, 95% CI 1.00 to 1.38, P = 0.05) (Analysis 2.30).
2.4.2 Studies without sponsorship bias
All studies were funded by a pharmaceutical company.
3. Mirtazapine versus serotonin-noradrenaline reuptake inhibitors (SNRIs)
Primary analysis
3.1 Primary outcome: response
Mirtazapine was shown to be significantly more effective than venlafaxine at two weeks (2 studies; OR 2.29, 95% CI 1.45 to 3.59, P = 0.0003) (Analysis 3.1) and at the end of the acute-phase treatment (2 studies; OR 1.53, 95% CI 1.03 to 2.25, P = 0.03) (Analysis 3.2). No substantial heterogeneity was observed.
3.2 Secondary outcomes
3.2.1 Remission
Mirtazapine was shown to be significantly more effective than venlafaxine in terms of the remission outcome at two weeks (2 studies; OR 2.34, 95% CI 1.07 to 5.13, P = 0.03) (Analysis 3.3). At the end of the acute-phase treatment, there was no robust evidence to detect a difference between mirtazapine and venlafaxine (2 studies; OR 1.55, 95% CI 0.98 to 2.47, P = 0.06) (Analysis 3.4).
3.2.2 Depression severity
No data were available for the meta-analysis.
3.2.3 Social adjustment
No data were available.
3.2.4 Health-related quality of life (HRQoL)
No data were available.
3.2.5 Withdrawal due to any reason
Mirtazapine was shown to be significantly more effective than venlafaxine in terms of withdrawal due to any reason (2 studies; OR 0.65, 95% CI 0.43 to 0.99, P = 0.04) during the acute-phase treatment (Analysis 3.5).
3.2.6 Withdrawal due to the development of adverse event
There was no robust evidence to detect a difference between mirtazapine and venlafaxine in terms of withdrawal due to the development of adverse event during the acute-phase treatment (2 studies; Analysis 3.6).
3.2.7 Having some adverse events
There was no robust evidence to detect a difference between mirtazapine and venlafaxine in terms of developing any adverse events during the acute-phase treatment (1 study; Analysis 3.7).
3.2.8 Individual adverse events
No data were available with regard to hypertension or tachycardia, diarrhoea, weight gain or increased appetite, weight loss or anorexia, sexual dysfunction, dizziness or vertigo, tremor and attempted suicide. In comparison with venlafaxine, mirtazapine was more likely to cause fatigue (1 study; OR 2.43, 95% CI 1.30 to 4.55, P = 0.006) (Analysis 3.14) but less likely to cause sweating (1 study; OR 0.03, 95% CI 0.00 to 0.45, P = 0.01) (Analysis 3.9), constipation (1 study; OR 0.22, 95% CI 0.06 to 0.83, P = 0.02) (Analysis 3.10) or sleep disturbance (1 study; OR 0.02, 95% CI 0.00 to 0.41, P = 0.01) (Analysis 3.16). There was no robust evidence to detect a difference between mirtazapine and venlafaxine in terms of developing other types of individual adverse events.
Secondary analysis
3.3 Subgroup analysis and investigation of heterogeneity
3.3.1 Individual comparator drugs
No studies focusing on SNRIs other than venlafaxine were identified.
3.3.2 Treatment settings
No studies focused on participants in primary care clinics only. In terms of inpatients, based on one study (Guelfi 2000) no robust evidence was observed to detect a difference between mirtazapine and venlafaxine at two weeks (OR 1.88, 95% CI 0.97 to 3.64, P = 0.06) (Analysis 3.1) or at the end of the acute-phase treatment (OR 1.64, 95% CI 0.87 to 3.10, P = 0.13) (Analysis 3.2).
3.3.3 Elderly participants
No studies focused on elderly participants only.
3.4 Sensitivity analysis
3.4.1 Studies without imputation
The two studies did not need any imputation method for the primary outcome at the end of the acute-phase treatment and the results of the analysis were the same as those from the primary analyses (Analysis 3.1; Analysis 3.2).
3.4.2 Studies without sponsorship bias
All studies were funded by a pharmaceutical company.
4. Mirtazapine versus heterocyclic antidepressants
Primary analysis
4.1 Primary outcome: response
There was no robust evidence to detect a difference between mirtazapine and an heterocyclic antidepressant (trazodone only) at two weeks (2 studies; OR 1.14, 95% CI 0.64 to 2.04, P = 0.66) (Analysis 4.1) or at the end of the acute-phase treatment (2 studies; OR 1.50, 95% CI 0.95 to 2.37, P = 0.08) (Analysis 4.2). No substantial heterogeneity was observed.
4.2 Secondary outcomes
4.2.1 Remission
There was no robust evidence to detect a difference between mirtazapine and trazodone at two weeks (2 studies; OR 1.00, 95% CI 0.36 to 2.80, P = 1.00) (Analysis 4.3) and at the end of the acute-phase treatment (2 studies; OR 1.38, 95% CI 0.76 to 2.52, P = 0.29) (Analysis 4.4).
4.2.2 Depression severity
No data were available for the meta-analysis.
4.2.3 Social adjustment
No data were available.
4.2.4 Health-related quality of life (HRQoL)
No data were available.
4.2.5 Withdrawal due to any reason
There was no robust evidence to detect a difference between mirtazapine and trazodone in terms of withdrawal due to any reason during the acute-phase treatment (2 studies; OR 0.90, 95% CI 0.47 to 1.72, P = 0.76) (Analysis 4.5).
4.2.6 Withdrawal due to the development of an adverse event
There was no robust evidence to detect a difference between mirtazapine and trazodone in terms of withdrawal due to the development of an adverse event during the acute-phase treatment (2 studies; OR 0.61, 95% CI 0.25 to 1.51, P = 0.29) (Analysis 4.6).
4.2.7 Having some adverse events
No data were available.
4.2.8 Individual adverse events
No data were available with regard to sweating, diarrhoea, nausea, anxiety or agitation, fatigue, tremor and somnolence. In comparison with trazodone, mirtazapine was less likely to cause hypotension or bradycardia (OR 0.17, 95% CI 0.03 to 1.00, P = 0.05) (Analysis 4.8). There was no robust evidence to detect a difference between mirtazapine and trazodone in terms of developing other types of individual adverse events.
Secondary analysis
4.3 Subgroup analysis and investigation of heterogeneity
4.3.1 Individual comparator drugs
No studies focusing on antidepressants other than trazodone were identified.
4.3.2 Treatment settings
No studies focused on participants in primary care clinics only. In terms of inpatients, based on one study (van Moffaert 1995) no robust evidence was identified to detect a difference between mirtazapine and trazodone in terms of the response at two weeks (OR 1.16, 95% CI 0.54 to 2.48, P = 0.70) (Analysis 4.1) or at the end of the acute-phase treatment (OR 1.50, 95% CI 0.86 to 2.63, P = 0.16) (Analysis 4.2).
4.3.3 Elderly participants
No studies focused on elderly participants only.
4.4 Sensitivity analysis
4.4.1 Studies without imputation
The two studies did not need any imputation method for the primary outcome at the end of the acute-phase treatment and the results of the analysis were the same as those from the primary analysis (Analysis 4.1; Analysis 4.2).
4.4.2 Studies without sponsorship bias
All studies were funded by a pharmaceutical company.
5. Mirtazapine versus newer antidepressants
Primary analysis
5.1 Primary outcome: response
There was no robust evidence to detect a difference between mirtazapine and a newer antidepressant (reboxetine only) in terms of the response outcome at two weeks (1 study; OR 1.00, 95% CI 0.18 to 5.67, P = 1.00) (Analysis 5.1) and at end of the acute-phase treatment (1 study; OR 1.00, 95% CI 0.27 to 3.67, P = 1.00) (Analysis 5.2).
5.2 Secondary outcomes
5.2.1 Remission
There was no robust evidence to detect a difference between mirtazapine and reboxetine in terms of the remission outcome at two weeks (1 study; OR 1.00, 95% CI 0.06 to 17.18, P = 1.00) (Analysis 5.3) and at the end of the acute-phase treatment (1 study; OR 1.26, 95% CI 0.33 to 4.73, P = 0.74) (Analysis 5.4).
5.2.2 Depression severity
There was no robust evidence to detect a difference between mirtazapine and reboxetine in terms of the severity of depression at two weeks (1 study; SMD −0.37, 95% CI −1.00 to 0.25, P = 0.24) (Analysis 5.5). At the end of the acute-phase treatment the data were skewed and presented in a descriptive table (1 study; Analysis 5.8).
5.2.3 Social adjustment
No data were available.
5.2.4 Health-related quality of life (HRQoL)
No data were available.
5.2.5 Withdrawal due to any reason
Not estimable because no participant in either group withdrew due to any reason (2 studies; Analysis 5.6).
5.2.6 Withdrawal due to the development of an adverse event
Not estimable, because no participant in either group withdrew due to the development of an adverse event (2 studies; Analysis 5.7).
5.2.7 Having some adverse events
No data were available.
5.2.8 Individual adverse events
No data were available with regard to any individual adverse event.
Secondary analysis
5.3 Subgroup analysis and investigation of heterogeneity
5.3.1 Individual comparator drugs
No studies focusing on antidepressants other than reboxetine were identified.
5.3.2 Treatment settings
The two studies focused on inpatients.
5.3.3 Elderly participants
No studies focused on elderly participants only.
5.4 Sensitivity analysis
5.4.1 Studies without imputation
One study needed the imputation method for the primary outcome at two weeks. The other provided no usable data for the primary outcome.
5.4.2 Studies without sponsorship bias
The two studies were funded by a pharmaceutical company.
6. Funnel plot analysis
There was no robust evidence of publication bias or other small study effects based on visual inspections of the funnel plot with regard to the primary outcome (response) at the primary time point (at the end of the acute-phase treatment) (Figure 8).
Figure 8. Funnel plot of comparison: 6 Funnel plot analysis: Primary outcome (response) at end of the acute-phase treatment, outcome: 6.1 vs all compounds.

DISCUSSION
Summary of main results
This systematic review and meta-analysis examined the efficacy, acceptability and tolerability of mirtazapine for the acute-phase treatment for depression, in comparison with other antidepressive agents.
In terms of the primary outcome, overall a response was achieved at two weeks in 616 (26.6%) of the 2316 participants treated with mirtazapine, and in 1413 (61.0%) out of the 2316 participants treated with mirtazapine at the end of the acute-phase treatment. In terms of one of the secondary outcomes, a remission was achieved in 222 (9.6%) of the 2316 participants treated with mirtazapine at two weeks, and in 847 (36.6%) out of the 2316 participants treated with mirtazapine at the end of acute-phase treatment. In terms of acceptability, 512 (25.2%) out of the 2030 participants treated with mirtazapine withdrew from treatment at some time point during the course of treatment.
We concluded from the results that there was no robust evidence to detect a difference between mirtazapine and other types of antidepressants in terms of the response outcome at the end of the acute-phase treatment, at approximately six weeks. At two weeks, mirtazapine is likely to be more effective than either SSRIs or SNRIs, especially paroxetine and venlafaxine. These results were confirmed even after an additional sensitivity analysis was conducted that excluded the two trials (Fava 2006; Thase 2000) that focused on treatment-resistant depression. In terms of tolerability, mirtazapine was not statistically significantly superior or inferior to other antidepressants.
Due to the unique pharmacological profile of mirtazapine, some anti-histaminergic effects have been thought to bring about drowsiness, sedation, dry mouth and an increase in appetite and body weight (Kent 2000). These side effects might result in a dropout of participants treated with mirtazapine. Approximately 70% of the participants treated with mirtazapine experienced at least one adverse event during the trials; and dry mouth, somnolence, weight or appetite increase, fatigue and headache were the most frequently observed. In comparison with SSRIs, treatment with mirtazapine was significantly more likely to lead to the development of dry mouth, weight gain or increased appetite, fatigue and somnolence but was significantly less likely to lead to the development of sweating, diarrhoea, nausea or vomiting, sexual dysfunction, headache, tremor and sleep disturbance.
Overall completeness and applicability of evidence
Participants
All but two studies (Fava 2006; Thase 2000) included in this review were not conducted for refractory depression. Psychiatric inpatients were included in five studies (Guelfi 2000; Organon 85146; Richou 1995; van Moffaert 1995; Zivkov 1995), participants in primary care were included in one study (Wade 2003) and the other 19 studies included both psychiatric inpatients and outpatients. The findings from this review, therefore, may not be representative of participants with refractory depression nor mildly affected participants frequently seen in primary care. Moreover, there was only one trial in which recruitment was wholly conducted for the depressive elderly (Schatzberg 2002. The findings from this review may therefore not be representative for the depressive elderly.
Interventions
Considering the often chronic and recurrence-prone presentation of major depression, long-term or follow-up interventions are often required for optimal treatment of this disorder. However, we could find only one study (Wade 2003) examining the long-term efficacy of mirtazapine for major depression.
Outcomes
Treatments for major depression should be assessed not only by psychiatric symptoms but also by general functioning and quality of life. However, no trials included in this review incorporated those outcomes. Considering that major depression is associated with a marked personal, social and economic morbidity, the under-investigation of these outcomes is a problem.
Quality of the evidence
No trials described methods of random sequence generation, and no trials reported the method of allocation concealment. It is conceivable that selection bias might have occurred in the trials included in this review. These methodologies should be adequately reported, as recommended in the CONSORT 2010 statement (Schulz 2010).
Physician and participant blinding were sought in all but one study (Fava 2006). However, no test of blinding success was conducted in any study. As a whole, there was little information on the outcome assessment process and the extent to which detection bias might have occurred was uncertain.
At the end of the acute-phase treatment, only four studies (Amini 2005; Bremner 1995; Brunnauer 2008; Leinonen 1999) reported the outcomes for an 80% or higher proportion of participants initially allocated to treatment conditions, and the other studies probably did not due to higher dropout rates. This high attrition rate could have influenced treatment outcomes.
All but two trials (Amini 2005; Fava 2006) included in our meta-analysis were funded or conducted under the advice of a manufacturer of mirtazapine. On the other hand, it has been repeatedly reported that industry sponsorship could influence trial outcomes in favour of a drug manufacturer (Als-Nielsen 2003; Lexchin 2003; Perlis 2005). The present review may suffer from sponsorship bias.
Potential biases in the review process
Some possible strengths and limitations of this review should be noted.
Strengths
First, we imputed the response and remission outcomes by applying the threshold of the most conventional and prevalent depression severity scales using a validated statistical method; we did not use the outcomes defined by the authors of the original trials. Although this methodology may appear arbitrary and to have possibly resulted in the loss of important information from the original trials, recent evidence has shown that in published RCTs the statistically significant outcomes for efficacy tend to be more fully reported than non-significant outcomes do, and that in 62% of trials at least one primary outcome was changed, introduced or omitted with reference to the protocols (Chan 2004; Furukawa 2007). For this reason, we decided to adhere to our criteria defined a priori for the response and remission outcomes and impute them if they were not unavailable from the original trials. We think that, as long as the selective reporting of outcomes remains prevalent, our methodology should be used in future systematic reviews.
Second, in addition to the response rate we took the remission rate into account as one of the outcomes. Previously reported metaanalyses have generally taken only the response outcome into account. However, a recent series of RCTs on the effectiveness of the sequential use of antidepressants and cognitive-behavioural therapy for depression, named STAR*D and one of which (Fava 2006) was included in our systematic review, revealed that the remission rate was more consistently associated with a better prognosis in terms of the long-term outcome than the response (Rush 2007). We, therefore, propose that all future studies on this subject should report on the remission outcome in addition to the response outcome for depression.
Limitations
First of all, industry sponsorship can influence trial outcomes in favour of a drug manufacturer. We were unable to rule out the possibility that the dosing of either mirtazapine or the comparator drug might have been designed in such a way as to induce differences in favour of mirtazapine because the doses of the comparator drugs seemed lower than the usual doses in clinical practice in some of the included trials, especially in some of the trials comparing mirtazapine with fluoxetine or paroxetine (see: Characteristics of included studies). We initially intended to conduct a sensitivity analysis by excluding trials sponsored by pharmaceutical companies, but did not because only two out of the 25 trials were free of industry sponsorship.
The second limitation of the review was the treatment durations in the included RCTs (see also: Quality of the evidence). Sixteen out of the 25 included trials followed up the participants for only six weeks. The STAR*D study revealed that one third of those showing a response to treatment with antidepressants did so only after six weeks of therapy (and half of those who showed remission did so after six weeks) (Rush 2007). In addition, the durations of the RCTs included in our analysis were not sufficiently long to address the long-term side effects of mirtazapine. Long-term side effects should be considered as much as those observed in the short term because they could play a big part in determining the burden and effective outcome of therapies (Hoyberg 1996; Trivedi 2006); and rare but otherwise crucial outcomes could occur only in the long term. Furthermore, some adverse events including nausea tend to subside rapidly (Mullin 1996), whereas other adverse events including weight gain might be an ongoing problem that has potentially serious health implications in the long term. Future studies should include long-term adverse events among their outcomes. Addressing them may require a systematic review of studies dealing with the long-term effects of the drug in study designs other than RCTs; this review focused only on RCTs.
We are also concerned about the representativeness of the populations recruited in the included trials. Most of the included trials were carried out to investigate the efficacy of mirtazapine. Generally speaking, efficacy trials tend to include only symptomatic volunteers with no concomitant medical or psychiatric disease as opposed to enrolling participants seeking health care in typical clinical treatment settings (Trivedi 2006). Thus, efficacy trials may eventually lead to results with only limited ecological validity and generalisability to clinical practice. Future research on mirtazapine should include effectiveness trials enrolling participants seen in everyday practice.
Lastly, only one author was involved in the first stage of selecting studies and in making the preliminary list of potentially eligible studies, due to shortage of initial human resources in the review procedure. This might have led to possible human error in selection. However, selecting studies in this stage was conducted as over-inclusively as possible, and all the full text articles in this preliminary list were assessed by two review authors independently (NW and IMO). The final rating were made by consensus.
Agreements and disagreements with other studies or reviews
There have been several other reviews published recently that investigate the efficacy of mirtazapine in comparison with other types of antidepressants.
Although the faster onset of therapeutic action of mirtazapine in comparison with that of the SSRIs has been reported previously from a non head-to-head review of the results from three RCTs (Quitkin 2001), our systematic review showed that this result on comparative efficacy was not the same for all SSRIs.
The most recent review compared the remission rates and time to remission in participants with major depression taking either mirtazapine or an SSRI through a meta-analysis of the individual participant data from 15 RCTs (Thase 2010). This review concluded that mirtazapine therapy resulted in significantly higher remission rates than SSRI therapy during six weeks of treatment, particularly within the first 15 days of treatment. Another recently published review compared the benefits and harms of 12 second-generation antidepressants for the treatment of depression in adults (Gartlehner 2008). It concluded that the clinical response and remission rates are similar among second-generation antidepressants, including mirtazapine, at the end of the acute-phase treatment. In terms of onset of action, this review concluded that mirtazapine has a significantly faster onset of action than citalopram, fluoxetine, paroxetine or sertraline.
The results from these reviews appear to be quite similar to those from the present review, although we have to be cautious to draw a definitive conclusion given the possibility of sponsorship bias and the width of confidence intervals of the outcomes.
We have recently published a multiple-treatments meta-analysis (MTM) in which our data for mirtazapine were merged with those for 11 other new generation antidepressants and both the direct and indirect comparisons were statistically pooled (Cipriani 2009a). The MTM offers a clinically meaningful synthesis when several competing treatments are available for one disease (Lu 2006; Lumley 2002; Salanti 2008), as is the case with major depression, while examining the overall strength and consistency of the network of evidence. In this MTM, mirtazapine emerged as one of the top four antidepressants in terms of response but not in terms of acceptability. The relative merits and drawbacks of direct versus MTM comparisons are still debatable (Bucher 1997; Ioannidis 2006; Song 2003) and we need to carefully weigh up and synthesise the direct with indirect comparisons.
AUTHORS’ CONCLUSIONS
Implications for practice
Although mirtazapine is more likely to have a better efficacy profile than paroxetine or venlafaxine in terms of response at two weeks, in view of the similar efficacy of mirtazapine and other antidepressant agents at the end of the acute-phase treatment (at approximately six weeks), and of possible sponsorship bias, the results of the study led us to conclude that clinicians should also focus on other practically or clinically relevant considerations, such as differences in the side-effect profiles, to tailor the treatment to best fit an individual participant’s needs.
Mirtazapine is less likely to cause tremor than TCAs, and nausea and sexual dysfunction than SSRIs, but is more likely to cause weight gain and somnolence.
Implications for research
Since the great majority of trials on the efficacy of mirtazapine are funded by the manufacturer, and thus might be subject to some sponsorship bias, future RCTs on the effectiveness of mirtazapine should be funded by non-profit organizations. Furthermore, future trials should include appropriate QoL outcome measures and should adequately report the method of generation of random sequence, allocation concealment, and blinding in accordance with the CONSORT 2010 statement. Finally, the effectiveness of mirtazapine should be investigated by conducting RCTs recruiting participants from populations seeking treatment in ordinary clinical practice settings.
PLAIN LANGUAGE SUMMARY.
Mirtazapine versus other antidepressive agents for depression
Major depression is characterised by a persistent low mood and loss of interest and pleasure. These symptoms are often accompanied by loss of appetite, insomnia, fatigue, poor concentration, inappropriate guilty feelings and even suicide. Depression was the third leading cause of disease burden among all diseases experienced by humankind in 2002. Antidepressants are used in treatment for major depression. They are the mainstay of treatment. Among them, mirtazapine is known to have a unique pharmacological profile and thus is supposed to differ in its efficacy and adverse effects profile in comparison with other antidepressants.
The evidence from this review, which included findings from 29 randomised controlled trials (4974 participants in total), suggests that mirtazapine is likely to have a faster onset of action than the most frequently used type of antidepressants, which are the selective serotonin reuptake inhibitors (SSRIs). It would appear that mirtazapine is superior to SSRIs at the end of treatment over 6 to 12 weeks. Mirtazapine causes adverse events that lead to a similar frequency of dropouts as SSRIs and tricyclic antidepressants, although adverse event profile of mirtazapine is unique. Mirtazapine is likely to cause weight gain or increased appetite and somnolence but is less likely to cause nausea or vomiting and sexual dysfunction than SSRIs.
ACKNOWLEDGEMENTS
The authors would like to thank Julian Higgins, Georgia Salanti and John Geddes for their helpful comments and feedback on the protocol. We would also like to thank Hugh McGuire for his assistance with this review.
CHARACTERISTICS OF STUDIES
Characteristics of included studies [ordered by study ID]
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: In- and outpatients |
|
| Interventions | 1. Mirtazapine: 30 mg/day, N = 18 2. Fluoxetine: 20 mg/day, N = 18 Fixed dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: None |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Low risk | Less than 20% of the participants dropped out and missing outcome data are balanced in numbers across intervention groups, with similar reasons of missing data across groups |
| Selective reporting (reporting bias) | Low risk | Both the response and the remission outcomes are reported in the figures with the actual numbers of the participants who achieved these |
| Free of Sponsorship bias? | Unclear risk | The funding source is not described. |
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Unclear |
|
| Interventions | 1. Mirtazapine: 30-45 mg/day, N = 176 2. Sertraline: 50-150 mg/day, N = 170 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, CGI-Improvement, CGI-Severity |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | Both the response and the remission outcomes are reported in the figures with the actual numbers of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 15-45 mg/day, N = 139 2. Paroxetine: 20-40 mg/day, N = 136 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: HAM-A, BDI, Welzil-Kohnen Colored Scales, Short Form-36, CGI-Improvement, CGI-severity |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | Both the response and the remission outcomes at end of the acute-phase treatment are reported with the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 45 mg/day, N = 130 2. Venlafaxine: 225 mg/day, N = 128 Fixed dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, CGI-Improvement, CGI-Severity |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | Both the response and the remission outcomes at end of the acute-phase treatment are reported with the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 5-35 mg/day, N = 50 2. Amitriptlyline: 40-280 mg/day, N = 50 3. Placebo, N = 50 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, CGI-Improvement, CGI-Severity, Zung Self-Rating Depression Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Low risk | Less than 20% of the participants dropped out and missing outcome data are balanced in numbers across intervention groups, with similar reasons of missing data across groups |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 2 week randomised study | |
| Participants | Diagnosis: DSM-IV major depressive disorder, single episode Setting: Psychiatric inpatients |
|
| Interventions | 1. Mirtazapine: mean 38.2 (SD 9.0) mg/day, N = 20 2. Reboxetine: mean 6.6 (SD 1.9) mg/day, N = 20 Flexible dosing scheduling |
|
| Outcomes | The measure used for primary outcome: Performance in driving simulator Other measures: HAM-D, BDI |
|
| Notes | Only information about attrition of the participants is available for the present review | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Low risk | Less than 20% of the participants dropped out and missing outcome data are balanced in numbers across intervention groups, with similar reasons of missing data across groups |
| Selective reporting (reporting bias) | Unclear risk | No useful information in terms of depression severity at the end of treatment is provide |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of the comparator drug |
| Methods | 4 week randomised study | |
| Participants | Diagnosis: DSM-IV major depression Setting: Unclear |
|
| Interventions | 1. Mirtazapine: 30-45 mg/day, N = 20 2. Paroxetine: 20-30 mg/day, N = 20 3. Combination of mirtazapine and paroxetine Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: Unclear Other measures: MADRS |
|
| Notes | We were unable to retrieve usable information for the meta-analysis and to contact the author because the principal author passed away | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Low risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | No information of attrition to permit judgement is provided. |
| Selective reporting (reporting bias) | Unclear risk | No information to permit judgement is provided. |
| Free of Sponsorship bias? | Unclear risk | No information to permit judgement is provided. |
| Methods | 14 week randomised study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 15-60 mg/day, N = 114 2. Nortriptyline: 25-150 mg/day, N = 121 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 16-item Quick Inventory of Depressive Symptomatology Other measures: 17-item HAM-D, Short-Form Health Survey, Work Productivity and Activity Impairment Questionnaire, Work and Social Adjustment Scale, Quality of Life Enjoyment and Satisfaction Questionnaire |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “random assignment” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
High risk | Comment: Both the participants and the clinicians knew the treatment status |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | No information of attrition to permit judgement is provided. |
| Selective reporting (reporting bias) | Low risk | Both the response and the remission outcomes are reported in figures with the proportion of the participants who achieved these |
| Free of Sponsorship bias? | Low risk | The funding source is National Institute of Mental Health. |
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Inpatients |
|
| Interventions | 1. Mirtazapine: 45-60 mg/day, N = 78 2. Venlafaxine: 75-375 mg/day, N = 79 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: The Quality of Life, Enjoyment, and Satisfaction Questionnaire and Quality of Life in Depression Scale, Quality of Life in Depression Scales |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Quote: “participants were randomised to receive treatment with either mirtazapine or venlafaxine orally for 8 weeks, prepared as indistinguishable capsules, according to a centrally prepared randomization list” Comment: No further information about actual central randomisation provided |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | Both the response and the remission outcomes at end of the acute-phase treatment are reported with the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: Outpatients over 55 years of age |
|
| Interventions | 1. Mirtazapine: 5-35 mg/day, N = 50 2. Trazodone: 40-280 mg/day, N = 50 3. Placebo, N = 50 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 21-item HAM-D Other measures: MADRS, CGI-Severity, Zung Self-Rating Scale for Depression |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 15-40 mg/day, N = 66 2. Fluoxetine: 20-40 mg/day, N = 66 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: CGI-Severity |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response and remission outcomes at the end of acute-phase treatment are provided as the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: In- and outpatients |
|
| Interventions | 1. Mirtazapine: 15-45 mg/day, N = 56 2. Amitriptyline: 30-90 mg/day, N = 59 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 21-item HAM-D Other measures: MADRS, CGI-Improvement, CGI-Severity, Brief Cognitive Rating Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to the mirtazapine arm dropped out during the study |
| Selective reporting (reporting bias) | High risk | Neither the response or remission outcomes at the end of acute-phase treatment are provided. They needed to be imputed in the analysis |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: In- and outpatients |
|
| Interventions | 1. Mirtazapine: 15-40 mg/day, N = 66 2. Fluoxetine: 20-40 mg/day, N = 66 Flexiblie dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: MADRS Other measures: HAM-A, CGI-Improvement, CGI-Severity, Leeds Sleep Evaluation Questionnaire -adapted, Quality of Life Enjoyment and Satisfaction Questionnaire |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Quote: “participants were allocated to treatment with either mirtazapine or citalopram, according to the centrally prepared randomization list” Comment: No further information about actual central randomisation provided |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Low risk | Less than 20% of the participants dropped out and missing outcome data are balanced in numbers across intervention groups, with similar reasons of missing data across groups |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: Research Diagnostic criteria major depressive disorder Setting: In- and outpatients |
|
| Interventions | 1. Mirtazapine: 20-60 mg/day, N =83 2. Doxepin: 75-300 mg/day, N = 80 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, Brief Psychiatric Rating Scale, Global Assessment Score, Beck Depression Inventory, Newcastle Endogenous / Reactive Depression Rating Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to the comparator arm dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 5 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: In- and outpatients |
|
| Interventions | 1. Mirtazapine: 20-60 mg/day, N = 79 2. Amitriptyline: 75-225 mg/day, N = 77 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, Brief Psychiatric Rating Scale, General Assessment Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to the comparator arm dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: Inpatients |
|
| Interventions | 1. Mirtazapine: 20-60 mg/day, N = 103 2. Amitriptyline: 75-225 mg/day, N = 104 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, CGI-Improvement |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomly allocated” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Inpatients |
|
| Interventions | 1. Mirtazapine: 20-80 mg/day, N = 87 2. Clomipramine: 50-200 mg/day, N = 87 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 21-item HAM-D Other measures: MADRS, Brief Psychiatric Rating Scale, General Assessment Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomly allocated” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 15-40 mg/day, N = 128 2. Paroxetine: 20-40 mg/day, N = 126 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: CGI-Severity, CGI-Improvement |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The information of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to the comparator arm dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response and remission outcomes at the end of acute-phase treatment are provided as the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 15-45 mg/day, N = 205 2. Fluvoxamine: 50-150 mg/day, N = 207 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: 21-item HAM-D |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to the comparator arm dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 5 week randomised study | |
| Participants | Diagnosis: DSM-IV major depressive episode (bipolar disorder not included) Setting: Psychiatric inpatients |
|
| Interventions | 1. Mirtazapine: 45 mg/day, N = 20 2. Reboxetine: 8 mg/day, N = 20 Fixed dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 21-item HAM-D Other measures: Hypothalamic-pituitary-adrenocortical axis activity |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
High risk | Quote: “We abstained from blinding the medication because the side effect profiles of reboxetine and mirtazapine markedly differ" |
| Incomplete outcome data (attrition bias) All outcomes |
Low risk | Less than 20% of the participants dropped out and missing outcome data are balanced in numbers across intervention groups, with similar reasons of missing data across groups |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the numbers of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mir-tazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: −35 mg/day, N = 50 2. Amitriptyline: −280 mg/day, N = 50 3. Placebo, N = 50 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, CGI-Improvement, CGI-Severity, Zung Self-Rating Depression Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | The numbers of dropouts in the both arms are not specified. |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients |
|
| Interventions | 1. Mirtazapine: 15-45 mg/day, N = 124 2. Sertraline: 50-200 mg/day, N = 126 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: Inventory of Depressive Symptomatology - Self-Report Scale, Social Adaptation Self-evaluation Scale, CGI-Severity, CGI-Efficacy |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response and remission outcomes at the end of acute-phase treatment are provided as the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 60 day randomised study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Unclear |
|
| Interventions | 1. Mirtazapine: unclear dose, N = 25 2. Amitriptyline: unclear dose, N = 27 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, CGI-I |
|
| Notes | We were unable to retrieve usable information for the meta-analysis and to obtain replies from the author | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of concealment is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | No information to permit judgement is provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Low risk | Less than 20% of the participants dropped out and missing outcome data are balanced in numbers across intervention groups, with similar reasons of missing data across groups |
| Selective reporting (reporting bias) | Unclear risk | No information to permit judgement is provided. |
| Free of Sponsorship bias? | Unclear risk | No information to permit judgement is provided. |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: Inpatients |
|
| Interventions | 1. Mirtazapine: 24-72 mg/day, N = 100 2. Trazodone: 150-450 mg/day, N = 100 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, Brief Psychiatric Rating Scale, General Psychiatric Impression Global Assessment Scale, Beck Depression Inventory |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: In- and outpatients |
|
| Interventions | 1. Mirtazapine: 30-60 mg/day, N = 147 2. Fluoxetine: 20-40 mg/day, N = 152 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: MADRS, CGI-Severity, Leeds Sleep Evaluation Questionnaire, Quality of Life, Enjoyment and Satisfaction Questionnaire, Changes in Sezual Functioning Questionnaire |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | The numbers of dropouts in the both arms are not specified. |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided in the figure as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 24 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Outpatients (in general practitioner clinics) |
|
| Interventions | 1. Mirtazapine: 30-45 mg/day, N = 99 2. Paroxetine: 20-30 mg/day, N = 98 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: CGI-Improvement, CGI-Severity, CGI-Patient Global Evaluation |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Quote: “Randomization was performed according to centrally prepared randomization lists” Comment: No further information about actual central randomization provided |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response and remission outcomes at the end of acute-phase treatment are provided in the figures as the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III-R major depressive disorder Setting: In- and outpatients |
|
| Interventions | 1. Mirtazapine: 15-60 mg/day, N = 66 2. Fluoxetine: 20-40 mg/day, N = 67 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 17-item HAM-D Other measures: CGI-Severity, the Visual Analogue Mood Rating Scale, Quality of Life Enjoyment and Satisfaction Questionnaire |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Quote: “participants were allocated to treatment with either mirtazapine or fluoxetine, according to the centrally prepared randomization list” Comment: No further information about actual central randomization provided |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response and remission outcomes at the end of acute-phase treatment are provided in the figures as the proportion of the participants who achieved these |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 8 week randomised double blind study | |
| Participants | Diagnosis: DSM-IV major depressive disorder Setting: Unclear |
|
| Interventions | 1. Mirtazapine: 45 mg/day, N = 9 2. Fluoxetine: 40 mg/day, N = 13 Fixed dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 21-item HAM-D Other measures: CGI-Severity, polysomnographic data, multiple sleep latency testing, performance vigilance testing, Epworth Sleepiness Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomly assigned” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to the comparator arm dropped out during the study |
| Selective reporting (reporting bias) | High risk | Neither the response nor remission outcomes at the end of acute-phase treatment are provided. They needed to be imputed in the analysis |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
| Methods | 6 week randomised double blind study | |
| Participants | Diagnosis: DSM-III major depressive disorder Setting: Inpatients |
|
| Interventions | 1. Mirtazapine: 20-60 mg/day, N = 125 2. Amitriptyline: 75-225 mg/day, N = 126 Flexible dosing scheduling |
|
| Outcomes | The measure used for response and remission in the review: 21-item HAM-D Other measures; Brief Psychiatric Rating Scale, General Assessment Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised” Comment: No further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: The method of allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Quote: “double-blind” Comment: No further information provided. |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | More than 20% of the allocated participants to both of the intervention arms dropped out during the study |
| Selective reporting (reporting bias) | Low risk | The response outcome at the end of acute-phase treatment is provided as the proportion of the participants who achieved this |
| Free of Sponsorship bias? | High risk | The funding source is the pharmaceutical company of mirtazapine |
Abbreviations: CGI = Clinical Global Impression, HAM-D = Hamilton Rating Scale for Depression, MADRS =Montgomery-Asberg Depression Rating Scale, RDC = Research Diagnostic Criteria
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Blier 2004 | Mirtazapine was combined with another antidepressant. |
| Bruijin 1996 | A review of other studies. |
| Kasper 1997a | A review of other studies. |
| Kasper 1997b | A review of other studies. |
| Kremer 1995 | Not a relevant diagnostic status. |
| Peyron 1996 | Not a relevant diagnostic status. |
| Tulen 1996 | Not a relevant diagnostic status. |
| Zourkova 2001 | Not employing random allocation. |
DATA AND ANALYSES
Comparison 1. Mirtazapine versus TCAs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome (response) at 2 weeks | 8 | 1294 | Odds Ratio (M-H, Random, 95% CI) | 0.85 [0.64, 1.13] |
| 1.1 vs Amitriptyline | 5 | 722 | Odds Ratio (M-H, Random, 95% CI) | 0.77 [0.54, 1.12] |
| 1.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.89 [0.46, 1.73] |
| 1.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 1.11 [0.58, 2.12] |
| 1.4 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 0.84 [0.22, 3.22] |
| 2 Primary outcome (response) at end of the acute-phase treatment | 9 | 1501 | Odds Ratio (M-H, Random, 95% CI) | 0.89 [0.72, 1.10] |
| 2.1 vs Amitriptyline | 6 | 929 | Odds Ratio (M-H, Random, 95% CI) | 0.90 [0.69, 1.17] |
| 2.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.90 [0.47, 1.71] |
| 2.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 0.85 [0.44, 1.63] |
| 2.4 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 0.91 [0.53, 1.55] |
| 3 Secondary outcome (remission) at 2 weeks | 8 | 1294 | Odds Ratio (M-H, Random, 95% CI) | 0.85 [0.55, 1.32] |
| 3.1 vs Amitriptyline | 5 | 722 | Odds Ratio (M-H, Random, 95% CI) | 0.67 [0.35, 1.29] |
| 3.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.89 [0.34, 2.31] |
| 3.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 1.27 [0.54, 3.00] |
| 3.4 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 0.70 [0.12, 4.28] |
| 4 Secondary outcome (remission) at end of the acute-phase treatment | 9 | 1501 | Odds Ratio (M-H, Random, 95% CI) | 0.86 [0.69, 1.08] |
| 4.1 vs Amitriptyline | 6 | 929 | Odds Ratio (M-H, Random, 95% CI) | 0.87 [0.65, 1.16] |
| 4.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.77 [0.41, 1.45] |
| 4.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 0.94 [0.50, 1.74] |
| 4.4 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 0.85 [0.46, 1.56] |
| 5 Secondary outcome (depression severity) at 2 weeks | 2 | 361 | Std. Mean Difference (IV, Random, 95% CI) | 0.10 [−0.11, 0.31] |
| 5.1 vs Amitritpyline | 2 | 361 | Std. Mean Difference (IV, Random, 95% CI) | 0.10 [−0.11, 0.31] |
| 6 Secondary outcome (depression severity) at end of the acute-phase treatment | 1 | 144 | Std. Mean Difference (IV, Random, 95% CI) | 0.12 [−0.21, 0.45] |
| 6.1 vs Amitritpyline | 1 | 144 | Std. Mean Difference (IV, Random, 95% CI) | 0.12 [−0.21, 0.45] |
| 7 Secondary outcome (Social adjustment) at 2 weeks | 1 | 138 | Mean Difference (IV, Random, 95% CI) | 1.60 [−1.98, 5.18] |
| 7.1 vs Amitriptyline | 1 | 138 | Mean Difference (IV, Random, 95% CI) | 1.60 [−1.98, 5.18] |
| 8 Secondary outcome (Social adjustment) at end of the acute-phase treatment | 3 | 440 | Std. Mean Difference (IV, Random, 95% CI) | 0.02 [−0.17, 0.21] |
| 8.1 vs Amitriptyline | 1 | 114 | Std. Mean Difference (IV, Random, 95% CI) | 0.03 [−0.34, 0.40] |
| 8.2 vs Clomipramine | 1 | 163 | Std. Mean Difference (IV, Random, 95% CI) | 0.12 [−0.19, 0.43] |
| 8.3 vs Doxepin | 1 | 163 | Std. Mean Difference (IV, Random, 95% CI) | −0.09 [−0.39, 0.22] |
| 9 Secondary outcome (withdrawal due to any reason) | 7 | 1166 | Odds Ratio (M-H, Random, 95% CI) | 0.83 [0.63, 1.10] |
| 9.1 vs Amitriptyline | 5 | 829 | Odds Ratio (M-H, Random, 95% CI) | 0.90 [0.65, 1.25] |
| 9.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.80 [0.42, 1.54] |
| 9.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 0.51 [0.22, 1.19] |
| 10 Secondary outcome (withdrawal due to adverse events) | 8 | 1266 | Odds Ratio (M-H, Random, 95% CI) | 0.65 [0.41, 1.03] |
| 10.1 vs Amitriptyline | 6 | 929 | Odds Ratio (M-H, Random, 95% CI) | 0.60 [0.35, 1.03] |
| 10.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 1.14 [0.42, 3.10] |
| 10.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 0.30 [0.06, 1.56] |
| 11 Secondary outcome (having some adverse events) | 2 | 442 | Odds Ratio (M-H, Random, 95% CI) | 1.06 [0.54, 2.10] |
| 11.1 vs Amitriptyline | 1 | 207 | Odds Ratio (M-H, Random, 95% CI) | 0.74 [0.16, 3.37] |
| 11.2 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 1.17 [0.55, 2.49] |
| 12 Hypertension/Tachycardia | 4 | 522 | Odds Ratio (M-H, Random, 95% CI) | 0.44 [0.24, 0.81] |
| 12.1 vs Amitriptyline | 4 | 522 | Odds Ratio (M-H, Random, 95% CI) | 0.44 [0.24, 0.81] |
| 13 Hypotension/Bradycardia | 2 | 215 | Odds Ratio (M-H, Random, 95% CI) | 0.46 [0.12, 1.81] |
| 13.1 vs Amitriptyline | 2 | 215 | Odds Ratio (M-H, Random, 95% CI) | 0.46 [0.12, 1.81] |
| 14 Sweating | 2 | 458 | Odds Ratio (M-H, Random, 95% CI) | 0.42 [0.05, 3.24] |
| 14.1 vs Amitriptyline | 2 | 458 | Odds Ratio (M-H, Random, 95% CI) | 0.42 [0.05, 3.24] |
| 15 Constipation | 6 | 1003 | Odds Ratio (M-H, Random, 95% CI) | 0.72 [0.46, 1.12] |
| 15.1 vs Amitriptyline | 5 | 829 | Odds Ratio (M-H, Random, 95% CI) | 0.72 [0.40, 1.29] |
| 15.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.62 [0.26, 1.48] |
| 16 Dry mouth/Decreased salivation | 8 | 1266 | Odds Ratio (M-H, Random, 95% CI) | 0.52 [0.24, 1.14] |
| 16.1 vs Amitriptyline | 6 | 929 | Odds Ratio (M-H, Random, 95% CI) | 0.36 [0.14, 0.92] |
| 16.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.81 [0.39, 1.69] |
| 16.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 2.86 [1.18, 6.94] |
| 17 Nausea/Vomiting/Gastric distress | 4 | 581 | Odds Ratio (M-H, Random, 95% CI) | 0.29 [0.05, 1.59] |
| 17.1 vs Amitriptyline | 3 | 407 | Odds Ratio (M-H, Random, 95% CI) | 0.33 [0.04, 2.59] |
| 17.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.16 [0.02, 1.33] |
| 18 Weight gain/Increased appetite | 3 | 463 | Odds Ratio (M-H, Random, 95% CI) | 1.04 [0.58, 1.86] |
| 18.1 vs Amitriptyline | 3 | 463 | Odds Ratio (M-H, Random, 95% CI) | 1.04 [0.58, 1.86] |
| 19 Sexual dysfunction | 2 | 351 | Odds Ratio (M-H, Random, 95% CI) | 0.41 [0.06, 2.61] |
| 19.1 vs Amitriptyline | 2 | 351 | Odds Ratio (M-H, Random, 95% CI) | 0.41 [0.06, 2.61] |
| 20 Anxiety/Agitation | 2 | 307 | Odds Ratio (M-H, Random, 95% CI) | 0.87 [0.34, 2.19] |
| 20.1 vs Amitriptyline | 2 | 307 | Odds Ratio (M-H, Random, 95% CI) | 0.87 [0.34, 2.19] |
| 21 Dizziness/Vertigo/Faintness | 7 | 1166 | Odds Ratio (M-H, Random, 95% CI) | 0.75 [0.43, 1.28] |
| 21.1 vs Amitriptyline | 5 | 829 | Odds Ratio (M-H, Random, 95% CI) | 0.64 [0.35, 1.17] |
| 21.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.70 [0.21, 2.29] |
| 21.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 3.04 [0.59, 15.53] |
| 22 Fatigue/Tiredness/Asthenia | 4 | 673 | Odds Ratio (M-H, Random, 95% CI) | 1.25 [0.71, 2.21] |
| 22.1 vs Amitriptyline | 4 | 673 | Odds Ratio (M-H, Random, 95% CI) | 1.25 [0.71, 2.21] |
| 23 Headache | 4 | 522 | Odds Ratio (M-H, Random, 95% CI) | 0.74 [0.31, 1.74] |
| 23.1 vs Amitriptyline | 4 | 522 | Odds Ratio (M-H, Random, 95% CI) | 0.74 [0.31, 1.74] |
| 24 Tremor | 7 | 1103 | Odds Ratio (M-H, Random, 95% CI) | 0.36 [0.22, 0.57] |
| 24.1 vs Amitriptyline | 6 | 929 | Odds Ratio (M-H, Random, 95% CI) | 0.36 [0.20, 0.62] |
| 24.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.36 [0.15, 0.88] |
| 25 Sleep disturbance | 1 | 207 | Odds Ratio (M-H, Random, 95% CI) | 1.43 [0.69, 2.98] |
| 25.1 vs Amitriptyline | 1 | 207 | Odds Ratio (M-H, Random, 95% CI) | 1.43 [0.69, 2.98] |
| 26 Sleepiness/Drowsiness/Somnolence | 6 | 841 | Odds Ratio (M-H, Random, 95% CI) | 0.92 [0.66, 1.27] |
| 26.1 vs Amitriptyline | 5 | 678 | Odds Ratio (M-H, Random, 95% CI) | 0.81 [0.58, 1.14] |
| 26.2 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 1.86 [0.82, 4.21] |
| 27 Suicide attempt | 5 | 935 | Odds Ratio (M-H, Random, 95% CI) | 1.77 [0.47, 6.58] |
| 27.1 vs Amitriptyline | 2 | 363 | Odds Ratio (M-H, Random, 95% CI) | 2.08 [0.27, 16.29] |
| 27.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.06, 16.25] |
| 27.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 0.32 [0.01, 7.91] |
| 27.4 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 9.90 [0.53, 185.90] |
| 28 Subgroup analysis: Response at 2 weeks: Treatment settings: Psychiatric inpatients | 2 | 425 | Odds Ratio (M-H, Random, 95% CI) | 0.81 [0.50, 1.32] |
| 28.1 vs Amitriptyline | 1 | 251 | Odds Ratio (M-H, Random, 95% CI) | 0.73 [0.36, 1.48] |
| 28.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.89 [0.46, 1.73] |
| 29 Subgroup analysis: Response at end of the acute-phase treatment: Treatment settings: Psychiatric inpatients | 3 | 632 | Odds Ratio (M-H, Random, 95% CI) | 0.89 [0.64, 1.23] |
| 29.1 vs Amitriptyline | 2 | 458 | Odds Ratio (M-H, Random, 95% CI) | 0.88 [0.60, 1.29] |
| 29.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.90 [0.47, 1.71] |
| 30 Sensitivity analysis: Response at 2 weeks: Studies without imputation | 7 | 1179 | Odds Ratio (M-H, Random, 95% CI) | 0.85 [0.64, 1.15] |
| 30.1 vs Amitriptyline | 4 | 607 | Odds Ratio (M-H, Random, 95% CI) | 0.76 [0.51, 1.13] |
| 30.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.89 [0.46, 1.73] |
| 30.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 1.11 [0.58, 2.12] |
| 30.4 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 0.84 [0.22, 3.22] |
| 31 Sensitivity analysis: Response at end of the acute-phase treatment: Studies without imputation | 8 | 1386 | Odds Ratio (M-H, Random, 95% CI) | 0.93 [0.74, 1.15] |
| 31.1 vs Amitriptyline | 5 | 814 | Odds Ratio (M-H, Random, 95% CI) | 0.95 [0.72, 1.26] |
| 31.2 vs Clomipramine | 1 | 174 | Odds Ratio (M-H, Random, 95% CI) | 0.90 [0.47, 1.71] |
| 31.3 vs Doxepin | 1 | 163 | Odds Ratio (M-H, Random, 95% CI) | 0.85 [0.44, 1.63] |
| 31.4 vs Nortriptyline | 1 | 235 | Odds Ratio (M-H, Random, 95% CI) | 0.91 [0.53, 1.55] |
| 32 Secondary outcome (SKEWED DATA: depression severity) at 2 weeks | Other data | No numeric data | ||
| 33 Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment | Other data | No numeric data |
Comparison 2. Mirtazapine versus SSRIs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome (response) at 2 weeks | 12 | 2626 | Odds Ratio (M-H, Random, 95% CI) | 1.57 [1.30, 1.88] |
| 1.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 2.01 [0.93, 4.35] |
| 1.2 vs Fluoxetine | 5 | 622 | Odds Ratio (M-H, Random, 95% CI) | 1.26 [0.86, 1.85] |
| 1.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 2.39 [1.42, 4.02] |
| 1.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 1.45 [1.04, 2.02] |
| 1.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.38 [0.90, 2.13] |
| 2 Primary outcome (response) at end of the acute-phase treatment | 12 | 2626 | Odds Ratio (M-H, Random, 95% CI) | 1.19 [1.01, 1.39] |
| 2.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.76 [0.38, 1.52] |
| 2.2 vs Fluoxetine | 5 | 622 | Odds Ratio (M-H, Random, 95% CI) | 1.55 [1.07, 2.23] |
| 2.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 1.27 [0.94, 1.70] |
| 2.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 0.97 [0.70, 1.35] |
| 2.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.14 [0.76, 1.70] |
| 3 Primary outcome (response) at end of the continuation treatment | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 1.60 [0.91, 2.81] |
| 3.1 vs Paroxetine | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 1.60 [0.91, 2.81] |
| 4 Secondary outcome (remission) at 2 weeks | 12 | 2626 | Odds Ratio (M-H, Random, 95% CI) | 1.82 [1.36, 2.44] |
| 4.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 2.48 [0.47, 13.02] |
| 4.2 vs Fluoxetine | 5 | 622 | Odds Ratio (M-H, Random, 95% CI) | 1.63 [0.81, 3.27] |
| 4.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 2.31 [1.04, 5.11] |
| 4.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 1.94 [1.19, 3.15] |
| 4.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.46 [0.77, 2.76] |
| 5 Secondary outcome (remission) at end of the acute-phase treatment | 12 | 2626 | Odds Ratio (M-H, Random, 95% CI) | 1.17 [0.98, 1.40] |
| 5.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.92 [0.55, 1.52] |
| 5.2 vs Fluoxetine | 5 | 622 | Odds Ratio (M-H, Random, 95% CI) | 1.12 [0.80, 1.57] |
| 5.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 1.58 [1.16, 2.15] |
| 5.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 1.18 [0.82, 1.71] |
| 5.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 0.84 [0.57, 1.23] |
| 6 Secondary outcome (remission) at end of the continuation treatment | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 1.89 [1.01, 3.54] |
| 6.1 vs Paroxetine | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 1.89 [1.01, 3.54] |
| 7 Secondary outcome (withdrawal due to any reason) | 11 | 2327 | Odds Ratio (M-H, Random, 95% CI) | 1.12 [0.89, 1.40] |
| 7.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 2.36 [0.99, 5.64] |
| 7.2 vs Fluoxetine | 4 | 323 | Odds Ratio (M-H, Random, 95% CI) | 1.09 [0.67, 1.78] |
| 7.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.80 [0.58, 1.10] |
| 7.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 1.47 [1.01, 2.13] |
| 7.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.20 [0.75, 1.93] |
| 8 Secondary outcome (withdrawal due to adverse events) | 11 | 2604 | Odds Ratio (M-H, Random, 95% CI) | 1.26 [0.85, 1.86] |
| 8.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 2.0 [0.59, 6.81] |
| 8.2 vs Fluoxetine | 4 | 600 | Odds Ratio (M-H, Random, 95% CI) | 1.05 [0.62, 1.78] |
| 8.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.74 [0.45, 1.21] |
| 8.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 2.88 [1.43, 5.77] |
| 8.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.66 [0.86, 3.21] |
| 9 Secondary outcome (having some adverse events) | 7 | 1773 | Odds Ratio (M-H, Random, 95% CI) | 1.01 [0.81, 1.26] |
| 9.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.82 [0.49, 1.37] |
| 9.2 vs Fluoxetine | 2 | 431 | Odds Ratio (M-H, Random, 95% CI) | 1.42 [0.97, 2.09] |
| 9.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.94 [0.66, 1.32] |
| 9.4 vs Sertraline | 1 | 346 | Odds Ratio (M-H, Random, 95% CI) | 0.86 [0.55, 1.34] |
| 10 Hypotension/Bradycardia | 1 | 133 | Odds Ratio (M-H, Random, 95% CI) | 5.41 [0.61, 47.62] |
| 10.1 vs Fluoxetine | 1 | 133 | Odds Ratio (M-H, Random, 95% CI) | 5.41 [0.61, 47.62] |
| 11 Sweating | 5 | 1342 | Odds Ratio (M-H, Random, 95% CI) | 0.25 [0.15, 0.44] |
| 11.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.13 [0.04, 0.44] |
| 11.2 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.32 [0.17, 0.62] |
| 11.3 vs Sertraline | 1 | 346 | Odds Ratio (M-H, Random, 95% CI) | 0.21 [0.04, 0.97] |
| 12 Constipation | 5 | 1109 | Odds Ratio (M-H, Random, 95% CI) | 1.20 [0.79, 1.82] |
| 12.1 vs Fluoxetine | 2 | 168 | Odds Ratio (M-H, Random, 95% CI) | 2.14 [0.81, 5.66] |
| 12.2 vs Paroxetine | 2 | 529 | Odds Ratio (M-H, Random, 95% CI) | 1.07 [0.59, 1.95] |
| 12.3 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.01 [0.48, 2.12] |
| 13 Diarrhoea | 8 | 2040 | Odds Ratio (M-H, Random, 95% CI) | 0.57 [0.41, 0.80] |
| 13.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.47 [0.14, 1.60] |
| 13.2 vs Fluoxetine | 1 | 36 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.06, 17.33] |
| 13.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.89 [0.55, 1.46] |
| 13.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 0.37 [0.21, 0.67] |
| 13.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 0.34 [0.13, 0.87] |
| 14 Dry mouth/Decreased salivation | 10 | 2305 | Odds Ratio (M-H, Random, 95% CI) | 1.80 [1.37, 2.36] |
| 14.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 1.72 [0.81, 3.68] |
| 14.2 vs Fluoxetine | 3 | 301 | Odds Ratio (M-H, Random, 95% CI) | 3.68 [1.52, 8.91] |
| 14.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 1.73 [0.81, 3.70] |
| 14.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 1.53 [0.92, 2.55] |
| 14.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.48 [0.77, 2.85] |
| 15 Nausea/Vomiting/Gastric distress | 11 | 2604 | Odds Ratio (M-H, Random, 95% CI) | 0.33 [0.26, 0.43] |
| 15.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.45 [0.22, 0.90] |
| 15.2 vs Fluoxetine | 4 | 600 | Odds Ratio (M-H, Random, 95% CI) | 0.34 [0.14, 0.81] |
| 15.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.27 [0.16, 0.44] |
| 15.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 0.27 [0.16, 0.48] |
| 15.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 0.29 [0.16, 0.53] |
| 16 Weight gain/Increased appetite | 11 | 2604 | Odds Ratio (M-H, Random, 95% CI) | 4.23 [2.93, 6.11] |
| 16.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 3.83 [1.49, 9.82] |
| 16.2 vs Fluoxetine | 4 | 600 | Odds Ratio (M-H, Random, 95% CI) | 5.23 [2.15, 12.76] |
| 16.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 3.92 [1.19, 12.92] |
| 16.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 6.67 [3.30, 13.49] |
| 16.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 2.51 [0.87, 7.26] |
| 17 Weight loss/Anorexia | 4 | 576 | Odds Ratio (M-H, Random, 95% CI) | 0.35 [0.10, 1.18] |
| 17.1 vs Fluoxetine | 3 | 301 | Odds Ratio (M-H, Random, 95% CI) | 0.42 [0.11, 1.62] |
| 17.2 vs Paroxetine | 1 | 275 | Odds Ratio (M-H, Random, 95% CI) | 0.14 [0.01, 2.67] |
| 18 Sexual dysfunction | 4 | 907 | Odds Ratio (M-H, Random, 95% CI) | 0.31 [0.13, 0.74] |
| 18.1 vs Fluoxetine | 1 | 36 | Odds Ratio (M-H, Random, 95% CI) | 0.15 [0.02, 1.47] |
| 18.2 vs Paroxetine | 1 | 275 | Odds Ratio (M-H, Random, 95% CI) | 0.19 [0.06, 0.59] |
| 18.3 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 0.43 [0.10, 1.82] |
| 19 Anxiety/Agitation | 4 | 1134 | Odds Ratio (M-H, Random, 95% CI) | 1.46 [0.59, 3.65] |
| 19.1 vs Paroxetine | 2 | 472 | Odds Ratio (M-H, Random, 95% CI) | 0.71 [0.32, 1.60] |
| 19.2 vs Sertraline | 1 | 250 | Odds Ratio (M-H, Random, 95% CI) | 1.65 [0.69, 3.98] |
| 19.3 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 5.76 [1.65, 20.07] |
| 20 Dizziness/Vertigo/Faintness | 10 | 2568 | Odds Ratio (M-H, Random, 95% CI) | 1.04 [0.77, 1.41] |
| 20.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 2.03 [0.74, 5.58] |
| 20.2 vs Fluoxetine | 3 | 564 | Odds Ratio (M-H, Random, 95% CI) | 0.92 [0.54, 1.56] |
| 20.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.84 [0.47, 1.50] |
| 20.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 1.16 [0.36, 3.70] |
| 20.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.31 [0.69, 2.48] |
| 21 Fatigue/Tiredness/Asthenia | 8 | 2137 | Odds Ratio (M-H, Random, 95% CI) | 1.53 [1.08, 2.15] |
| 21.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.91 [0.44, 1.84] |
| 21.2 vs Fluoxetine | 1 | 133 | Odds Ratio (M-H, Random, 95% CI) | 1.38 [0.30, 6.40] |
| 21.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 1.71 [0.94, 3.11] |
| 21.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 2.31 [1.32, 4.04] |
| 21.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 0.94 [0.44, 2.00] |
| 22 Headache | 11 | 2604 | Odds Ratio (M-H, Random, 95% CI) | 0.69 [0.56, 0.86] |
| 22.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.63 [0.30, 1.33] |
| 22.2 vs Fluoxetine | 4 | 600 | Odds Ratio (M-H, Random, 95% CI) | 0.86 [0.54, 1.36] |
| 22.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 0.57 [0.36, 0.89] |
| 22.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 0.67 [0.44, 1.01] |
| 22.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 0.84 [0.46, 1.53] |
| 23 Tremor | 5 | 996 | Odds Ratio (M-H, Random, 95% CI) | 0.34 [0.18, 0.66] |
| 23.1 vs Fluoxetine | 3 | 467 | Odds Ratio (M-H, Random, 95% CI) | 0.42 [0.17, 1.07] |
| 23.2 vs Paroxetine | 2 | 529 | Odds Ratio (M-H, Random, 95% CI) | 0.27 [0.11, 0.70] |
| 24 Sleep disturbance | 5 | 1346 | Odds Ratio (M-H, Random, 95% CI) | 0.52 [0.31, 0.86] |
| 24.1 vs Fluoxetine | 1 | 299 | Odds Ratio (M-H, Random, 95% CI) | 0.53 [0.21, 1.38] |
| 24.2 vs Paroxetine | 2 | 451 | Odds Ratio (M-H, Random, 95% CI) | 0.68 [0.25, 1.85] |
| 24.3 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 0.37 [0.16, 0.85] |
| 25 Sleepiness/Drowsiness/Somnolence | 11 | 2604 | Odds Ratio (M-H, Random, 95% CI) | 1.81 [1.39, 2.37] |
| 25.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 1.36 [0.53, 3.51] |
| 25.2 vs Fluoxetine | 4 | 600 | Odds Ratio (M-H, Random, 95% CI) | 1.59 [0.97, 2.61] |
| 25.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 1.24 [0.84, 1.83] |
| 25.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 3.07 [1.89, 4.99] |
| 25.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 2.34 [1.45, 3.77] |
| 26 Suicide attempt | 1 | 346 | Odds Ratio (M-H, Random, 95% CI) | 4.89 [0.23, 102.51 |
| 26.1 vs Sertraline | 1 | 346 | Odds Ratio (M-H, Random, 95% CI) | 4.89 [0.23, 102.51 |
| 27 Subgroup analysis: Response at 2 weeks: Treatment settings: Outpatients in primary care | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 4.38 [1.69, 11.35] |
| 27.1 vs Paroxetine | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 4.38 [1.69, 11.35] |
| 28 Subgroup analysis: Response at end of the acute-phase treatment: Treatment settings: Outpatients in primary care | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 1.17 [0.66, 2.10] |
| 28.1 vs Paroxetine | 1 | 197 | Odds Ratio (M-H, Random, 95% CI) | 1.17 [0.66, 2.10] |
| 29 Sensitivity analysis: Response at 2 weeks: Studies without imputation | 11 | 2604 | Odds Ratio (M-H, Random, 95% CI) | 1.58 [1.31, 1.90] |
| 29.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 2.01 [0.93, 4.35] |
| 29.2 vs Fluoxetine | 4 | 600 | Odds Ratio (M-H, Random, 95% CI) | 1.27 [0.86, 1.88] |
| 29.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 2.39 [1.42, 4.02] |
| 29.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 1.45 [1.04, 2.02] |
| 29.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.38 [0.90, 2.13] |
| 30 Sensitivity analysis: Response at end of the acute-phase treatment: Studies without imputation | 11 | 2604 | Odds Ratio (M-H, Random, 95% CI) | 1.17 [1.00, 1.38] |
| 30.1 vs Citalolpram | 1 | 270 | Odds Ratio (M-H, Random, 95% CI) | 0.76 [0.38, 1.52] |
| 30.2 vs Fluoxetine | 4 | 600 | Odds Ratio (M-H, Random, 95% CI) | 1.46 [1.04, 2.04] |
| 30.3 vs Paroxetine | 3 | 726 | Odds Ratio (M-H, Random, 95% CI) | 1.27 [0.94, 1.70] |
| 30.4 vs Sertraline | 2 | 596 | Odds Ratio (M-H, Random, 95% CI) | 0.97 [0.70, 1.35] |
| 30.5 vs Fluvoxamine | 1 | 412 | Odds Ratio (M-H, Random, 95% CI) | 1.14 [0.76, 1.70] |
| 31 Secondary outcome (SKEWED DATA: depression severity) at 2 weeks | Other data | No numeric data | ||
| 32 Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment | Other data | No numeric data |
Comparison 3. Mirtazapine versus SNRIs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome (response) at 2 weeks | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 2.29 [1.45, 3.59] |
| 1.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 2.29 [1.45, 3.59] |
| 2 Primary outcome (response) at end of the acute-phase treatment | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 1.53 [1.03, 2.25] |
| 2.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 1.53 [1.03, 2.25] |
| 3 Secondary outcome (remission) at 2 weeks | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 2.34 [1.07, 5.13] |
| 3.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 2.34 [1.07, 5.13] |
| 4 Secondary outcome (remission) at end of the acute-phase treatment | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 1.55 [0.98, 2.47] |
| 4.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 1.55 [0.98, 2.47] |
| 5 Secondary outcome (withdrawal due to any reason) | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.65 [0.43, 0.99] |
| 5.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.65 [0.43, 0.99] |
| 6 Secondary outcome (withdrawal due to adverse events) | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.55 [0.24, 1.24] |
| 6.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.55 [0.24, 1.24] |
| 7 Secondary outcome (having some adverse events) | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 1.51 [0.76, 3.00] |
| 7.1 vs Venlafaxine | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 1.51 [0.76, 3.00] |
| 8 Hypotension/Bradycardia | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.19 [0.02, 1.68] |
| 8.1 vs Venlafaxine | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.19 [0.02, 1.68] |
| 9 Sweating | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.03 [0.00, 0.45] |
| 9.1 vs Venlafaxine | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.03 [0.00, 0.45] |
| 10 Constipation | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.22 [0.06, 0.83] |
| 10.1 vs Venlafaxine | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.22 [0.06, 0.83] |
| 11 Dry mouth/Decreased salivation | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 8.61 [0.35, 211.85] |
| 11.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 8.61 [0.35, 211.85] |
| 12 Nausea/Vomiting/Gastric distress | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.11 [0.00, 9.34] |
| 12.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.11 [0.00, 9.34] |
| 13 Anxiety/Agitation | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 1.01 [0.24, 4.20] |
| 13.1 vs Venlafaxine | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 1.01 [0.24, 4.20] |
| 14 Fatigue/Tiredness/Asthenia | 1 | 258 | Odds Ratio (M-H, Random, 95% CI) | 2.43 [1.30, 4.55] |
| 14.1 vs Venlafaxine | 1 | 258 | Odds Ratio (M-H, Random, 95% CI) | 2.43 [1.30, 4.55] |
| 15 Headache | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.95 [0.53, 1.72] |
| 15.1 vs Venlafaxine | 2 | 415 | Odds Ratio (M-H, Random, 95% CI) | 0.95 [0.53, 1.72] |
| 16 Sleep disturbance | 1 | 258 | Odds Ratio (M-H, Random, 95% CI) | 0.02 [0.00, 0.41] |
| 16.1 vs Venlafaxine | 1 | 258 | Odds Ratio (M-H, Random, 95% CI) | 0.02 [0.00, 0.41] |
| 17 Sleepiness/Drowsiness/Somnolence | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 1.56 [0.42, 5.77] |
| 17.1 vs Venlafaxine | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 1.56 [0.42, 5.77] |
| 18 Completed suicide | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.33 [0.01, 8.31] |
| 18.1 vs Venlafaxine | 1 | 157 | Odds Ratio (M-H, Random, 95% CI) | 0.33 [0.01, 8.31] |
Comparison 4. Mirtazapine versus heterocyclic antidepressants.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome (response) at 2 weeks | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.14 [0.64, 2.04] |
| 1.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.14 [0.64, 2.04] |
| 2 Primary outcome (response) at end of the acute-phase treatment | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.50 [0.95, 2.37] |
| 2.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.50 [0.95, 2.37] |
| 3 Secondary outcome (remission) at 2 weeks | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.36, 2.80] |
| 3.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.36, 2.80] |
| 4 Secondary outcome (remission) at end of the acute-phase treatment | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.38 [0.76, 2.52] |
| 4.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 1.38 [0.76, 2.52] |
| 5 Secondary outcome (withdrawal due to any reason) | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.90 [0.47, 1.72] |
| 5.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.90 [0.47, 1.72] |
| 6 Secondary outcome (withdrawal due to adverse events) | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.61 [0.25, 1.51] |
| 6.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.61 [0.25, 1.51] |
| 7 Hypertension/Tachycardia | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.31 [0.06, 1.59] |
| 7.1 vs Trazodone | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.31 [0.06, 1.59] |
| 8 Hypotension/Bradycardia | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.17 [0.03, 1.00] |
| 8.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.17 [0.03, 1.00] |
| 9 Constipation | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.70 [0.26, 1.83] |
| 9.1 vs Trazodone | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.70 [0.26, 1.83] |
| 10 Dry mouth/Decreased salivation | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.39 [0.11, 1.37] |
| 10.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.39 [0.11, 1.37] |
| 11 Nausea/Vomiting/Gastric distress | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.68 [0.20, 2.32] |
| 11.1 vs Trazodone | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.68 [0.20, 2.32] |
| 12 Weight gain/Increased appetite | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 4.95 [1.30, 18.81] |
| 12.1 vs Trazodone | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 4.95 [1.30, 18.81] |
| 13 Weight loss/Anorexia | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.33 [0.01, 8.21] |
| 13.1 vs Trazodone | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.33 [0.01, 8.21] |
| 14 Anxiety/Agitation | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.62 [0.19, 1.96] |
| 14.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.62 [0.19, 1.96] |
| 15 Dizziness/Vertigo/Faintness | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.64 [0.30, 1.39] |
| 15.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.64 [0.30, 1.39] |
| 16 Headache | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.65 [0.23, 1.87] |
| 16.1 vs Trazodone | 1 | 100 | Odds Ratio (M-H, Random, 95% CI) | 0.65 [0.23, 1.87] |
| 17 Sleep disturbance | 1 | 200 | Odds Ratio (M-H, Random, 95% CI) | 0.42 [0.13, 1.42] |
| 17.1 vs Trazodone | 1 | 200 | Odds Ratio (M-H, Random, 95% CI) | 0.42 [0.13, 1.42] |
| 18 Sleepiness/Drowsiness/Somnolence | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.62 [0.29, 1.36] |
| 18.1 vs Trazodone | 2 | 300 | Odds Ratio (M-H, Random, 95% CI) | 0.62 [0.29, 1.36] |
| 19 Completed suicide | 1 | 200 | Odds Ratio (M-H, Random, 95% CI) | 3.03 [0.12, 75.28] |
| 19.1 vs Trazodone | 1 | 200 | Odds Ratio (M-H, Random, 95% CI) | 3.03 [0.12, 75.28] |
| 20 Suicide attempt | 1 | 200 | Odds Ratio (M-H, Random, 95% CI) | 2.02 [0.18, 22.65] |
| 20.1 vs Trazodone | 1 | 200 | Odds Ratio (M-H, Random, 95% CI) | 2.02 [0.18, 22.65] |
Comparison 5. Mirtazapine versus newer antidepressants.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome (response) at 2 weeks | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.18, 5.67] |
| 1.1 vs Reboxetine | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.18, 5.67] |
| 2 Primary outcome (response) at end of the acute-phase treatment | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.27, 3.67] |
| 2.1 vs Reboxetine | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.27, 3.67] |
| 3 Secondary outcome (remission) at 2 weeks | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.06, 17.18] |
| 3.1 vs Reboxetine | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.0 [0.06, 17.18] |
| 4 Secondary outcome (remission) at end of the acute-phase treatment | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.26 [0.33, 4.73] |
| 4.1 vs Reboxetine | 1 | 40 | Odds Ratio (M-H, Random, 95% CI) | 1.26 [0.33, 4.73] |
| 5 Secondary outcome (depression severity) at 2 weeks | 1 | 40 | Std. Mean Difference (IV, Random, 95% CI) | −0.37 [−1.00, 0.25] |
| 5.1 vs Reboxetine | 1 | 40 | Std. Mean Difference (IV, Random, 95% CI) | −0.37 [−1.00, 0.25] |
| 6 Secondary outcome (withdrawal due to any reason) | 2 | 80 | Odds Ratio (M-H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 6.1 vs Reboxetine | 2 | 80 | Odds Ratio (M-H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Secondary outcome (withdrawal due to adverse events) | 2 | 80 | Odds Ratio (M-H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7.1 vs Reboxetine | 2 | 80 | Odds Ratio (M-H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment | Other data | No numeric data |
Comparison 6. Funnel plot analysis: primary outcome (response) at end of the acute-phase treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 vs all compounds | 26 | 4882 | Risk Ratio (M-H, Fixed, 99% CI) | 1.05 [0.99, 1.12] |
| 1.1 vs Amitriptyline | 6 | 929 | Risk Ratio (M-H, Fixed, 99% CI) | 0.95 [0.82, 1.11] |
| 1.2 vs Clomipramine | 1 | 174 | Risk Ratio (M-H, Fixed, 99% CI) | 0.97 [0.74, 1.26] |
| 1.3 vs Doxepin | 1 | 163 | Risk Ratio (M-H, Fixed, 99% CI) | 0.95 [0.71, 1.26] |
| 1.4 vs Nortriptyline | 1 | 235 | Risk Ratio (M-H, Fixed, 99% CI) | 0.94 [0.59, 1.49] |
| 1.5 vs Citalolpram | 1 | 270 | Risk Ratio (M-H, Fixed, 99% CI) | 0.96 [0.85, 1.09] |
| 1.6 vs Fluoxetine | 5 | 622 | Risk Ratio (M-H, Fixed, 99% CI) | 1.18 [1.00, 1.39] |
| 1.7 vs Paroxetine | 3 | 726 | Risk Ratio (M-H, Fixed, 99% CI) | 1.13 [0.93, 1.38] |
| 1.8 vs Sertraline | 2 | 596 | Risk Ratio (M-H, Fixed, 99% CI) | 0.99 [0.83, 1.17] |
| 1.9 vs Fluvoxamine | 1 | 412 | Risk Ratio (M-H, Fixed, 99% CI) | 1.05 [0.86, 1.28] |
| 1.10 vs Venlafaxine | 2 | 415 | Risk Ratio (M-H, Fixed, 99% CI) | 1.24 [0.96, 1.60] |
| 1.11 vs Trazodone | 2 | 300 | Risk Ratio (M-H, Fixed, 99% CI) | 1.21 [0.91, 1.61] |
| 1.12 vs Reboxetine | 1 | 40 | Risk Ratio (M-H, Fixed, 99% CI) | 1.0 [0.55, 1.82] |
Analysis 1.1. Comparison 1 Mirtazapine versus TCAs, Outcome 1 Primary outcome (response) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 1 Primary outcome (response) at 2 weeks
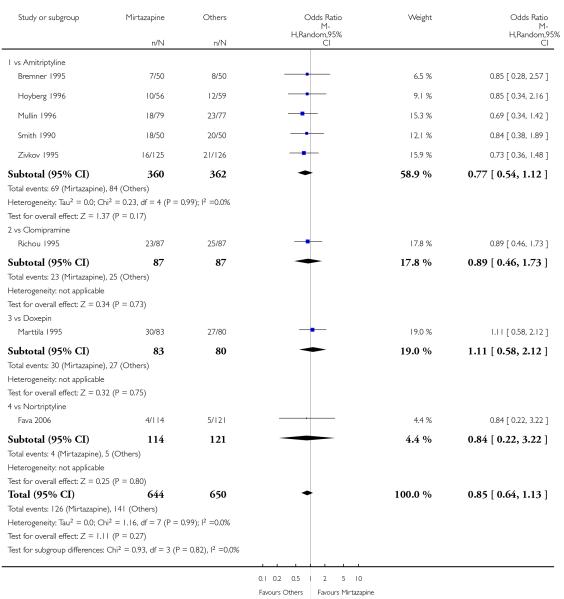 |
Analysis 1.2. Comparison 1 Mirtazapine versus TCAs, Outcome 2 Primary outcome (response) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 2 Primary outcome (response) at end of the acute-phase treatment
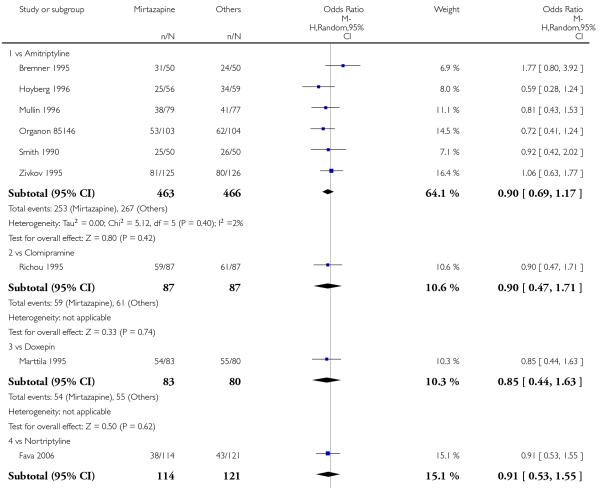 |
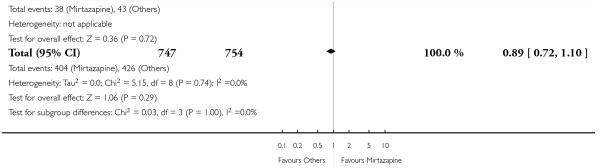 |
Analysis 1.3. Comparison 1 Mirtazapine versus TCAs, Outcome 3 Secondary outcome (remission) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 3 Secondary outcome (remission) at 2 weeks
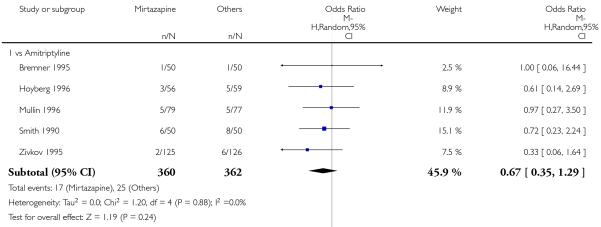 |
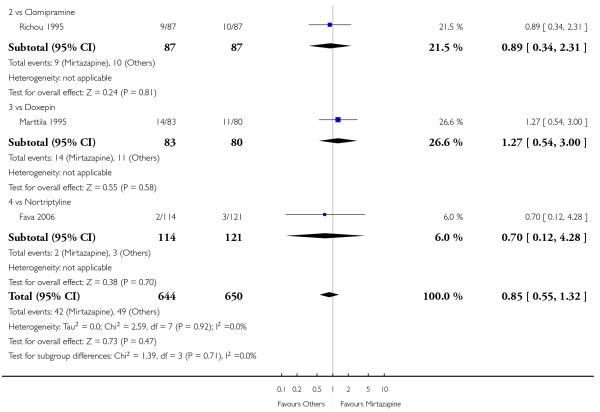 |
Analysis 1.4. Comparison 1 Mirtazapine versus TCAs, Outcome 4 Secondary outcome (remission) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 4 Secondary outcome (remission) at end of the acute-phase treatment
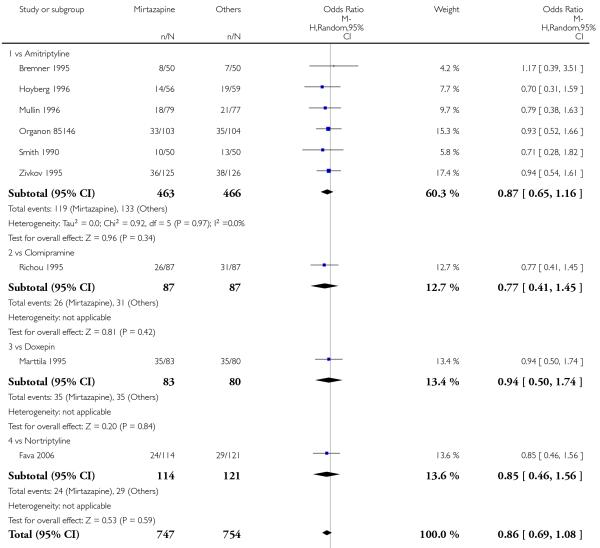 |
 |
Analysis 1.5. Comparison 1 Mirtazapine versus TCAs, Outcome 5 Secondary outcome (depression severity) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 5 Secondary outcome (depression severity) at 2 weeks
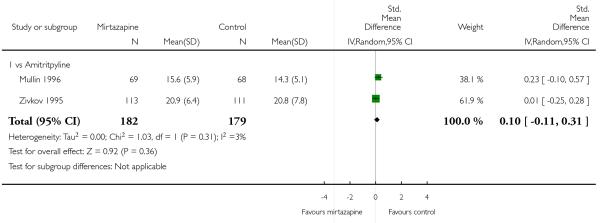 |
Analysis 1.6. Comparison 1 Mirtazapine versus TCAs, Outcome 6 Secondary outcome (depression severity) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 6 Secondary outcome (depression severity) at end of the acute-phase treatment
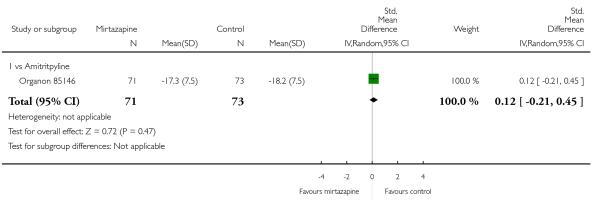 |
Analysis 1.7. Comparison 1 Mirtazapine versus TCAs, Outcome 7 Secondary outcome (Social adjustment) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 7 Secondary outcome (Social adjustment) at 2 weeks
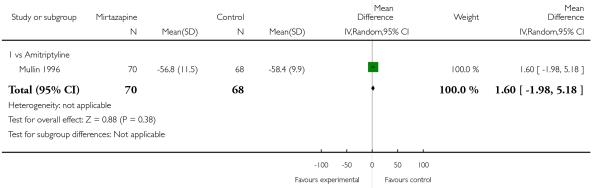 |
Analysis 1.8. Comparison 1 Mirtazapine versus TCAs, Outcome 8 Secondary outcome (Social adjustment) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 8 Secondary outcome (Social adjustment) at end of the acute-phase treatment
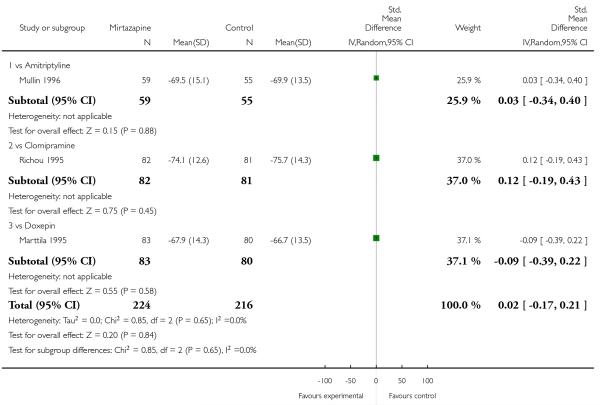 |
Analysis 1.9. Comparison 1 Mirtazapine versus TCAs, Outcome 9 Secondary outcome (withdrawal due to any reason).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 9 Secondary outcome (withdrawal due to any reason)
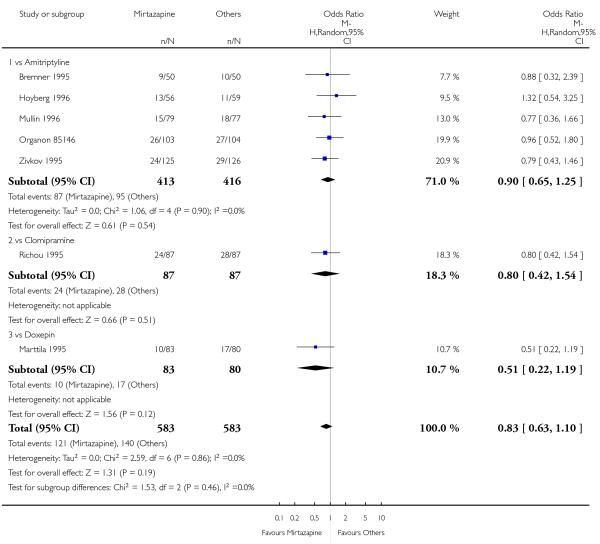 |
Analysis 1.10. Comparison 1 Mirtazapine versus TCAs, Outcome 10 Secondary outcome (withdrawal due to adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 10 Secondary outcome (withdrawal due to adverse events)
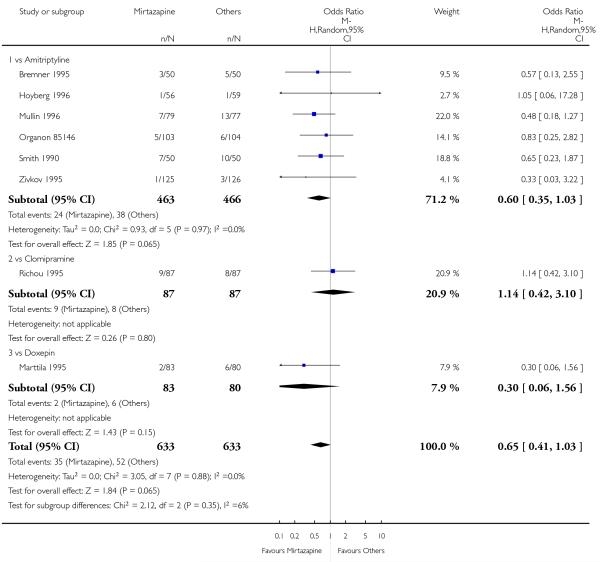 |
Analysis 1.11. Comparison 1 Mirtazapine versus TCAs, Outcome 11 Secondary outcome (having some adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 11 Secondary outcome (having some adverse events)
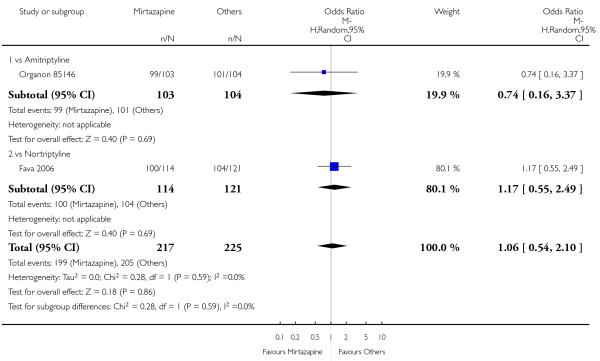 |
Analysis 1.12. Comparison 1 Mirtazapine versus TCAs, Outcome 12 Hypertension/Tachycardia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 12 Hypertension/Tachycardia
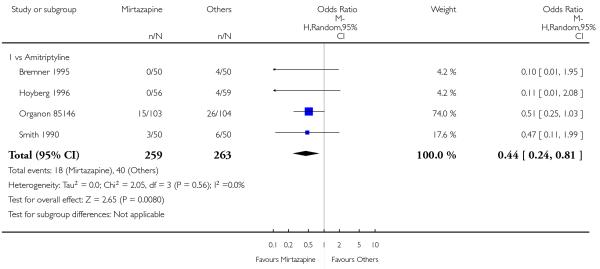 |
Analysis 1.13. Comparison 1 Mirtazapine versus TCAs, Outcome 13 Hypotension/Bradycardia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 13 Hypotension/Bradycardia
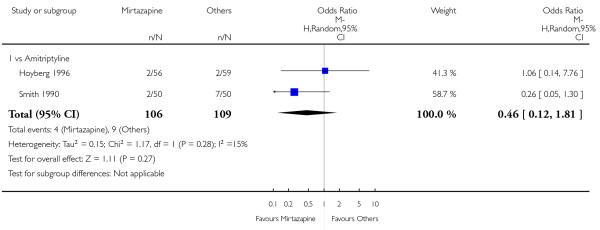 |
Analysis 1.14. Comparison 1 Mirtazapine versus TCAs, Outcome 14 Sweating.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 14 Sweating
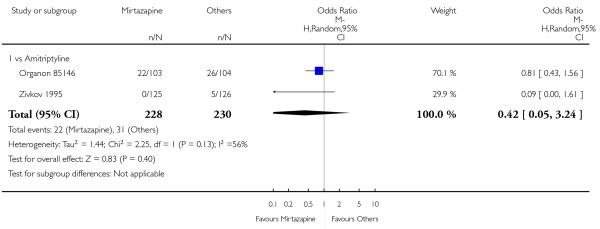 |
Analysis 1.15. Comparison 1 Mirtazapine versus TCAs, Outcome 15 Constipation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 15 Constipation
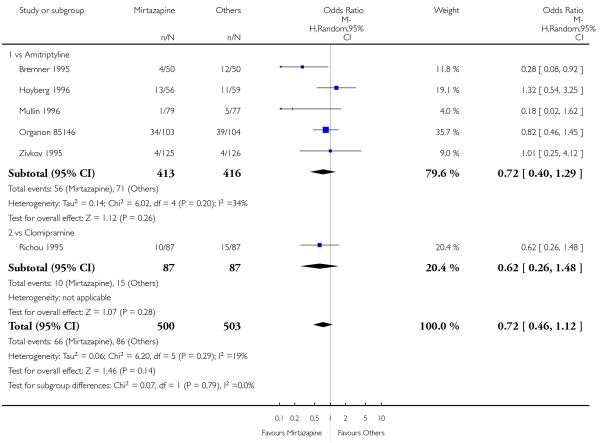 |
Analysis 1.16. Comparison 1 Mirtazapine versus TCAs, Outcome 16 Dry mouth/Decreased salivation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 16 Dry mouth/Decreased salivation
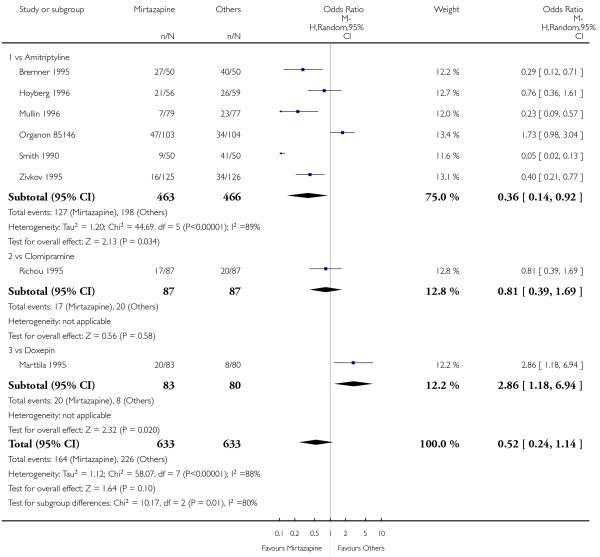 |
Analysis 1.17. Comparison 1 Mirtazapine versus TCAs, Outcome 17 Nausea/Vomiting/Gastric distress.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 17 Nausea/Vomiting/Gastric distress
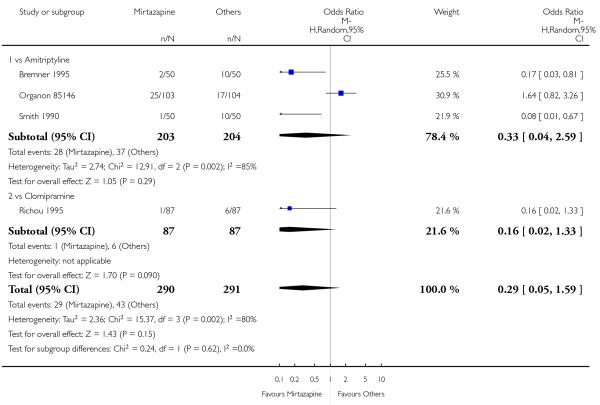 |
Analysis 1.18. Comparison 1 Mirtazapine versus TCAs, Outcome 18 Weight gain/Increased appetite.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 18 Weight gain/Increased appetite
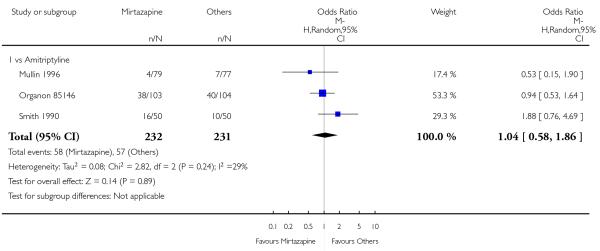 |
Analysis 1.19. Comparison 1 Mirtazapine versus TCAs, Outcome 19 Sexual dysfunction.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 19 Sexual dysfunction
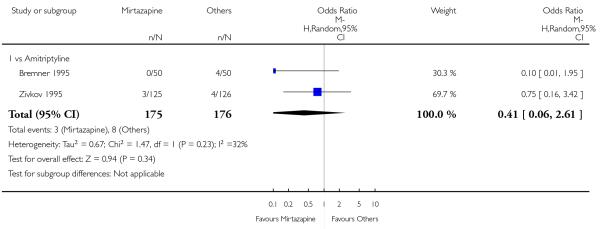 |
Analysis 1.20. Comparison 1 Mirtazapine versus TCAs, Outcome 20 Anxiety/Agitation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 20 Anxiety/Agitation
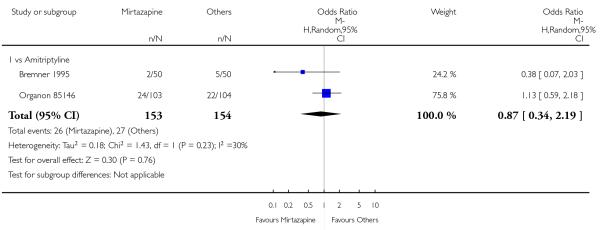 |
Analysis 1.21. Comparison 1 Mirtazapine versus TCAs, Outcome 21 Dizziness/Vertigo/Faintness.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 21 Dizziness/Vertigo/Faintness
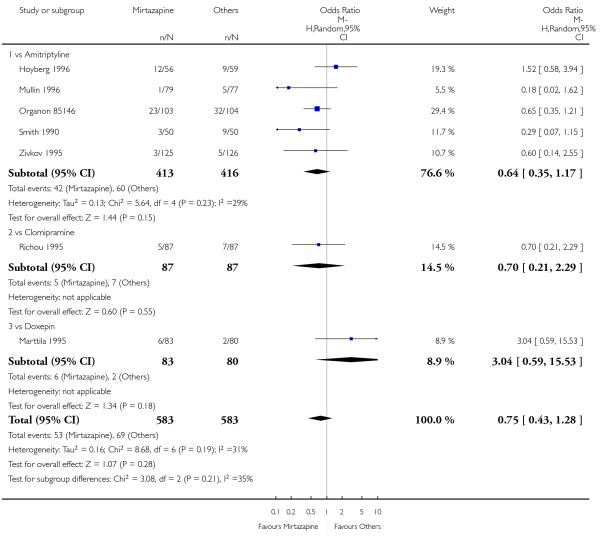 |
Analysis 1.22. Comparison 1 Mirtazapine versus TCAs, Outcome 22 Fatigue/Tiredness/Asthenia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 22 Fatigue/Tiredness/Asthenia
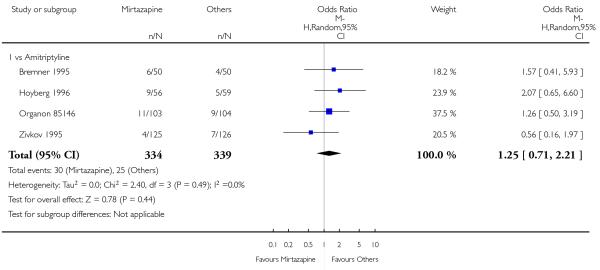 |
Analysis 1.23. Comparison 1 Mirtazapine versus TCAs, Outcome 23 Headache.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 23 Headache
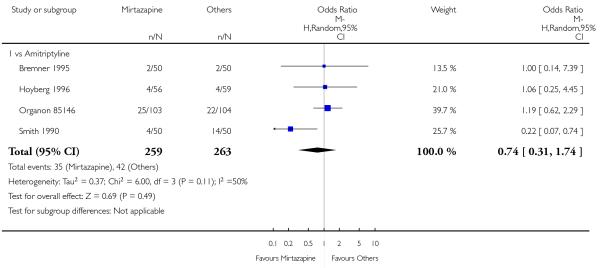 |
Analysis 1.24. Comparison 1 Mirtazapine versus TCAs, Outcome 24 Tremor.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 24 Tremor
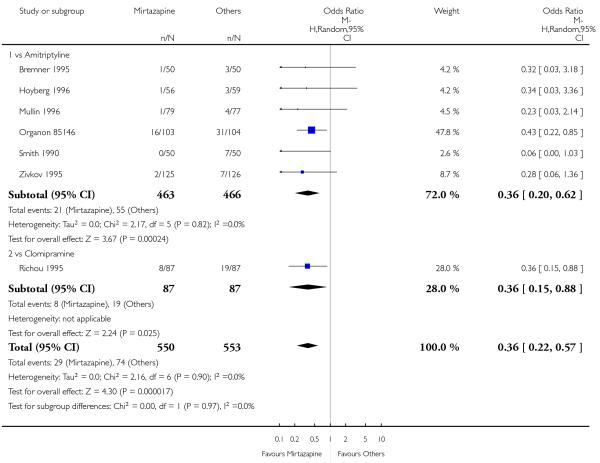 |
Analysis 1.25. Comparison 1 Mirtazapine versus TCAs, Outcome 25 Sleep disturbance.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 25 Sleep disturbance
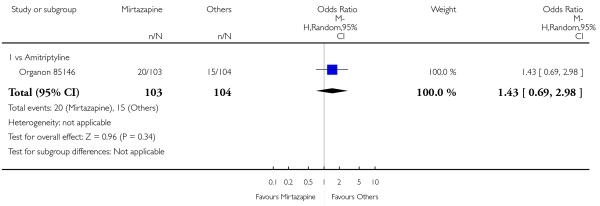 |
Analysis 1.26. Comparison 1 Mirtazapine versus TCAs, Outcome 26 Sleepiness/Drowsiness/Somnolence.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 26 Sleepiness/Drowsiness/Somnolence
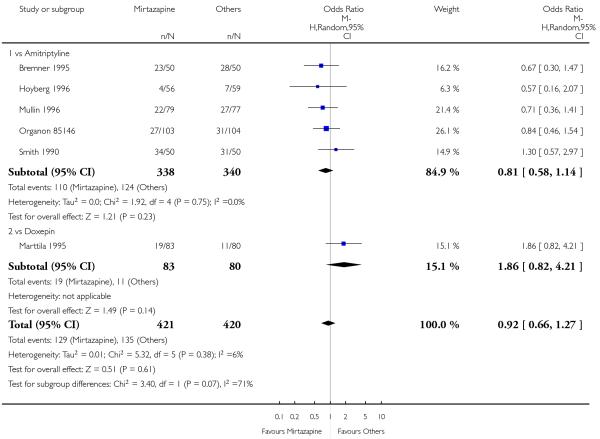 |
Analysis 1.27. Comparison 1 Mirtazapine versus TCAs, Outcome 27 Suicide attempt.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 27 Suicide attempt
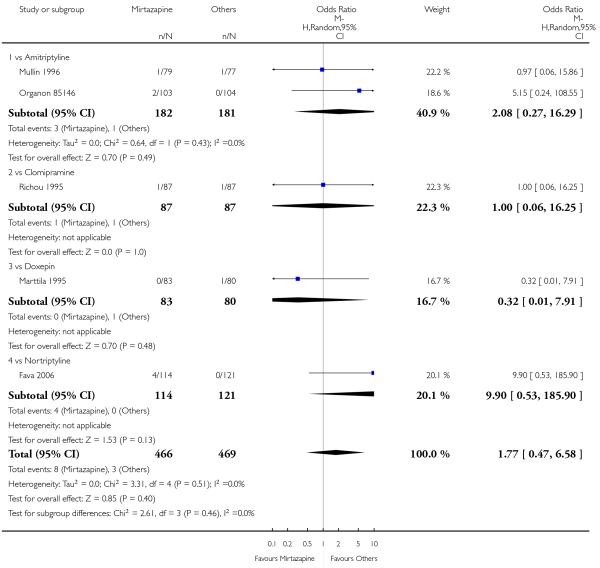 |
Analysis 1.28. Comparison 1 Mirtazapine versus TCAs, Outcome 28 Subgroup analysis: Response at 2 weeks: Treatment settings: Psychiatric inpatients.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 28 Subgroup analysis: Response at 2 weeks: Treatment settings: Psychiatric inpatients
 |
Analysis 1.29. Comparison 1 Mirtazapine versus TCAs, Outcome 29 Subgroup analysis: Response at end of the acute-phase treatment: Treatment settings: Psychiatric inpatients.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 29 Subgroup analysis: Response at end of the acute-phase treatment: Treatment settings: Psychiatric inpatients
 |
Analysis 1.30. Comparison 1 Mirtazapine versus TCAs, Outcome 30 Sensitivity analysis: Response at 2 weeks: Studies without imputation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 30 Sensitivity analysis: Response at 2 weeks: Studies without imputation
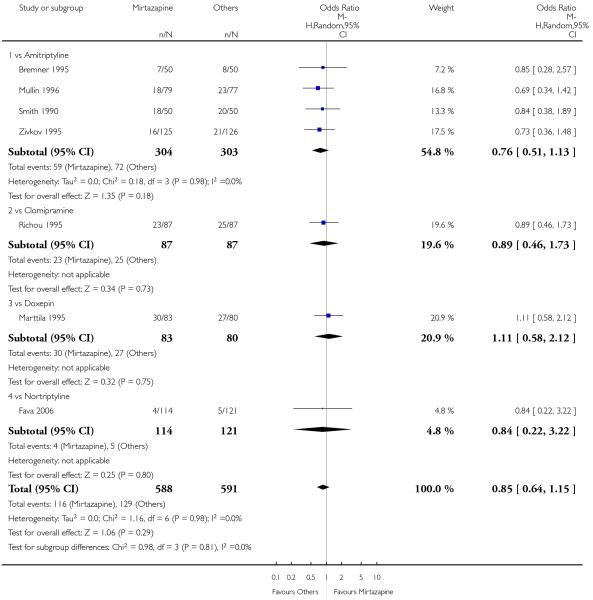 |
Analysis 1.31. Comparison 1 Mirtazapine versus TCAs, Outcome 31 Sensitivity analysis: Response at end of the acute-phase treatment: Studies without imputation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 1 Mirtazapine versus TCAs
Outcome: 31 Sensitivity analysis: Response at end of the acute-phase treatment: Studies without imputation
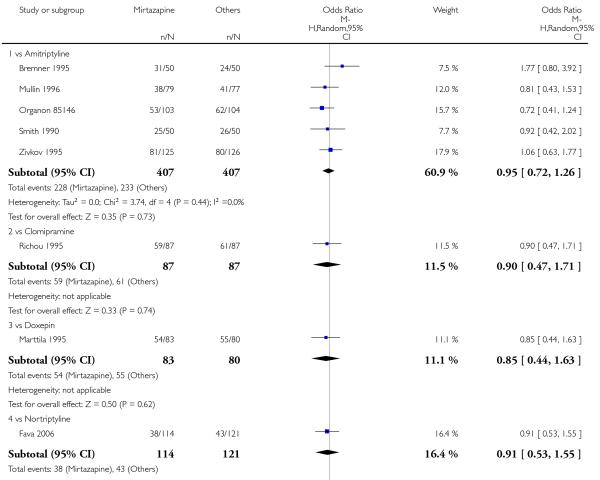 |
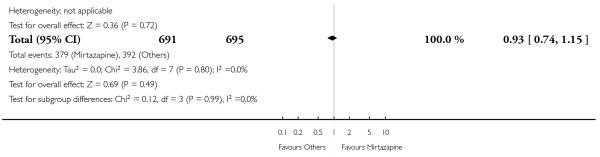 |
Analysis 1.32. Comparison 1 Mirtazapine versus TCAs, Outcome 32 Secondary outcome (SKEWED DATA: depression severity) at 2 weeks.
Secondary outcome (SKEWED DATA: depression severity) at 2 weeks
| Study | Comparator drug | Measurement | Mirtazapine: mean | SD | N | Comparator: mean | SD | N | note |
|---|---|---|---|---|---|---|---|---|---|
| Hoyberg 1996 | Amitripty-line | HAMD-21 change score | −4.7 | 4.5 | 54 | −5.2 | 4.4 | 59 |
Analysis 1.33. Comparison 1 Mirtazapine versus TCAs, Outcome 33 Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment.
Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment
| Study | Comparator | Measurement | Mirtazapine: mean | SD | N | Comparator: mean | SD | N | Note |
|---|---|---|---|---|---|---|---|---|---|
| Hoyberg 1996 | Amitripty-line | HAMD-21 change score | −11.1 | 7.9 | 54 | −13.1 | 7.5 | 59 | |
| Marttila 1995 | Doxepin | HAMD-17 | 9.15 | 7.5 | 83 | 9.0 | 6.35 | 80 | |
| Mullin 1996 | Amitripty-line | HAMD-17 | 11.7 | 7.3 | 71 | 10.7 | 6.8 | 71 | |
| Zivkov 1995 | Amitripty-line | HAMD-21 | 12.8 | 10.1 | 113 | 12.0 | 10.0 | 111 |
Analysis 2.1. Comparison 2 Mirtazapine versus SSRIs, Outcome 1 Primary outcome (response) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 1 Primary outcome (response) at 2 weeks
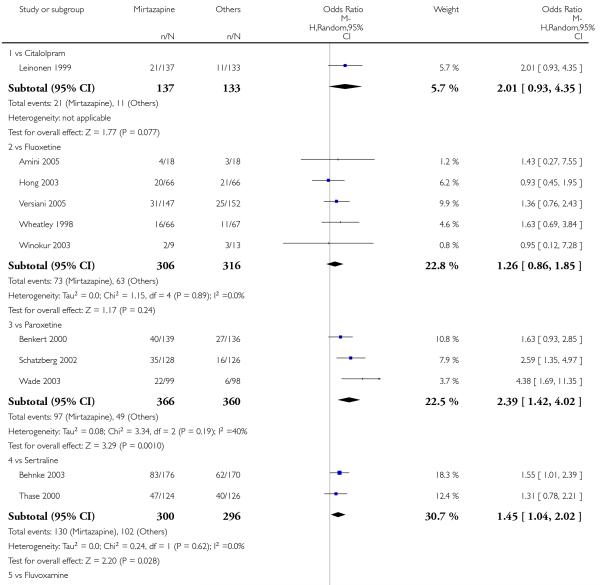 |
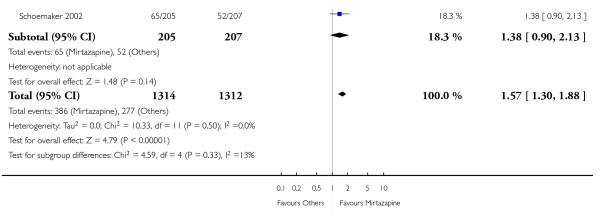 |
Analysis 2.2. Comparison 2 Mirtazapine versus SSRIs, Outcome 2 Primary outcome (response) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 2 Primary outcome (response) at end of the acute-phase treatment
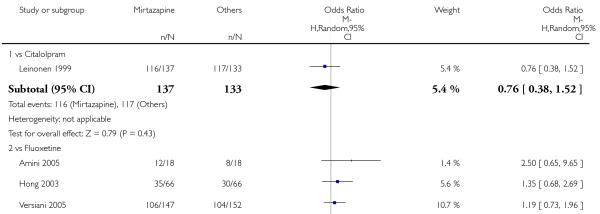 |
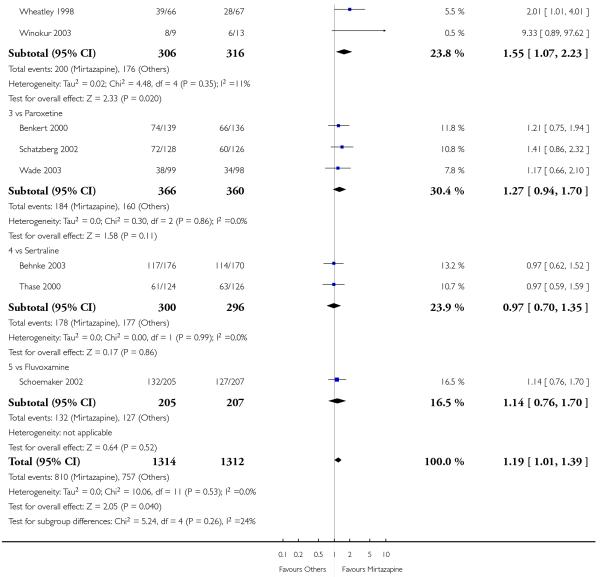 |
Analysis 2.3. Comparison 2 Mirtazapine versus SSRIs, Outcome 3 Primary outcome (response) at end of the continuation treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 3 Primary outcome (response) at end of the continuation treatment
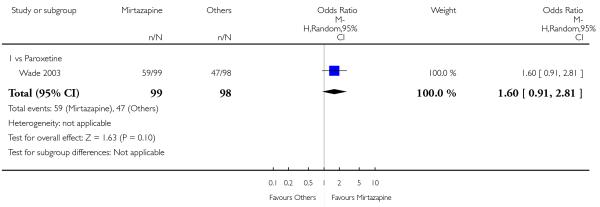 |
Analysis 2.4. Comparison 2 Mirtazapine versus SSRIs, Outcome 4 Secondary outcome (remission) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 4 Secondary outcome (remission) at 2 weeks
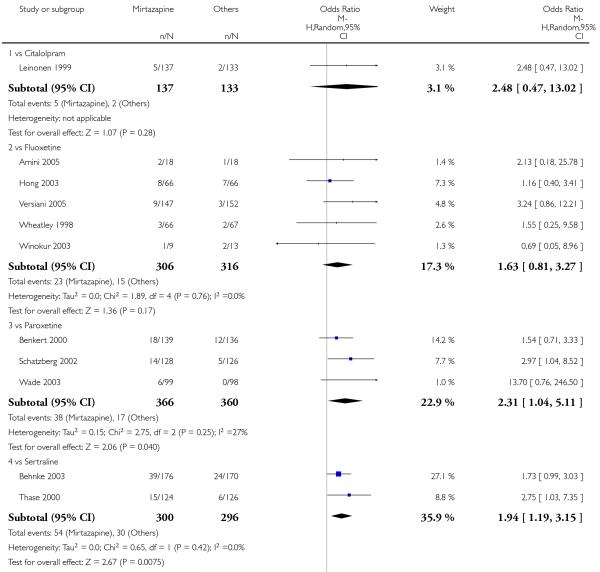 |
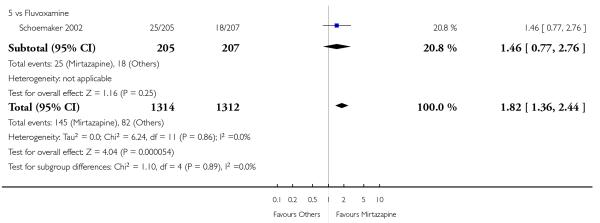 |
Analysis 2.5. Comparison 2 Mirtazapine versus SSRIs, Outcome 5 Secondary outcome (remission) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 5 Secondary outcome (remission) at end of the acute-phase treatment
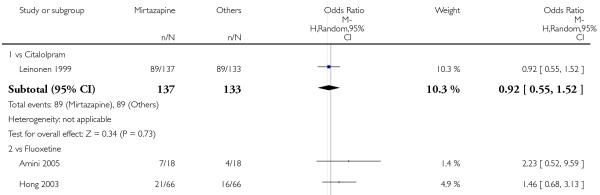 |
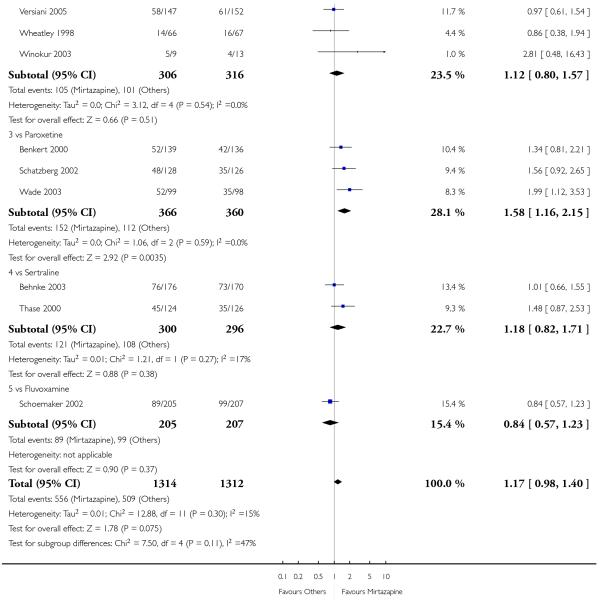 |
Analysis 2.6. Comparison 2 Mirtazapine versus SSRIs, Outcome 6 Secondary outcome (remission) at end of the continuation treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 6 Secondary outcome (remission) at end of the continuation treatment
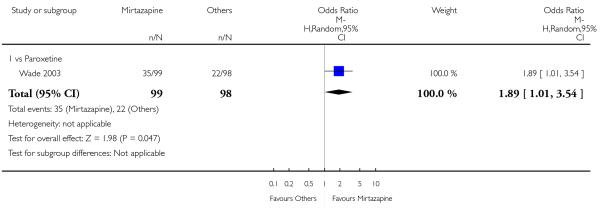 |
Analysis 2.7. Comparison 2 Mirtazapine versus SSRIs, Outcome 7 Secondary outcome (withdrawal due to any reason).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 7 Secondary outcome (withdrawal due to any reason)
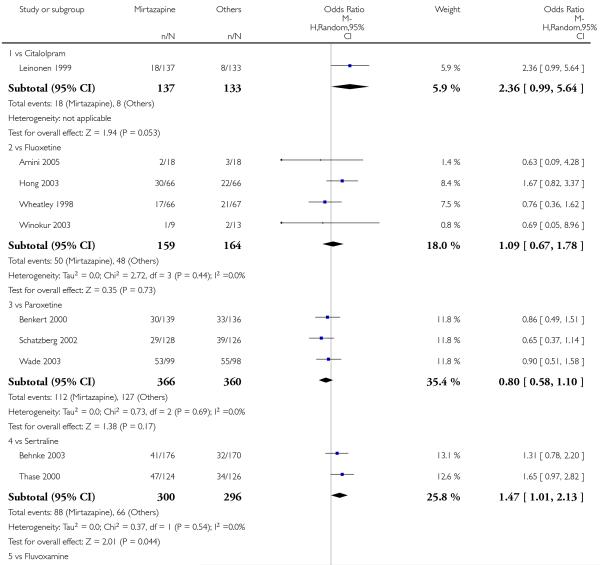 |
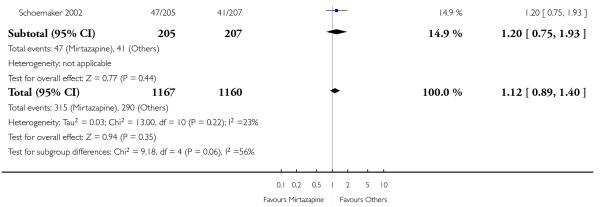 |
Analysis 2.8. Comparison 2 Mirtazapine versus SSRIs, Outcome 8 Secondary outcome (withdrawal due to adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 8 Secondary outcome (withdrawal due to adverse events)
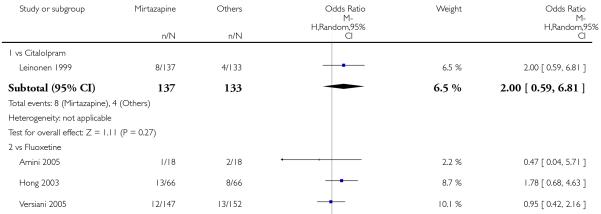 |
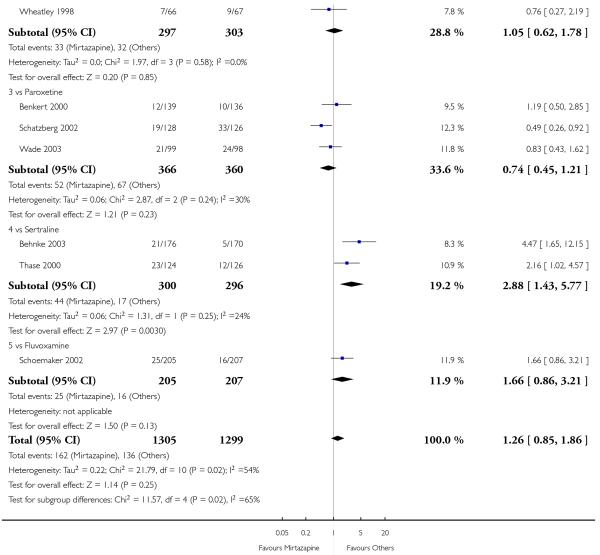 |
Analysis 2.9. Comparison 2 Mirtazapine versus SSRIs, Outcome 9 Secondary outcome (having some adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 9 Secondary outcome (having some adverse events)
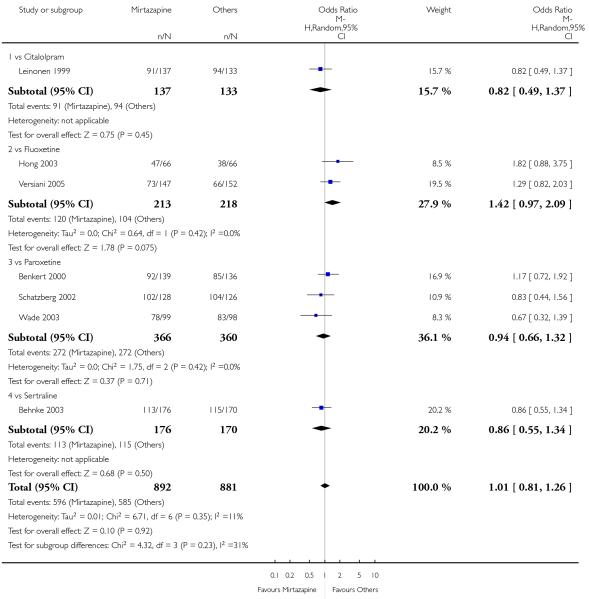 |
Analysis 2.10. Comparison 2 Mirtazapine versus SSRIs, Outcome 10 Hypotension/Bradycardia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 10 Hypotension/Bradycardia
 |
Analysis 2.11. Comparison 2 Mirtazapine versus SSRIs, Outcome 11 Sweating.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 11 Sweating
 |
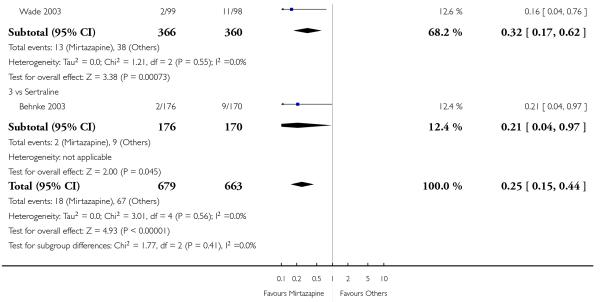 |
Analysis 2.12. Comparison 2 Mirtazapine versus SSRIs, Outcome 12 Constipation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 12 Constipation
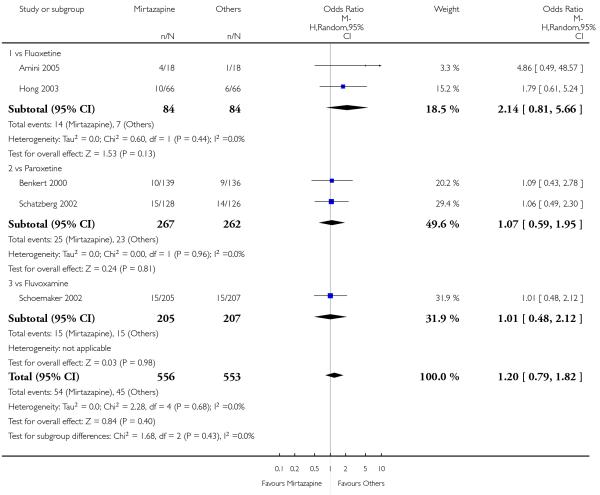 |
Analysis 2.13. Comparison 2 Mirtazapine versus SSRIs, Outcome 13 Diarrhoea.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 13 Diarrhoea
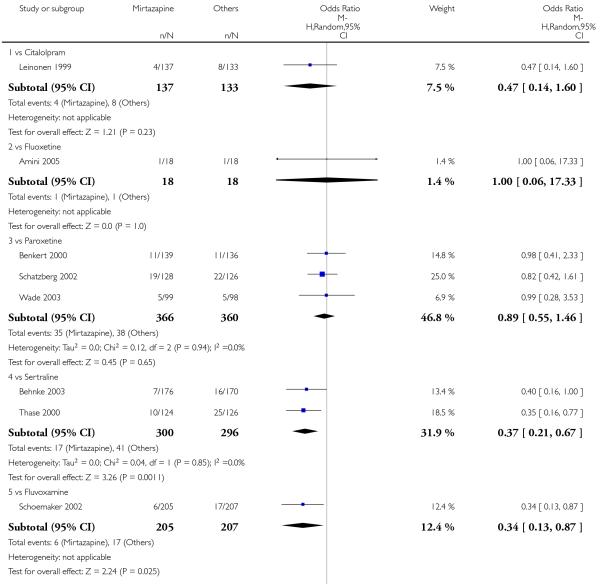 |
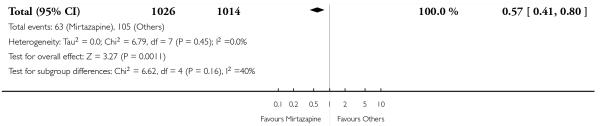 |
Analysis 2.14. Comparison 2 Mirtazapine versus SSRIs, Outcome 14 Dry mouth/Decreased salivation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 14 Dry mouth/Decreased salivation
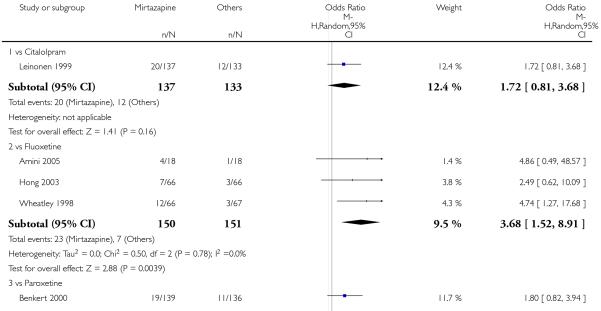 |
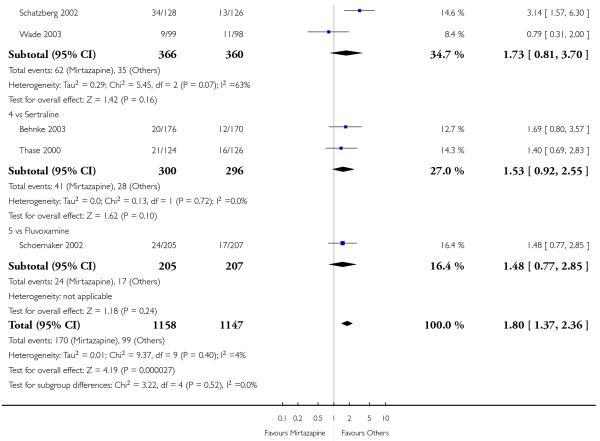 |
Analysis 2.15. Comparison 2 Mirtazapine versus SSRIs, Outcome 15 Nausea/Vomiting/Gastric distress.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 15 Nausea/Vomiting/Gastric distress
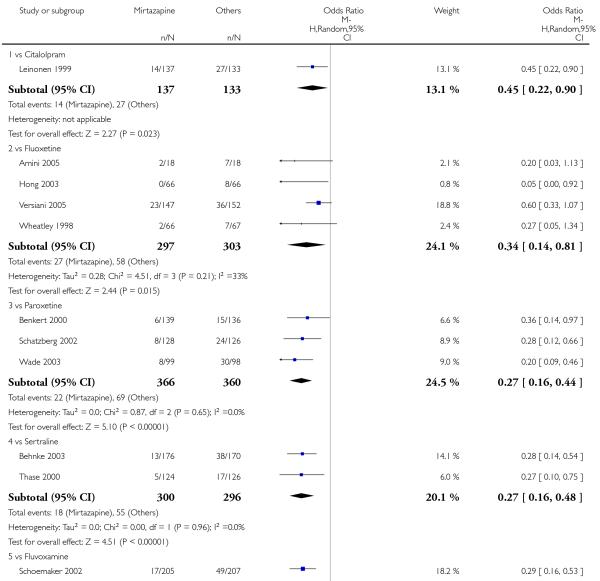 |
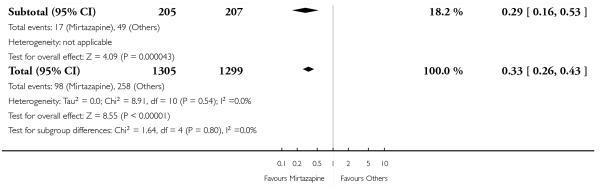 |
Analysis 2.16. Comparison 2 Mirtazapine versus SSRIs, Outcome 16 Weight gain/Increased appetite.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 16 Weight gain/Increased appetite
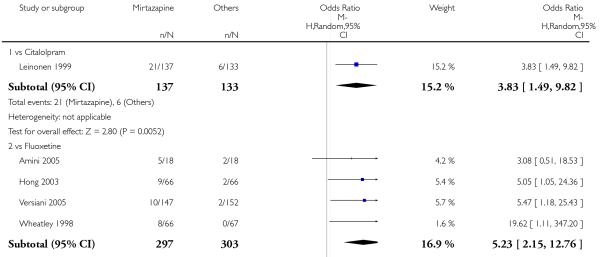 |
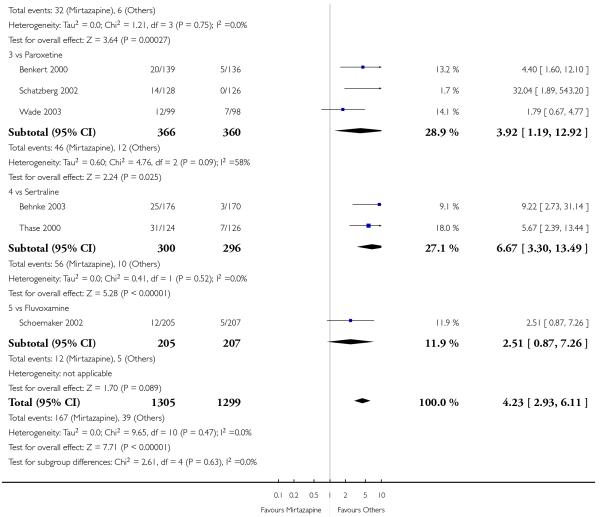 |
Analysis 2.17. Comparison 2 Mirtazapine versus SSRIs, Outcome 17 Weight loss/Anorexia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 17 Weight loss/Anorexia
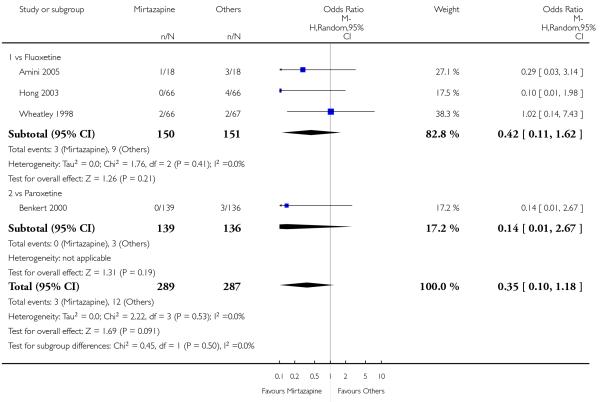 |
Analysis 2.18. Comparison 2 Mirtazapine versus SSRIs, Outcome 18 Sexual dysfunction.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 18 Sexual dysfunction
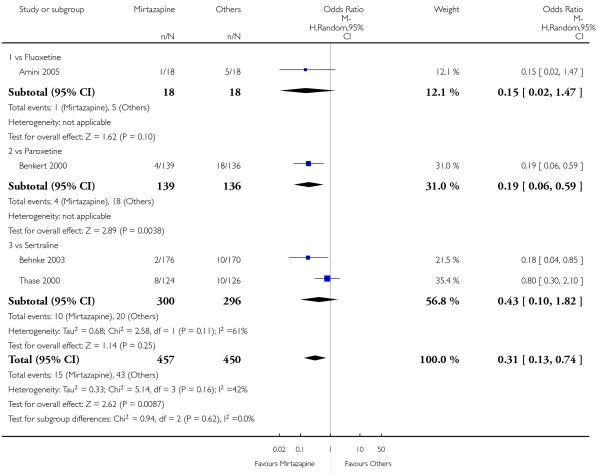 |
Analysis 2.19. Comparison 2 Mirtazapine versus SSRIs, Outcome 19 Anxiety/Agitation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 19 Anxiety/Agitation
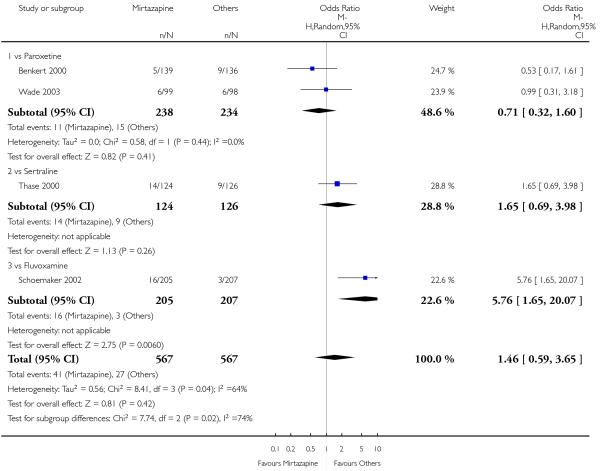 |
Analysis 2.20. Comparison 2 Mirtazapine versus SSRIs, Outcome 20 Dizziness/Vertigo/Faintness.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 20 Dizziness/Vertigo/Faintness
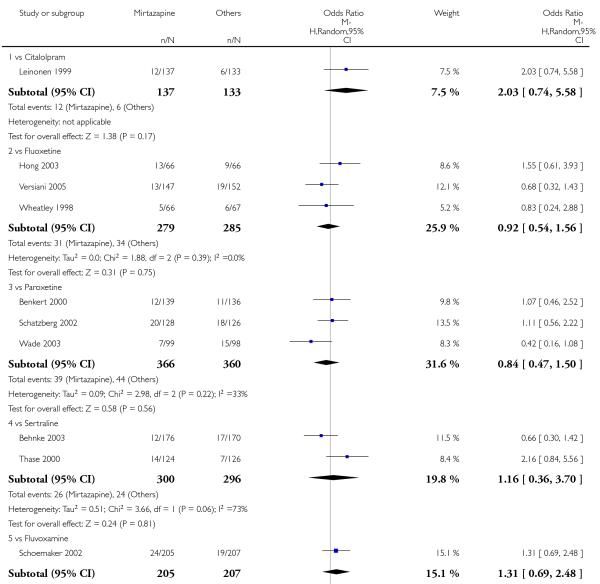 |
 |
Analysis 2.21. Comparison 2 Mirtazapine versus SSRIs, Outcome 21 Fatigue/Tiredness/Asthenia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 21 Fatigue/Tiredness/Asthenia
 |
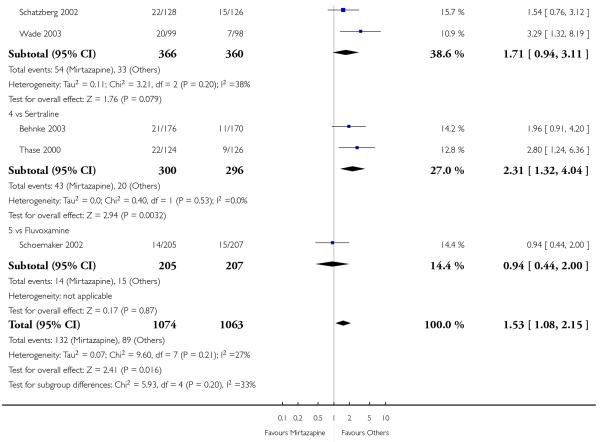 |
Analysis 2.22. Comparison 2 Mirtazapine versus SSRIs, Outcome 22 Headache.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 22 Headache
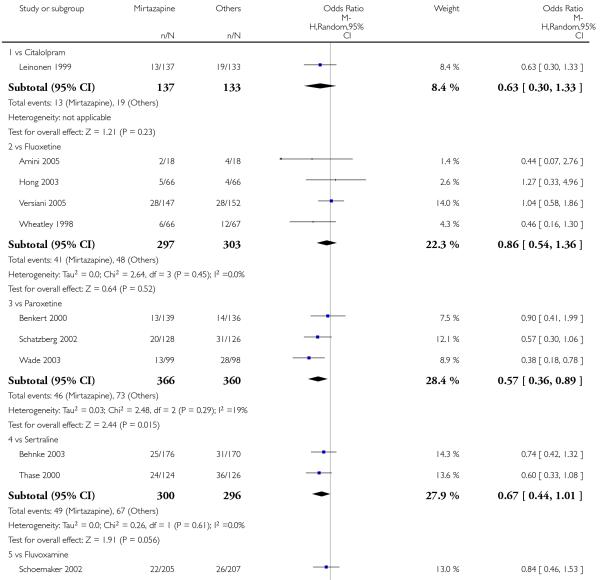 |
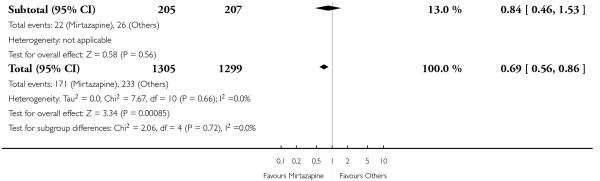 |
Analysis 2.23. Comparison 2 Mirtazapine versus SSRIs, Outcome 23 Tremor.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 23 Tremor
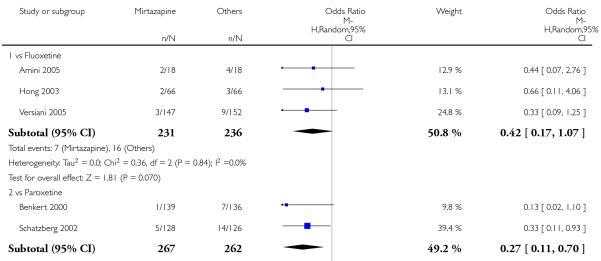 |
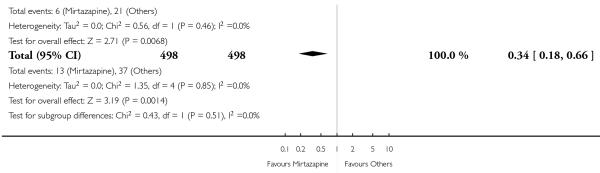 |
Analysis 2.24. Comparison 2 Mirtazapine versus SSRIs, Outcome 24 Sleep disturbance.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 24 Sleep disturbance
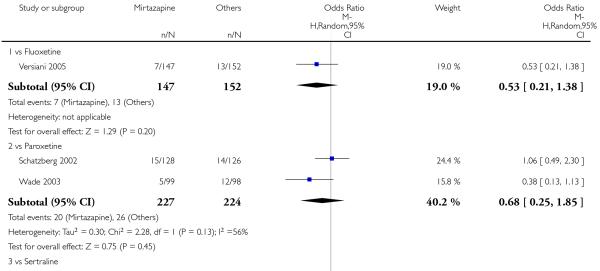 |
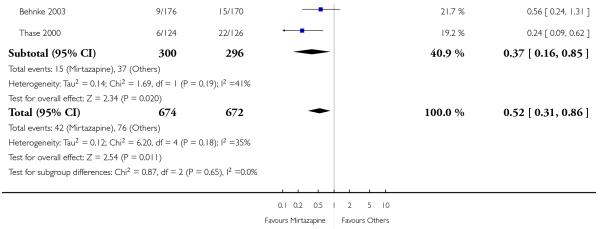 |
Analysis 2.25. Comparison 2 Mirtazapine versus SSRIs, Outcome 25 Sleepiness/Drowsiness/Somnolence.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 25 Sleepiness/Drowsiness/Somnolence
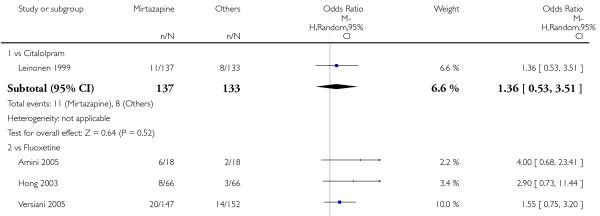 |
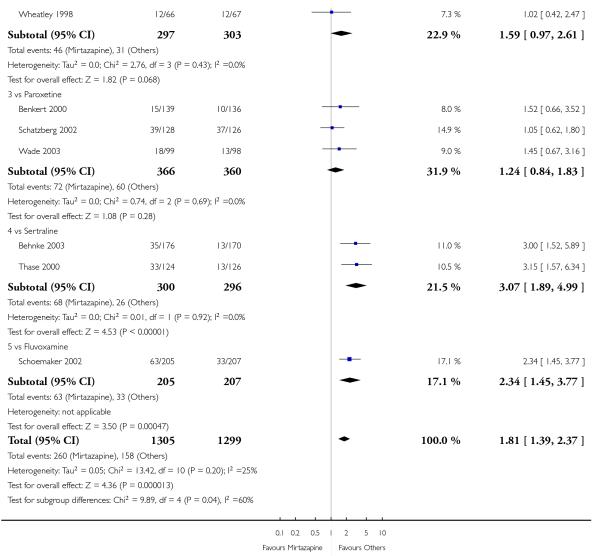 |
Analysis 2.26. Comparison 2 Mirtazapine versus SSRIs, Outcome 26 Suicide attempt.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 26 Suicide attempt
 |
Analysis 2.27. Comparison 2 Mirtazapine versus SSRIs, Outcome 27 Subgroup analysis: Response at 2 weeks: Treatment settings: Outpatients in primary care.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 27 Subgroup analysis: Response at 2 weeks: Treatment settings: Outpatients in primary care
 |
Analysis 2.28. Comparison 2 Mirtazapine versus SSRIs, Outcome 28 Subgroup analysis: Response at end of the acute-phase treatment: Treatment settings: Outpatients in primary care.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 28 Subgroup analysis: Response at end of the acute-phase treatment: Treatment settings: Outpatients in primary care
 |
Analysis 2.29. Comparison 2 Mirtazapine versus SSRIs, Outcome 29 Sensitivity analysis: Response at 2 weeks: Studies without imputation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 29 Sensitivity analysis: Response at 2 weeks: Studies without imputation
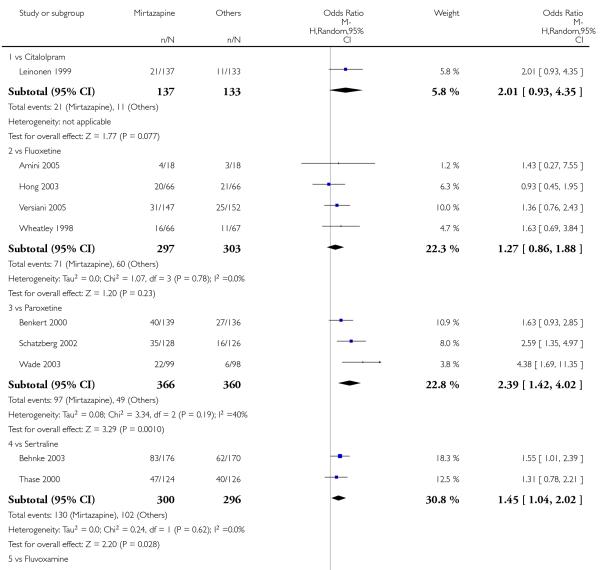 |
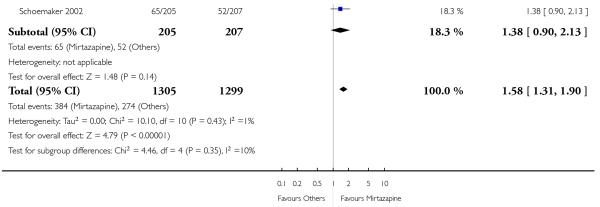 |
Analysis 2.30. Comparison 2 Mirtazapine versus SSRIs, Outcome 30 Sensitivity analysis: Response at end of the acute-phase treatment: Studies without imputation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 2 Mirtazapine versus SSRIs
Outcome: 30 Sensitivity analysis: Response at end of the acute-phase treatment: Studies without imputation
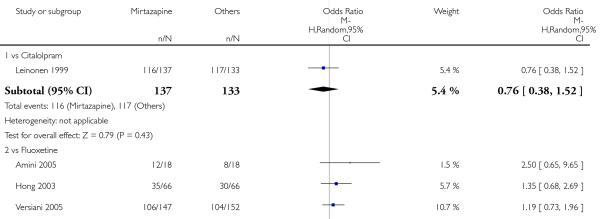 |
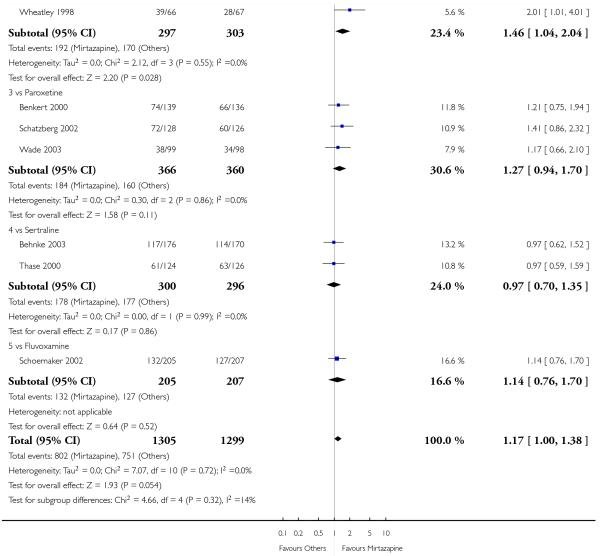 |
Analysis 2.31. Comparison 2 Mirtazapine versus SSRIs, Outcome 31 Secondary outcome (SKEWED DATA: depression severity) at 2 weeks.
Secondary outcome (SKEWED DATA: depression severity) at 2 weeks
| Study | Comparator drug | Measurement | Mirtazapine: mean | SD | N | Comparator: mean | SD | N | note |
|---|---|---|---|---|---|---|---|---|---|
| Schoemaker 2002 | Fluvoxamine | HAMD-17 | −9.2 | 5.86 | 199 | −7.3 | 6.11 | 203 | |
| Winokur 2003 | Fluoxetine | HAMD-21 | 16.1 | 5.7 | 8 | 18.0 | 9.6 | 11 |
Analysis 2.32. Comparison 2 Mirtazapine versus SSRIs, Outcome 32 Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment.
Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment
| Study | Comparator drug | Measurement | Mirtazapine: mean | SD | N | Comparator: mean | SD | N | note |
|---|---|---|---|---|---|---|---|---|---|
| Schoemaker 2002 | Fluvoxamine | HAMD-17 | −14.3 | 7.33 | 199 | −13.7 | 7.68 | 203 | |
| Thase 2000 | Sertraline | HAMD-17 | 8.7 | 7.6 | 119 | 10.5 | 7.2 | 124 | |
| Winokur 2003 | Fluoxetine | HAMD-21 | 7.1 | 3.7 | 8 | 12.2 | 9.2 | 11 |
Analysis 3.1. Comparison 3 Mirtazapine versus SNRIs, Outcome 1 Primary outcome (response) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 1 Primary outcome (response) at 2 weeks
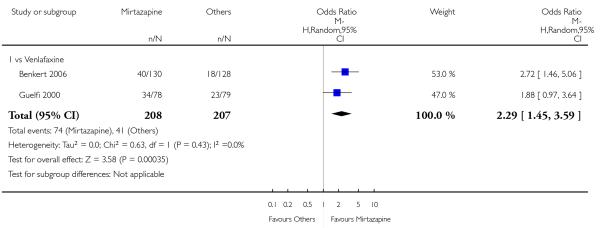 |
Analysis 3.2. Comparison 3 Mirtazapine versus SNRIs, Outcome 2 Primary outcome (response) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 2 Primary outcome (response) at end of the acute-phase treatment
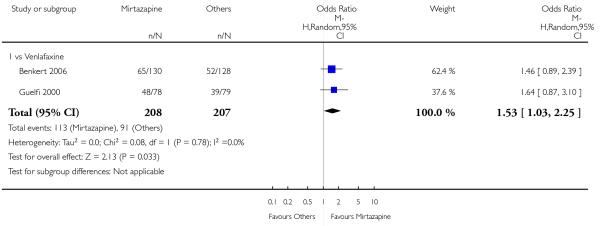 |
Analysis 3.3. Comparison 3 Mirtazapine versus SNRIs, Outcome 3 Secondary outcome (remission) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 3 Secondary outcome (remission) at 2 weeks
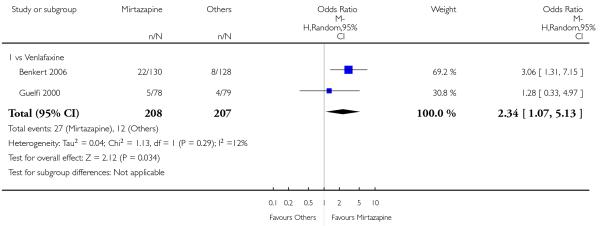 |
Analysis 3.4. Comparison 3 Mirtazapine versus SNRIs, Outcome 4 Secondary outcome (remission) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 4 Secondary outcome (remission) at end of the acute-phase treatment
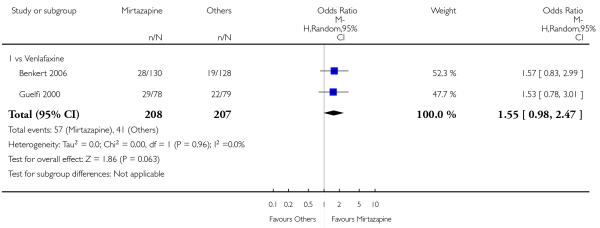 |
Analysis 3.5. Comparison 3 Mirtazapine versus SNRIs, Outcome 5 Secondary outcome (withdrawal due to any reason).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 5 Secondary outcome (withdrawal due to any reason)
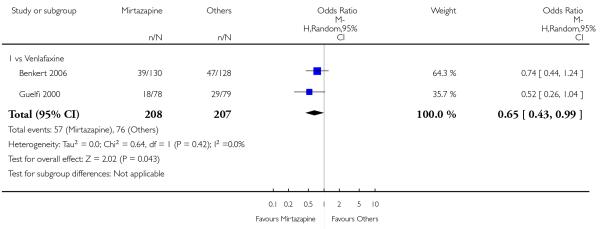 |
Analysis 3.6. Comparison 3 Mirtazapine versus SNRIs, Outcome 6 Secondary outcome (withdrawal due to adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 6 Secondary outcome (withdrawal due to adverse events)
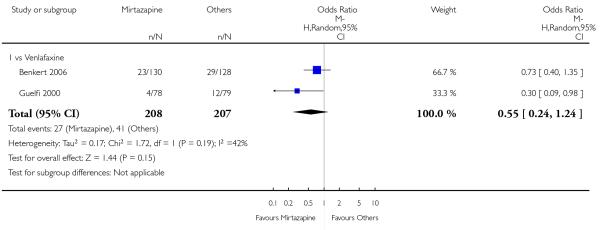 |
Analysis 3.7. Comparison 3 Mirtazapine versus SNRIs, Outcome 7 Secondary outcome (having some adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 7 Secondary outcome (having some adverse events)
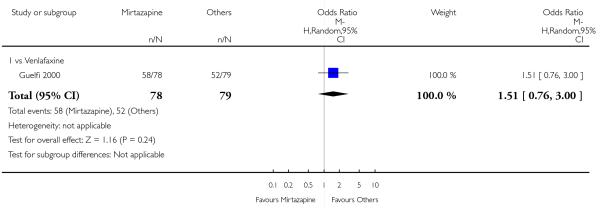 |
Analysis 3.8. Comparison 3 Mirtazapine versus SNRIs, Outcome 8 Hypotension/Bradycardia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 8 Hypotension/Bradycardia
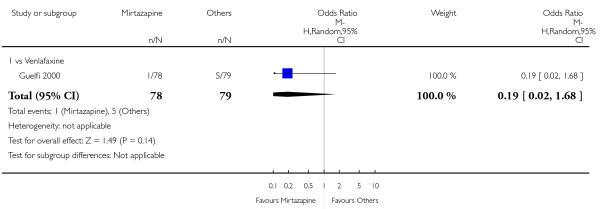 |
Analysis 3.9. Comparison 3 Mirtazapine versus SNRIs, Outcome 9 Sweating.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 9 Sweating
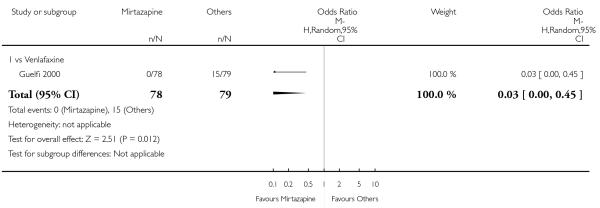 |
Analysis 3.10. Comparison 3 Mirtazapine versus SNRIs, Outcome 10 Constipation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 10 Constipation
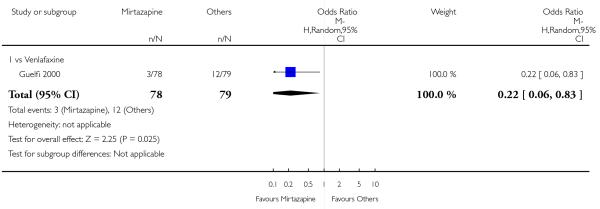 |
Analysis 3.11. Comparison 3 Mirtazapine versus SNRIs, Outcome 11 Dry mouth/Decreased salivation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 11 Dry mouth/Decreased salivation
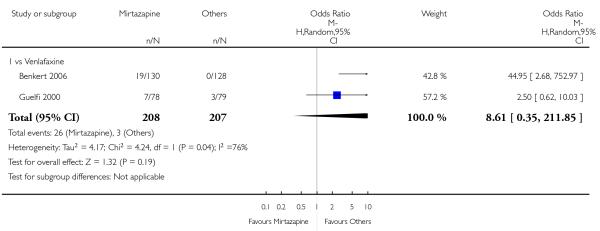 |
Analysis 3.12. Comparison 3 Mirtazapine versus SNRIs, Outcome 12 Nausea/Vomiting/Gastric distress.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 12 Nausea/Vomiting/Gastric distress
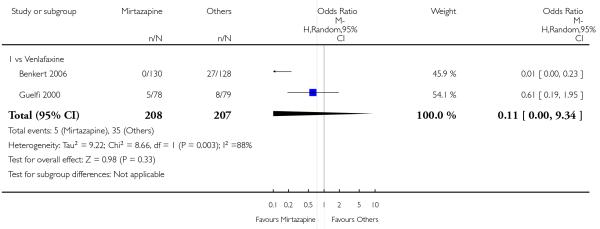 |
Analysis 3.13. Comparison 3 Mirtazapine versus SNRIs, Outcome 13 Anxiety/Agitation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 13 Anxiety/Agitation
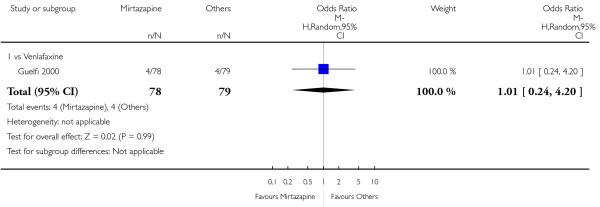 |
Analysis 3.14. Comparison 3 Mirtazapine versus SNRIs, Outcome 14 Fatigue/Tiredness/Asthenia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 14 Fatigue/Tiredness/Asthenia
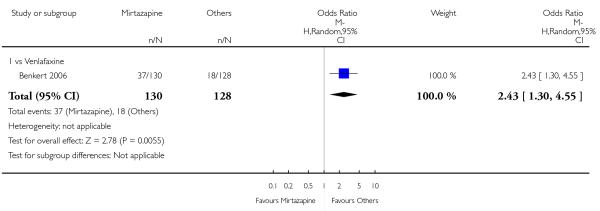 |
Analysis 3.15. Comparison 3 Mirtazapine versus SNRIs, Outcome 15 Headache.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 15 Headache
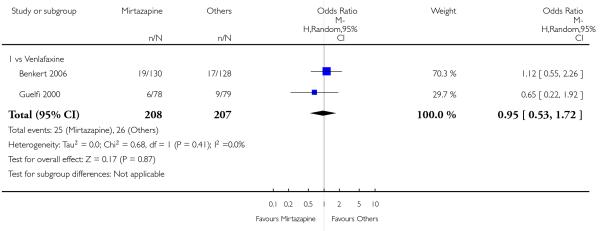 |
Analysis 3.16. Comparison 3 Mirtazapine versus SNRIs, Outcome 16 Sleep disturbance.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 16 Sleep disturbance
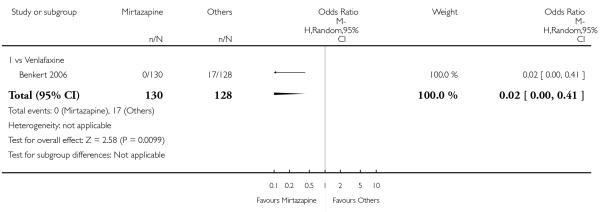 |
Analysis 3.17. Comparison 3 Mirtazapine versus SNRIs, Outcome 17 Sleepiness/Drowsiness/Somnolence.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 17 Sleepiness/Drowsiness/Somnolence
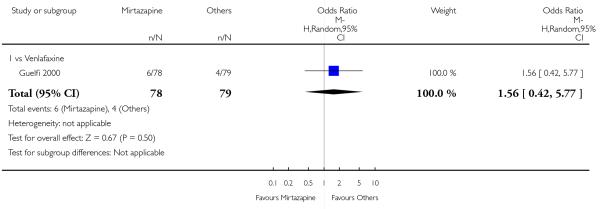 |
Analysis 3.18. Comparison 3 Mirtazapine versus SNRIs, Outcome 18 Completed suicide.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 3 Mirtazapine versus SNRIs
Outcome: 18 Completed suicide
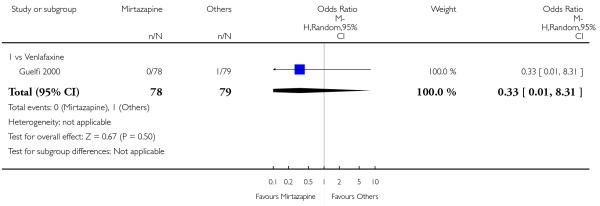 |
Analysis 4.1. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 1 Primary outcome (response) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 1 Primary outcome (response) at 2 weeks
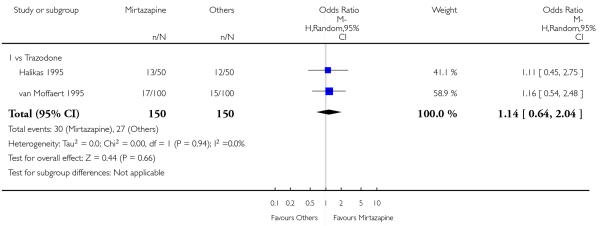 |
Analysis 4.2. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 2 Primary outcome (response) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 2 Primary outcome (response) at end of the acute-phase treatment
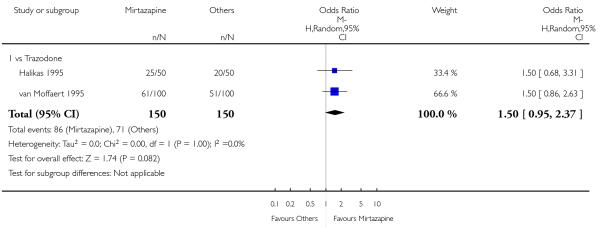 |
Analysis 4.3. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 3 Secondary outcome (remission) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 3 Secondary outcome (remission) at 2 weeks
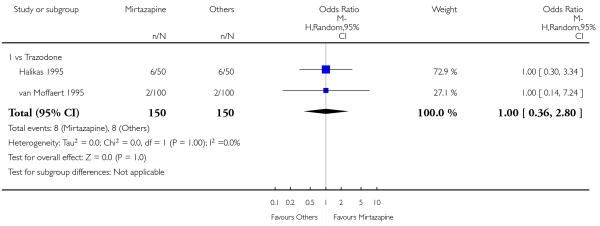 |
Analysis 4.4. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 4 Secondary outcome (remission) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 4 Secondary outcome (remission) at end of the acute-phase treatment
 |
Analysis 4.5. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 5 Secondary outcome (withdrawal due to any reason).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 5 Secondary outcome (withdrawal due to any reason)
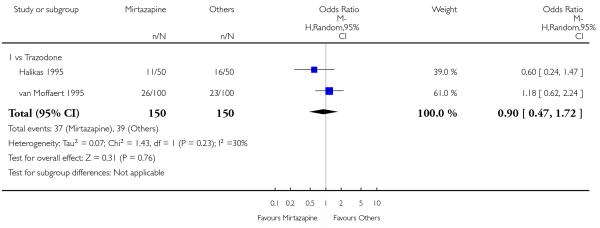 |
Analysis 4.6. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 6 Secondary outcome (withdrawal due to adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 6 Secondary outcome (withdrawal due to adverse events)
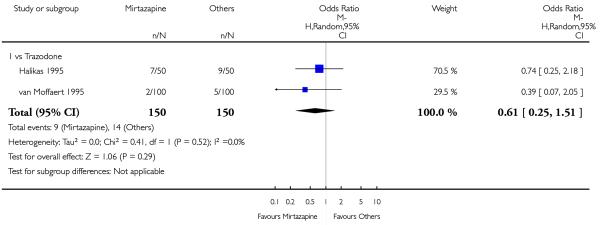 |
Analysis 4.7. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 7 Hypertension/Tachycardia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 7 Hypertension/Tachycardia
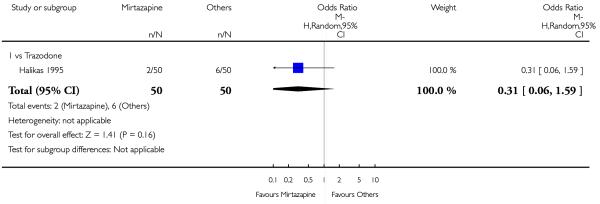 |
Analysis 4.8. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 8 Hypotension/Bradycardia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 8 Hypotension/Bradycardia
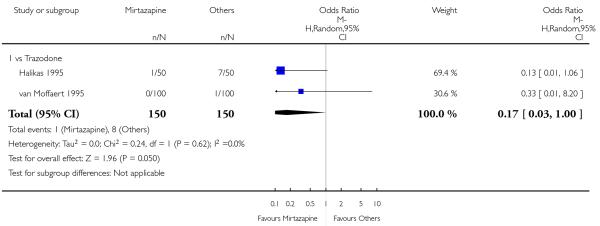 |
Analysis 4.9. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 9 Constipation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 9 Constipation
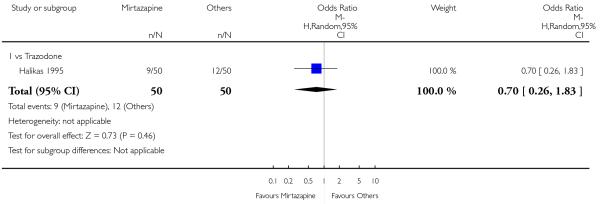 |
Analysis 4.10. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 10 Dry mouth/Decreased salivation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 10 Dry mouth/Decreased salivation
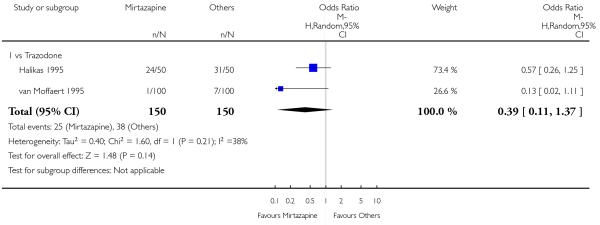 |
Analysis 4.11. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 11 Nausea/Vomiting/Gastric distress.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 11 Nausea/Vomiting/Gastric distress
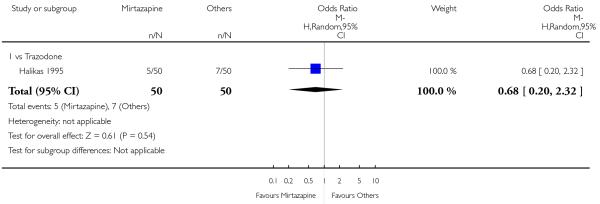 |
Analysis 4.12. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 12Weight gain/Increased appetite.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 12 Weight gain/Increased appetite
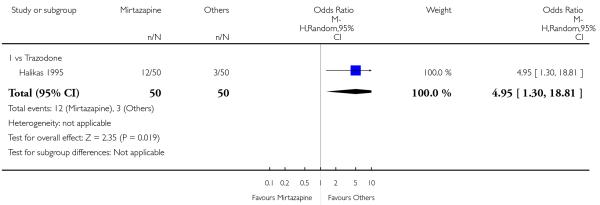 |
Analysis 4.13. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 13Weight loss/Anorexia.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 13 Weight loss/Anorexia
 |
Analysis 4.14. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 14 Anxiety/Agitation.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 14 Anxiety/Agitation
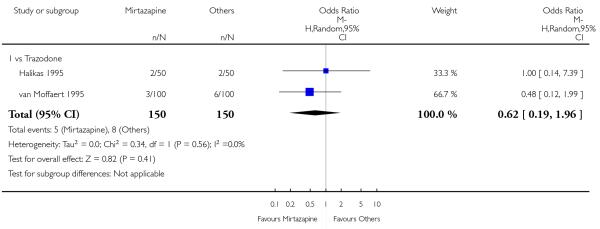 |
Analysis 4.15. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 15 Dizziness/Vertigo/Faintness.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 15 Dizziness/Vertigo/Faintness
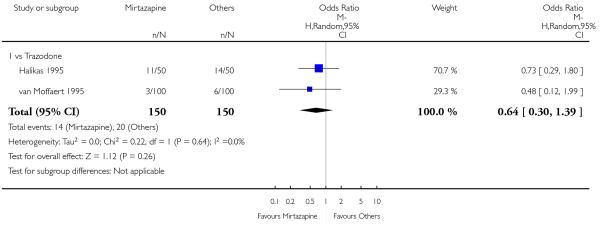 |
Analysis 4.16. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 16 Headache.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 16 Headache
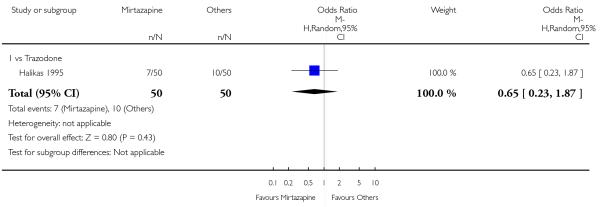 |
Analysis 4.17. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 17 Sleep disturbance.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 17 Sleep disturbance
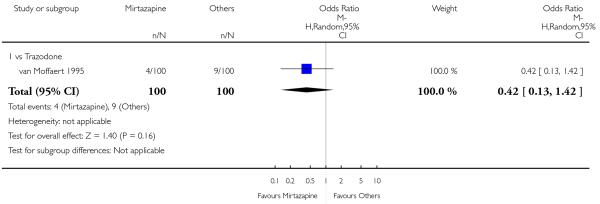 |
Analysis 4.18. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 18 Sleepiness/Drowsiness/Somnolence.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 18 Sleepiness/Drowsiness/Somnolence
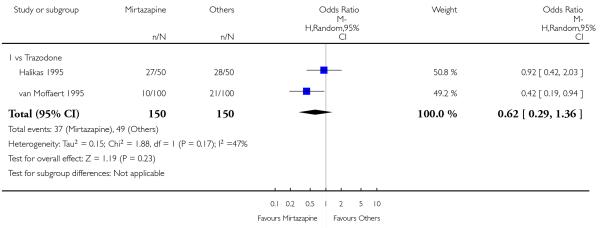 |
Analysis 4.19. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 19 Completed suicide.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 19 Completed suicide
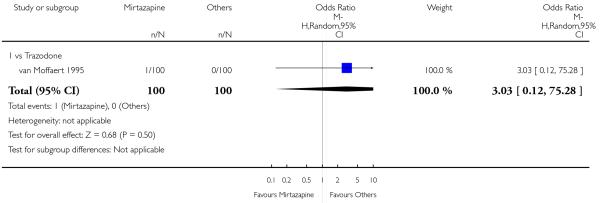 |
Analysis 4.20. Comparison 4 Mirtazapine versus heterocyclic antidepressants, Outcome 20 Suicide attempt.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 4 Mirtazapine versus heterocyclic antidepressants
Outcome: 20 Suicide attempt
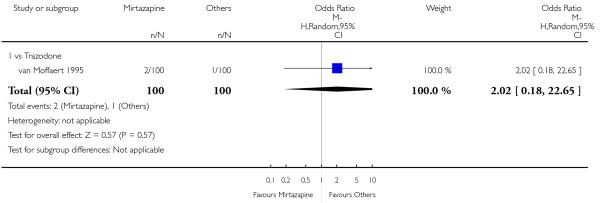 |
Analysis 5.1. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 1 Primary outcome (response) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 5 Mirtazapine versus newer antidepressants
Outcome: 1 Primary outcome (response) at 2 weeks
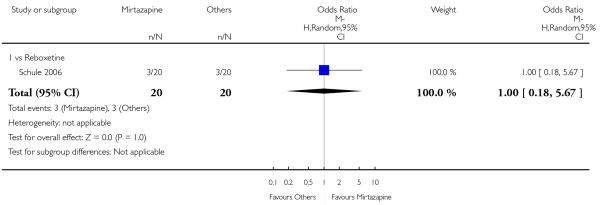 |
Analysis 5.2. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 2 Primary outcome (response) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 5 Mirtazapine versus newer antidepressants
Outcome: 2 Primary outcome (response) at end of the acute-phase treatment
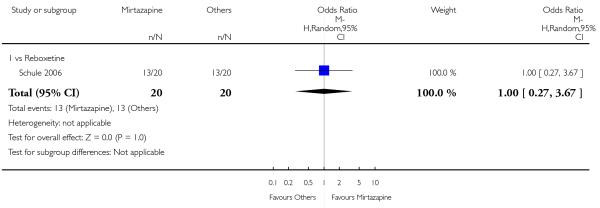 |
Analysis 5.3. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 3 Secondary outcome (remission) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 5 Mirtazapine versus newer antidepressants
Outcome: 3 Secondary outcome (remission) at 2 weeks
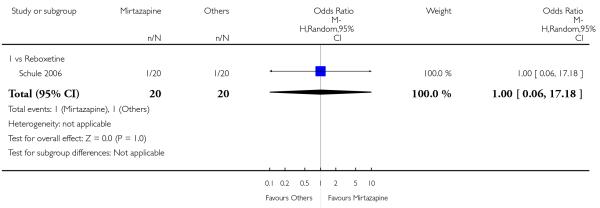 |
Analysis 5.4. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 4 Secondary outcome (remission) at end of the acute-phase treatment.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 5 Mirtazapine versus newer antidepressants
Outcome: 4 Secondary outcome (remission) at end of the acute-phase treatment
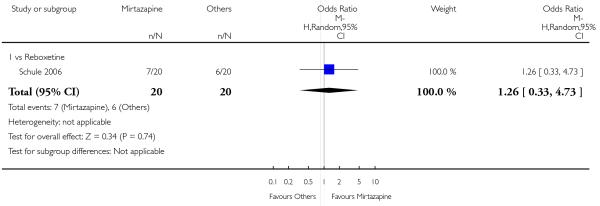 |
Analysis 5.5. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 5 Secondary outcome (depression severity) at 2 weeks.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 5 Mirtazapine versus newer antidepressants
Outcome: 5 Secondary outcome (depression severity) at 2 weeks
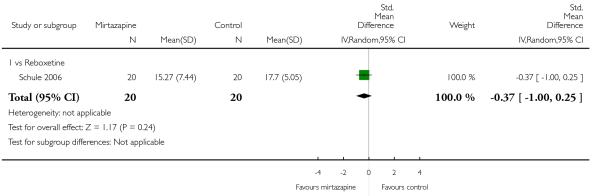 |
Analysis 5.6. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 6 Secondary outcome (withdrawal due to any reason).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 5 Mirtazapine versus newer antidepressants
Outcome: 6 Secondary outcome (withdrawal due to any reason)
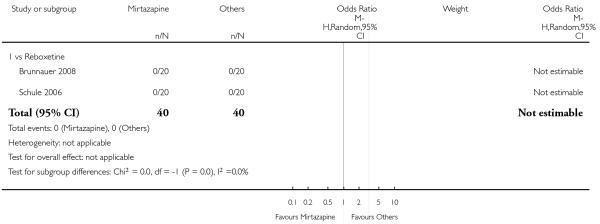 |
Analysis 5.7. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 7 Secondary outcome (withdrawal due to adverse events).
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 5 Mirtazapine versus newer antidepressants
Outcome: 7 Secondary outcome (withdrawal due to adverse events)
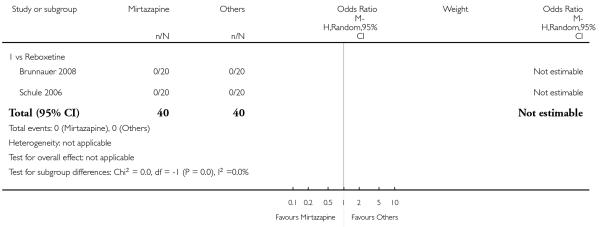 |
Analysis 5.8. Comparison 5 Mirtazapine versus newer antidepressants, Outcome 8 Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment.
Secondary outcome (SKEWED DATA: depression severity) at end of the acute-phase treatment
| Study | Comparator drug | Measurement | Mirtazapine: mean | SD | N | Comparator: mean | SD | N | note |
|---|---|---|---|---|---|---|---|---|---|
| Schule 2006 | Reboxetine | 21-item HAM-D | 10.87 | 6.91 | 20 | 11.17 | 6.17 | 20 |
Analysis 6.1. Comparison 6 Funnel plot analysis: primary outcome (response) at end of the acute-phase treatment, Outcome 1 vs all compounds.
Review: Mirtazapine versus other antidepressive agents for depression
Comparison: 6 Funnel plot analysis: primary outcome (response) at end of the acute-phase treatment
Outcome: 1 vs all compounds
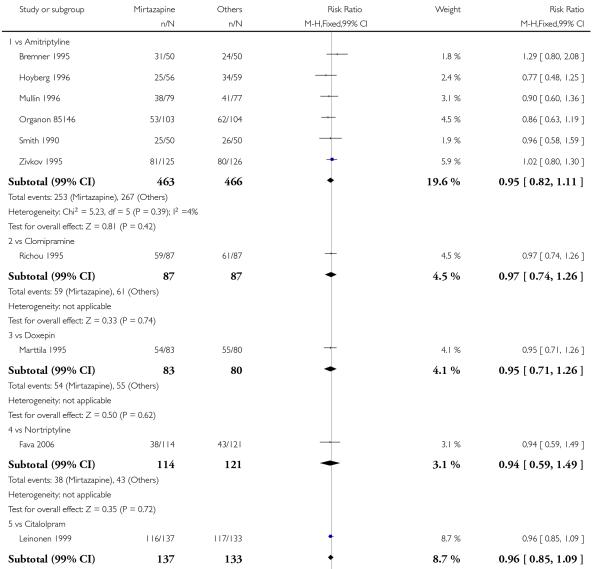 |
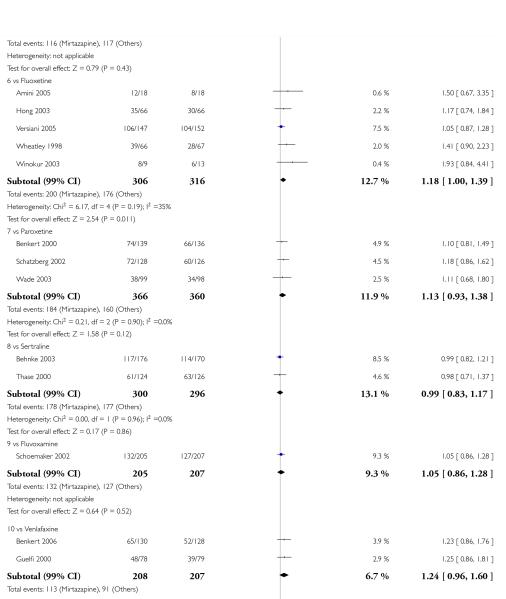 |
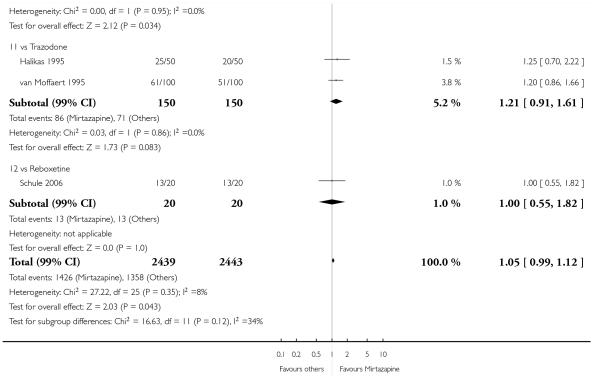 |
HISTORY
Protocol first published: Issue 2, 2007
Review first published: Issue 12, 2011
DIFFERENCES BETWEEN PROTOCOL AND REVIEW
The definition of the early response rates has been amended from that in the published protocol (“Early response rates: between 1 and 4 weeks, the time point closest to 2 weeks will be given preference”), because, after starting the review process, we recognised that all trials reported outcomes at 2 weeks when trials gave information about early response rate.
The subgroup analyses have been amended from the published protocol in which there are five subgroup analyses (mirtazapine dosing, comparator dosing, depression severity, treatment settings, elderly participants). The sensitivity analyses have been amended from the published protocol in which there are six sensitivity analyses (excluding trials with unclear concealment of random allocation or unclear double blinding, excluding trials whose dropout rate is greater than 20%, performing the worst case and best case scenario ITT, excluding trials for which the response rates had to be calculated based on the imputation method or borrowed from other trials, examination of ‘wish bias’ by comparing mirtazapine as investigational drug versus mirtazapine as comparator, excluding studies funded by the pharmaceutical company marketing mirtazapine).
These planned but not conducted analyses will be done in future updates of the review if adequate numbers of studies are available for the analyses.
Footnotes
DECLARATIONS OF INTEREST
NW has received research grants from the Japanese Ministry of Education, Science, Sports and Culture; and from the Japanese Ministry of the Health, Labour and Welfare. He has also received speaking fees and research funds from Asahi Kasei, Dai-Nippon Sumitomo, Eli Lilly, GlaxoSmithKline, Janssen, Otsuka, Pfizer and Schering-Plough.
TAF has received research funds and speaking fees from Astellas, Dai-Nippon Sumitomo, Eli Lilly, GlaxoSmithKline, Janssen, Meiji, Otsuka, Pfizer, Schering-Plough and Yoshitomi. He was on a research advisory board for Meiji and Mochida, and is currently on a research advisory board for Sekisui Chemicals and the Takeda Science Foundation. The Japanese Ministry of Education, Science, and Technology and the Japanese Ministry of Health Labor and Welfare have also funded his research.
IMO, AN, AC, CB, and RC have nothing to be declared.
This review is one of a number of separate reviews examining head-to-head comparisons as part of the multiple Meta-Analyses of New Generation Antidepressants (MANGA) Study. These individual reviews were combined in a multiple-treatments meta-analysis and published elsewhere (Cipriani 2009a).
References to studies included in this review
- Amini 2005 {published data only} .*; Amini H, Aghayan S, Jalili SA, Akhondzadeh S, Yahyazadeh O, Pakravan-Nejad M. Comparison of mirtazapine and fluoxetine in the treatment of major depressive disorder: a double-blind, randomized trial. Journal of Clinical Pharmacy and Therapeutics. 2005;30(2):133–8. doi: 10.1111/j.1365-2710.2004.00585.x. [DOI] [PubMed] [Google Scholar]
- Behnke 2003 {published data only} .Baker R, Schutte AJ. NR326 More rapid onset of sleep-improving effects with mirtazapine FDT versus sertraline; 158th Annual Meeting of the American Psychiatric Association; Atlanta, GA. 2005 May 21-26.2005. [Google Scholar]
- *; Behnke K, Sogaard J, Martin S, Bauml J, Ravindran AV, Agren H, et al. Mirtazapine orally disintegrating tablet versus sertraline: a prospective onset of action study. Journal of Clinical Psychopharmacology. 2003;23(4):358–64. doi: 10.1097/01.jcp.0000085408.08426.05. Erratum appears in Journal of Clinical Psychopharmacology 2003;23 (6):682. [DOI] [PubMed] [Google Scholar]
- Benkert 2000 {published data only} .*; Benkert O, Szegedi A, Kohnen R. Mirtazapine compared with paroxetine in major depression. Journal of Clinical Psychiatry. 2000;61(9):656–63. doi: 10.4088/jcp.v61n0911. [DOI] [PubMed] [Google Scholar]
- Benkert 2006 {published data only} .*; Benkert O, Szegedi A, Philipp M, Kohnen R, Heinrich C, Heukels A, et al. Mirtazapine orally disintegrating tablets versus venlafaxine extended release. Journal of Clinical Psychopharmacology. 2006;26(1):75–8. doi: 10.1097/01.jcp.0000194622.99986.d6. [DOI] [PubMed] [Google Scholar]
- Bremner 1995 {published data only} .*; Bremner JD. A double-blind comparison of Org 3770, amitriptyline, and placebo in major depression. Journal of Clinical Psychiatry. 1995;56(11):519–25. [PubMed] [Google Scholar]
- Brunnauer 2008 {published data only} .*; Brunnauer A, Laux G, David I, Fric M, Hermisson I, Moller HJ. The impact of reboxetine and mirtazapine on driving simulator performance and psychomotor function in depressed patients. Journal of Clinical Psychiatry. 2008;69(12):1880–6. doi: 10.4088/jcp.v69n1205. [DOI] [PubMed] [Google Scholar]
- Debonnel 2000a {published data only} .Debonnel G, Gobbi G, Turcotte J, Boucher N, Hebert C, de Montigny C, Blier P. Effects of mirtazapine, paroxetine and their combination: a double-blind study in major depression. European Neuropsychopharmacology. 2000;10(Suppl 3):S252. doi: 10.1016/j.euroneuro.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Fava 2006 {published data only} .*; Fava M, Rush AJ, Wisniewski SR, Nierenberg AA, Alpert JE, McGrath PJ, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. American Journal of Psychiatry. 2006;163(7):1161–72. doi: 10.1176/ajp.2006.163.7.1161. [DOI] [PubMed] [Google Scholar]
- Guelfi 2000 {published data only} .*; Guelfi JD, Ansseau M, Timmerman L, Korsgaard S. Mirtazapine versus venlafaxine in hospitalized severely depressed patients with melancholic features. Journal of Clinical Psychopharmacology. 2001;21(4):425–31. doi: 10.1097/00004714-200108000-00010. [DOI] [PubMed] [Google Scholar]
- Halikas 1995 {published data only} .*; Halikas JA. Org 3770 (mirtazapine) versus trazodone: A placebo controlled trial in depressed elderly patients. Human Psychopharmacology. 1995;10(Suppl):125–33. [Google Scholar]
- Hong 2003 {published data only} .*; Hong CJ, Hu WH, Chen CC, Hsiao CC, Tsai SJ, Ruwe FJL. A double-blind, randomized, group-comparative study of the tolerability and efficacy of 6 weeks’ treatment with mirtazapine or fluoxetine in depressed Chinese patients. Journal of Clinical Psychiatry. 2003;64(8):921–6. doi: 10.4088/jcp.v64n0810. [DOI] [PubMed] [Google Scholar]
- Hoyberg 1996 {published data only} .*; Hoyberg OJ, Maragakis B, Mullin J, Norum D, Stordall E, Ekdahl P, et al. A double-blind multicentre comparison of mirtazapine and amitriptyline in elderly depressed patients. Acta Psychiatrica Scandinavica. 1996;93(3):184–90. doi: 10.1111/j.1600-0447.1996.tb10629.x. [DOI] [PubMed] [Google Scholar]
- Leinonen 1999 {published data only} .Agren H, Skarstein J, Behke K, Schutte A-J, Leinonen E. Efficacy and tolerability of mirtazapine versus citalopram in major depression: a double-blind, randomized study; 152nd Annual Meeting of the American Psychiatric Association; Washington DC, USA. 1999.p. NR476. [Google Scholar]
- *; Leinonen E, Skarstein J, Behnke K, Agren H, Helsdingen JT. Efficacy and tolerability of mirtazapine versus citalopram: a double-blind, randomized study in patients with major depressive disorder. International Clinical Psychopharmacology. 1999;14(6):228. doi: 10.1097/00004850-199911000-00002. [DOI] [PubMed] [Google Scholar]
- Marttila 1995 {published data only} .*; Marttila M, Jaaskelainen J, Jarvi R, Romanov M, Miettinen E, Sorri P, et al. A double-blind study comparing the efficacy and tolerability of mirtazapine and doxepin in patients with major depression. European Neuropsychopharmacology. 1995;5(4):441–6. doi: 10.1016/0924-977x(95)00016-i. [DOI] [PubMed] [Google Scholar]
- Mullin 1996 {published data only} .*; Mullin J, Lodge A, Bennie E, McCreadie R, Bhatt GS, Fenton G. A multicentre, double-blind, amitriptyline-controIled study of mirtazapine in patients with major depression. Journal of Psychopharmacology. 1996;10(3):235–40. doi: 10.1177/026988119601000310. [DOI] [PubMed] [Google Scholar]
- Roland A, Margakis P, Mullin J, Haug JO, Stordal E, Ekdal P, et al. Double-blind, multicentre comparison of remergon and amitriptyline in elderly depressed patients. Clinical Neuropharmacology. 1992;15(1 Pt B):182. [Google Scholar]
- Organon 85146 {unpublished data only} .Zivkov M, Roes KCB, Pols AG. Efficacy of Org 3770 (mirtazapine) vs amitriptyline in patients with major depressive disorder: a meta-analysis. Human Psychopharmacology. 1995;10(suppl 2):S135–S145. [Google Scholar]
- Richou 1995 {published data only} .*; Richou H, Ruimy P, Charbaut J, Delisle JP, Brunner H, Patris M, et al. A multicentre, double-blind, clomipramine-controlled efficacy and safety study of Org 3770. Human Psychopharmacology. 1995;10(4):263–71. [Google Scholar]
- Ruimy P, Delisle J, Richou H, Charbaut J, Patris M, Brunner H, De Jongh GD. Remergon efficacy in major depressive episode: A randomized, double-blind, clomipramine-controlled study in 174 French hospitalized patients. Clinical Neuropharmacology. 1992;15(suppl 1 pt B):237. [Google Scholar]
- Schatzberg 2002 {published data only} .Murphy GM. Pharmacogenetics of mirtazapine and paroxetine in the treatment of geriatric major depression; 39th Annual Meeting of the American College of Neuropsychopharmacology; San Juan; Puerto Rico. 2000; Dec 10-14.2000. p. 235. [Google Scholar]
- Murphy GM, Kremer C, Rodrigues H, Schatzberg AF. The apolipoprotein E epsilon4 allele and antidepressant efficacy in cognitively intact elderly depressed patients. Biological Psychiatry. 2003;54(7):665–73. doi: 10.1016/s0006-3223(03)00174-4. [DOI] [PubMed] [Google Scholar]
- Murphy GM., Jr. Pharmacokinetic and pharmacodynamic genetic predictors of antidepressant tolerability and efficacy; 45th Annual NCDEU (New Clinical Drug Evaluation Unit) Meeting; Boca Raton, FL. 2005 June 6 - 9.2005. p. 50. [Google Scholar]
- Murphy GM, Jr, Hollander SB, Rodrigues HE, Kremer C, Schatzberg AF. Effects of the serotonin transporter gene promoter polymorphism on mirtazapine and paroxetine efficacy and adverse events in geriatric major depression. Archives of General Psychiatry. 2004;61(11):1163–9. doi: 10.1001/archpsyc.61.11.1163. [DOI] [PubMed] [Google Scholar]
- Murphy GM, Jr, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. American Journal of Psychiatry. 2003;160(10):1830–5. doi: 10.1176/appi.ajp.160.10.1830. [DOI] [PubMed] [Google Scholar]
- O’Hara R, Schatzberg AF, Murphy GM. The impact on cognition of pharmacological treatment for late-life depression; 155th Annual Meeting of the American Psychiatric Association; Philadelphia, PA. 2002 May 18-23; 2002. No. 15B. [Google Scholar]
- O’Hara R, Hollander SB, Lapp W, Boyle L, Rodrigues H, Kraemer HC, et al. ApoE4 Allele influences antidepressants’ cognitive effects in the elderly; 157th Annual Meeting of the American Psychiatric Association; New York, NY. 2004 May 1-6.2004. p. NR876. [Google Scholar]
- Sarginson JE, Lazzeroni LC, Ryan HS, Schatzberg AF, Murphy GM., Jr. FKBP5 polymorphisms and antidepressant response in geriatric depression. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics: the Official Publication of the International Society of Psychiatric Genetics. 2010;153B(2):554–60. doi: 10.1002/ajmg.b.31019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatzberg AF, Kremer C, Rodrigues H. Mirtazapine and paroxetine in elderly depressed patients; 52nd Institute on Psychiatric Services; Philadelphia, PA. 2000 October 25-29th.2000. [Google Scholar]
- Schatzberg AF, Kremer C, Rodrigues H. Mirtazapine versus paroxetine in elderly depressed patients; 14th Annual Meeting of the American Association for Geriatric Psychiatry; San Francisco, Ca, USA. 2001 23rd-26th February; 2001. [PubMed] [Google Scholar]
- Schatzberg AF, Kremer C, Rodrigues H. Mirtazapine versus paroxetine in elderly depressed patients; 154th Annual Meeting of the American Psychiatric Association; Orleans LA. 2001 May 5-10; 2001. p. NR478. [PubMed] [Google Scholar]
- Schatzberg AF, Kremer C, Rodrigues HE. Mirtazapine versus paroxetine in elderly depressed patients; 39th Annual Meeting of the American College of Neuropsychopharmacology; San Juan; Puerto Rico. 2000; Dec 10-14.2000. p. 97. [Google Scholar]
- *; Schatzberg AF, Kremer C, Rodrigues HE, Murphy GM., Jr The Mirtazapine vs. Paroxetine Study Group. Double-blind, randomized comparison of mirtazapine and paroxetine in elderly depressed patients. American Journal of Geriatric Psychiatry. 2002;10(5):541–50. [PubMed] [Google Scholar]
- Schoemaker 2002 {unpublished data only} .Schoemaker J, Gailledreau J, Hoyberg OJ. First, randomized, double-blind comparison of mirtazapine (15-45 mg) and fluvoxamine (50-150 mg) in the treatment of depression. International Journal of Neuropsychopharmacology. 2002;5(suppl 1):140. [Google Scholar]
- *; Schoemaker JH, The Mirtazapine Bridging Study Group . Double-blind comparison of mirtazapine versus fluvoxamine in patients with major depressive disorder (DSM-IV) In preparation. [Google Scholar]
- Schule 2006 {published data only} .*; Schule C, Baghai TC, Eser D, Zwanzger P, Jordan M, Buechs R, et al. Time course of hypothalamic-pituitary-adrenocortical axis activity during treatment with reboxetine and mirtazapine in depressed patients. Psychopharmacology. 2006;186(4):601–11. doi: 10.1007/s00213-006-0382-7. [DOI] [PubMed] [Google Scholar]
- Smith 1990 {published data only} .*; Smith WT, Glaudin V, Panagides J, Gilvary E. Mirtazapine vs amitriptyline vs placebo in the treatment of major depressive disorder. Psychopharmacology Bulletin. 1990;26(2):191–6. [PubMed] [Google Scholar]
- Thase 2000 {published data only} .Thase ME, Kremer C, Rodrigues H. Mirtazapine versus sertraline after SSRI non-response; 39th Annual Meeting of the American College of Neuropsychopharmacology; San Juan; Puerto Rico. 2000; Dec 10-14.2000. p. 260. [Google Scholar]
- Thase ME, Kremer C, Rodrigues H. Mirtazapine versus sertraline after SSRI nonresponse; 52nd Institute on Psychiatric Services; Philadelphia, PA. 2000 October 25-29th.2000. [Google Scholar]
- *; Thase ME, Simmons JH, Howland RH, Fava M. Doubleblind, randomized comparison of mirtazapine and sertraline in depressed patients who had not responded to SSRI treatment. In preparation. [Google Scholar]
- Turan 2000a {published data only} .Turan M, Askin R, Telcioglu M, Cilli AS. Mirtazapine versus amitriptyline in treatment of major depressive disorder. European Neuropsychopharmacology. 2000;10(Suppl 3):S228. [Google Scholar]
- van Moffaert 1995 {published data only} .de Wilde J, Dierick M, Van Moffaert M, Vereecken A, Mendlewicz J, Evrard J, et al. Remergon efficacy in major depressive episode: A randomized, double-blind, trazodon-controlled study in 200 Belgian patients. Clinical Neuropharmacology. 1992;15(1 Pt B):237. [Google Scholar]
- Van Moffaert M. Efficacy and safety of mirtazapine vs trazodone in hospitalised depressed patients; 8th ECNP (European College of Neuropsychopharmacology) Congress; Venice, Italy. 1995. [Google Scholar]
- *; van Moffaert M, de Wilde J, Vereecken A, Dierick M, Evrard JL, Wilmotte J, et al. Mirtazapine is more effective than trazodone: a double-blind controlled study in hospitalized patients with major depression. International Clinical Psychopharmacology. 1995;10(1):3–9. [PubMed] [Google Scholar]
- Versiani 2005 {published data only} .*; Versiani M, Moreno R, Ramakers-van Moorsel CJ, Schutte AJ. Comparison of the effects of mirtazapine and fluoxetine in severely depressed patients. CNS Drugs. 2005;19(2):137–46. doi: 10.2165/00023210-200519020-00004. [DOI] [PubMed] [Google Scholar]
- Wade 2003 {published data only} .*; Wade A, Crawford GM, Angus M, Wilson R, Hamilton L. A randomized, double-blind, 24-week study comparing the efficacy and tolerability of mirtazapine and paroxetine in depressed patients in primary care. International Clinical Psychopharmacology. 2003;18(3):133–41. doi: 10.1097/01.yic.0000068045.82050.00. [DOI] [PubMed] [Google Scholar]
- Wheatley 1998 {published data only} .*; Wheatley DP, van Moffaert M, Timmerman L, Kremer CM. Mirtazapine: efficacy and tolerability in comparison with fluoxetine in patients with moderate to severe major depressive disorder. Journal of Clinical Psychiatry. 1998;59(6):306–12. [PubMed] [Google Scholar]
- Winokur 2003 {published data only} .*; Winokur A, DeMartinis NA, 3rd, McNally DP, Gary EM, Cormier JL, Gary KA. Comparative effects of mirtazapine and fluoxetine on sleep physiology measures in patients with major depression and insomnia. Journal of Clinical Psychiatry. 2003;64(10):1224–9. doi: 10.4088/jcp.v64n1013. [DOI] [PubMed] [Google Scholar]
- Zivkov 1995 {published data only} .*; Zivkov M, De Jongh GD. Org 3770 versus amitriptyline: a 6-week randomized double-blind multicentre trial in hospitalized depressed patients. Human Psychopharmacology. 1995;10(3):173–80. [Google Scholar]
References to studies excluded from this review
- Blier 2004 {unpublished data only} .*; Blier P, Ward HE, Jacobs W, Herbert C, O’Hara SA, Pigott TA. Combining two antidepressants from treatment start: a preliminary analysis; New research program and abstracts, American Psychiatric Association 157th Annual Meeting; 2004.p. 157. [Google Scholar]
- Bruijin 1996 {published data only} .Bruijn JA, Moleman P, Mulder PG, van den Broek WW. Comparison of 2 treatment strategies for depressed inpatients: Imipramine and lithium addition or mirtazapine and lithium addition. Journal of Clinical Psychiatry. 1998;59(12):657–63. doi: 10.4088/jcp.v59n1203. [DOI] [PubMed] [Google Scholar]
- Bruijn JA, Moleman P, Mulder PG, van den Broek WW. Depressed in-patients respond differently to imipramine and mirtazapine. Pharmacopsychiatry. 1999;32(3):87–92. doi: 10.1055/s-2007-979200. [DOI] [PubMed] [Google Scholar]
- Bruijn JA, Moleman P, Mulder PG, Van den Broek WW. Treatment of mood-congruent psychotic depression with imipramine. Journal of Affective Disorders. 2001;66:165–74. doi: 10.1016/s0165-0327(00)00296-2. [DOI] [PubMed] [Google Scholar]
- *; Bruijn JA, Moleman P, Mulder PGH, van den Broek WW, van Hulst AM, van der Mast RC, et al. A double-blind, fixed blood-level study comparing mirtazapine with imipramine in depressed in-patients. Pharmacology. 1996;127:231–7. [PubMed] [Google Scholar]
- Bruijn JA, Moleman P, van den Broek WW, Mulder PG. Trait anxiety and the effect of a single high dose of diazepam in unipolar depression. Journal of Psychiatric Research. 2001;35(6):331–7. doi: 10.1016/s0022-3956(01)00035-8. [DOI] [PubMed] [Google Scholar]
- Kasper 1997a {published data only} .*; Kasper S. Efficacy of antidepressants in the treatment of severe depression: the place of mirtazapine. Journal of Clinical Psychopharmacology. 1997;17(Suppl 1):19S–28S. doi: 10.1097/00004714-199704001-00003. [DOI] [PubMed] [Google Scholar]
- Kasper 1997b {published data only} .*; Kasper S, Zivkov M, Roses KCB, Pols AG. Pharmacological treatment of severely depressed patients: a meta-analysis comparing efficacy of mirtazapine and amitriptyline. European Neuropsychopharmacology. 1997;7:115–24. doi: 10.1016/s0924-977x(96)00394-x. [DOI] [PubMed] [Google Scholar]
- Kremer 1995 {unpublished data only} .Kremer CME, Helsdingen JTH, Schutte JA, Vester E. Tolerability of mirtazapine vs SSRIs in short term treatment of major depression. European Neuropsychopharmacology. 1999;9(5):S229. [Google Scholar]
- Peyron 1996 {published data only} .*; Peyron E. Efficacy of mirtazapine vs clomipramine in severely depressed, hospitalized patients. European Neuropsychopharmacology. 1996;6(Suppl 3):46–7. [Google Scholar]
- Tulen 1996 {published data only} .*; Tulen JHM, Bruijn JA, de Man KJ, Pepplinkhuizen L, van den Meiracker AH, Man AJ, et al. Cardiovascular variability in major depressive disorder and effects of imipramine or mirtazapine. Journal of Clinical Psychopharmacology. 1996;16(2):135–45. doi: 10.1097/00004714-199604000-00006. [DOI] [PubMed] [Google Scholar]
- Zourkova 2001 {published data only} .*; Zourkova A. Effect of mirtazapine and paroxetine on residual symptoms of depressive disorders and their effect on P450 CYP 2D6 activity. Homeostasis in Health and Disease. 2001;41(6):242–9. [Google Scholar]
References to studies awaiting assessment
- Blier 2009 {published data only} .Blier P, Gobbi G, Turcotte JE, de Montigny C, Boucher N, Hebert C, et al. Mirtazapine and paroxetine in major depression: A comparison of monotherapy versus their combination from treatment initiation. European Neuropsychopharmacology. 2009;19(7):457–65. doi: 10.1016/j.euroneuro.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Catterson 1996a {unpublished data only} .Catterson ML, Preskorn SH. Double-blind crossover study of mirtazapine, amitriptyline and placebo in patient with major depression; 149th Annual Meeting of the American Psychiatric Association; New York, NY. 1996 May 4-9.1996. p. 0157. [Google Scholar]
- Chang 2006 {published data only} .Chang FW, Ma YP, Yan F, Wang CH. Comparative study of mirtazapine vs clomipramine in treatment of dysthymic disorder. Chinese Journal of New Drugs and Clinical Remedies. 2006;25(1):55–7. [Google Scholar]
- Chen 2002 {published data only} .Chen X, Tan L, Zhao J, Li L, Chen Y. Randomized controlled clinical trials of remeron and fluoxetine in depressive patients. Chinese Journal of Clinical Psychology. 2002;10(2):100–2. [Google Scholar]
- Chen 2003 {published data only} .Chen G, Gu G, Li C, et al. A study of mirtazapine and venlafaxine in the treatment of depression. Journal of Clinical Psychological Medicine. 2003;13(2):77–9. [Google Scholar]
- Chen 2004 {published data only} .Chen Z, Zhang J, Li Z, et al. Efficacy of mirtazapine and sertraline in the treatment of depression with anxiety symptoms. Chinese Mental Health Journal. 2004;18(5):358–9. [Google Scholar]
- Chen 2004a {published data only} .Chen Z, Zhang J, Li Z, Zhang H, Mu X. Mirtazapine vs. sertraline in treating depressive disorder associated with anxiety. Chinese Journal of New Drugs and Clinical Remedies. 2004;23(2):90–2. [Google Scholar]
- Chen 2005 {published data only} .Chen Q. The control study of mirtazapine versus fluoxetine in the treatment of depression. Chinese Journal of Health Psychology. 2005;13(1):25–6. [Google Scholar]
- Fang 2010 {published data only} .Fang Y, Yuan C, Xu Y, Chen J, Wu Z, Cao L, et al. Comparisons of the Efficacy and Tolerability of Extended-Release Venlafaxine, Mirtazapine, and Paroxetine in Treatment-Resistant Depression: A Double-Blind, Randomized Pilot Study in a Chinese Population. Journal of Clinical Psychopharmacology. 2010;30(4):357–64. doi: 10.1097/JCP.0b013e3181e7784f. [DOI] [PubMed] [Google Scholar]
- Gong 2005 {published data only} .Gong C, Xu H, Xiang D, Zhou X. Comparative study on depression treated by citalopram or mirtazapine. Journal of Clinical Psychological Medicine. 2005;15(3):154–5. [Google Scholar]
- Guo 2005 {published data only} .Guo HR, Ren YM, Li SY. Controlled study of mirtazapine and mianserine in the treatment of senile depression. Chinese Mental Health Journal. 2005;19(7):489–91. [Google Scholar]
- Guo 2005a {published data only} .Guo P, Guo H, Jia J. Comparison of mirtazapine versus clomipramine in maintenance treatment of depression. Journal of Clinical Psychological Medicine. 2005;15(3):152–3. [Google Scholar]
- Guo 2006 {published data only} .Guo P. Clinical evaluation of mirtazapine and clomipramine for the treatment of depressive disorder associated with anxiety. Evaluation and Analysis of Drug-Use in Hospitals of China. 2006;6(2):79–81. [Google Scholar]
- Guo 2006a {published data only} .Guo JH, Cao CA, Liao CP. Effect of Mirtazapine and Clomipramine on the Life Quality of Patients. Chinese Mental Health Journal. 2006;20(6):413–5. [Google Scholar]
- Hang 2005 {published data only} .Hang R, Xu P, Wang R. A comparative study of mirtazapine and paroxetine in the treatment of depression. Shandong Archives of Psychiatry. 2005;18(4):225–6. [Google Scholar]
- Hu 2004 {published data only} .Hu S, Xu Y, Wei N, et al. A comparative study of treatment of depression: Mirtazapine vs. fluoxetine. Zhejiang Medical Journal. 2004;26(12):885–7. [Google Scholar]
- Huang 2007 {published data only} .Huang X-J, Gong M-E, Tang Z-Y. Comparison of mirtazapine and paroxetine in patients with first-episode climacteric depression. Chinese Mental Health Journal. 2007;21(6):428–30. [Google Scholar]
- Kang 2009 {published data only} .Kang E-H, Lee I-S, Chung S-K, Lee S-Y, Kim E-J, Hong J-P, et al. Mirtazapine versus venlafaxine for the treatment of somatic symptoms associated with major depressive disorder: A randomized, open-labelled trial. Psychiatry Research. 2009;169(2):118–23. doi: 10.1016/j.psychres.2008.06.021. [DOI] [PubMed] [Google Scholar]
- Kim 2011 {published data only} .Kim JE, Yoon SJ, Kim J, Jung JY, Jeong HS, Cho HB, et al. Efficacy and tolerability of mirtazapine in treating major depressive disorder with anxiety symptoms: An 8-week open-label randomised paroxetine-controlled trial. International Journal of Clinical Practice. 2011;65(3):323–9. doi: 10.1111/j.1742-1241.2010.02624.x. [DOI] [PubMed] [Google Scholar]
- Li 2005 {published data only} .Li Jing, Meng Hua-Qing, Deng Wei. Mirtazapine and fluoxetine in the treatment of cardiovascular neurosis with depression. Chinese Mental Health Journal. 2005;19(9):637–9. [Google Scholar]
- Liang 2006 {published data only} .Liang K. Comparative study of mirtazapine and sertraline in treatment of elderly depressive patient. Journal of Heze Medical College. 2006;18(2):5–7. [Google Scholar]
- Lin 2005 {published data only} .Lin Z, Chen Y. A study of mirtazapine and paroxetine in the treatment of anxiety. Sichuan Medical Journal. 2005;26(11):1229–30. [Google Scholar]
- Liu 2004 {published data only} .Liu P. A study of mirtazapine and venlafaxine in the treatment of depression. Health Psychology Journal. 2004;12(1):15–6. [Google Scholar]
- Ma 2003 {published data only} .Ma ZW, Li MX, Yang FS, et al. Comparative study on the efficacy and safety of mirtazapine and amitriptyline in treatment of depression. Chinese Journal of New Drugs. 2003;12(10):858–60. [Google Scholar]
- Ning 2004 {published data only} .Ning J, Lu D. Observation of the clinical efficacy and safety of mirtazapine on elder depression treatment. Clnical Pharmaceuticals. 2004;13(5):63–4. [Google Scholar]
- Niu 2004 {published data only} .Niu F, Shen X, Sun S. Comparison of efficacy of mirtazapine and fluoxetine in treatment of depression with generalized anxiety disorder. Chinese Journal of New Drugs and Clinical Remedies. 2004;23(12):853–5. [Google Scholar]
- Paslakis 2010 {published data only} .Paslakis G, Luppa P, Gilles M, Kopf D, Hamann Weber B, Lederbogen F, et al. Venlafaxine and mirtazapine treatment lowers serum concentrations of dehydroepiandrosterone-sulfate in depressed patients remitting during the course of treatment. Journal of Psychiatric Research. 2010;44(8):556–60. doi: 10.1016/j.jpsychires.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Peng 2004 {published data only} .Peng J, Xu Y, Piao S, et al. A comparative study of mirtazapine and paroxetine for depressed patients. Medical Journal of Chinese People Health. 2004;16(11):664–5. [Google Scholar]
- Ren 2004 {published data only} .Ren H, Guo Q, Cheng M. A study of mirtazapine and paroxetine in the treatment of depression. Journal of Clinical Psychological Medicine. 2004;14(2):88–9. [Google Scholar]
- Sarginson 2010 {published data only} .Sarginson JE, Lazzeroni LC, Ryan HS, Ershoff BD, Schatzberg AF, Murphy GM., Jr. ABCB1 (MDR1) polymorphisms and antidepressant response in geriatric depression. Pharmacogenetics and Genomics. 2010;20(8):467–75. doi: 10.1097/FPC.0b013e32833b593a. [DOI] [PubMed] [Google Scholar]
- Scharnholz 2010 {published data only} .Scharnholz B, Weber-Hamann B, Lederbogen F, Schilling C, Gilles M, Onken V, et al. Antidepressant treatment with mirtazapine, but not venlafaxine, lowers cortisol concentrations in saliva: A randomised open trial. Psychiatry Research. 2010;177(1-2):109–2. doi: 10.1016/j.psychres.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Su 2005 {published data only} .Su H, Lei D, Yu H. Comparative study of mirtazapine and paroxetine in the treatment of depression. Modern Medicine and Health. 2005;21(12):1480–1. [Google Scholar]
- Tang 2005 {published data only} .Tang Z, Li M. Mirtazapine compared with paroxetine in depressed patients with anxiety symptoms. Chinese Mental Health Journal. 2005;19(9):643–5. [Google Scholar]
- Tao 2004 {published data only} .Tao M, Gao JF, Tang WX, et al. Clinical efficacy and compliance of mirtazapine in the treatment of depression. Chinese Mental Health Journal. 2004;18(5):360–2. [Google Scholar]
- Wang 2006 {published data only} .Wang A, Zhu W. Efficacy and safety of mirtazapine versus clomipramine in patients with depression and anxiety. Shanghai Archives of Psychiatry. 2006;18(1):24–6. [Google Scholar]
- Weng 2001 {published data only} .Weng S, Li H, Zhao J, Zhang H, Li T, Shu L, et al. Mirtazapine vs. fluoxetine in treatment of major depressive disorder: a multicenter clinical trial. Chinese Journal of New Drugs and Clinical Remedies. 2001;20(5):329–33. [Google Scholar]
- Wu 2006 {published data only} .Wu Y. Controlled study of mirtazapine vs. venlafaxine in the treatment of depression. Journal of Clinical Psychosomatic Diseases. 2006;12(2):111–2. [Google Scholar]
- Xie 2002 {published data only} .Xie K. Venlafaxine vs buspirone in treating depression associated with anxiety. Health Psychology Journal. 2002;10(4):252–3. [Google Scholar]
- Xie 2004 {published data only} .Xie N, Yan Y, Di L, et al. A comparative study of mirtazapine and efexor XR for depressive patients. Medical Journal of Chinese People Health. 2004;16(2):75–6. [Google Scholar]
- Xie 2004a {published data only} .Xie N, Zhao H, Di L, et al. A comparative study of mirtazapine and fluoxetine for depressive patients. Sichuan Mental Health. 2004;17(1):32–4. [Google Scholar]
- Xie 2005 {published data only} .Xie K, Hai Y, Zhang D, et al. A comparative study of venlafaxine, maprotiline and buspirone in the treatment of depression. Chinese Journal of Health Psychology. 2005;13(1):71–3. [Google Scholar]
- Xiong 2003 {published data only} .Xiong P, Xuan X, Wang J. A comparative study of mirtazapine and imipramine for depressive patients. Shanghai Archives of Psychiatry. 2003;15(2):93–5. [Google Scholar]
- Yang 2005 {published data only} .Yang L, Yang K, Li Y. Clinical comparative study on mirtazapine and venlafaxine used in treating depression. Nervous Diseases and Mental Health. 2005;5(4):271–3. [Google Scholar]
- Yao 2006 {published data only} .Yao X. Study of the treatment of depression with mirtazapine, trazodone and maprotiline. Chinese Journal of Pharmacoepidemiology. 2006;15(4):202–3. [Google Scholar]
- Ye 2005 {published data only} .Ye C, Zhou F. Mirtazapine in the treatment of depression associated with anxiety. Herald of Medicine. 2005;24(10):893–5. [Google Scholar]
- Yu 2004 {published data only} .Yu G, Ding G, Li X. Mirtazapine and amitriptyline in the maintenance treatment of depressed patients: Comparison of efficacy and compliance. Chinese Mental Health Journal. 2004;18(5):356–7. [Google Scholar]
- Zhang 2003 {published data only} .Zhang J, Liu T, Zhao J, et al. The control study of mirtazapine versus fluoxetine in the treatment of depression. Shanghai Archives of Psychiatry. 2003;15(6):351–4. [Google Scholar]
- Zhang 2003a {published data only} .Zhang J, Liu T, Zhao J, Hao W, Xie G, Su L, et al. Control study of mirtazapine and fluoxetine in therapy of depression. Chinese Journal of Clinical Rehabilitation. 2003;7(30):4102–4. [Google Scholar]
- Zhang 2004 {published data only} .Zhang J, Wang Y, Li Z. A controlled study of mirtazapine and amitriptyline in depression. Journal of Clinical Psychosomatic Diseases. 2004;10(3):184–6. [Google Scholar]
- Zheng 2005 {published data only} .Zheng H. Control study on clinical effect of mirtazapine in treatment depression. Chinese Journal of Health Psychology. 2005;13(6):440–2. [Google Scholar]
- Zhu 2003 {published data only} .Zhu H, Yu J, Zheng H. A study of switching to mirtazapine for treatment-resistant depression. Shanghai Archives of Psychiatry. 2003;15(6):355–7. [Google Scholar]
- Zhu 2005 {published data only} .Zhu J, Jiang X, Zhou D, Zhang F. Comparative study of mirtazapine vs. fluoxetine in treatment of elderly depressive patients. Journal of Clinical Psychological Medicine. 2005;15(5):277–8. [Google Scholar]
- Zhu 2006 {published data only} .Zhu J. Comparative study of mirtazapine and fluoxetine in the treatment of senile depression. China Journal of Health Psychology. 2006;14(5):546–7. [Google Scholar]
- Zou 2006 {published data only} .Zou J, Jing Y, Song K, Hu Y. Comparative study on mirtazapine and paroxetine in the treatment of depression. Journal of Clinical Psychological Medicine. 2006;16(4):235–6. [Google Scholar]
Additional references
- Als-Nielsen 2003 .Als-Nielsen B, Chen W, Gluud C, Kjaergard LL. Association of funding and conclusions in randomized drug trials: a reflection of treatment effect or adverse events? JAMA. 2003;290(7):921–8. doi: 10.1001/jama.290.7.921. [DOI] [PubMed] [Google Scholar]
- Altman 1996 .Altman DG, Bland JM. Detecting skewness from summary information. BMJ. 1996;313(7066):1200. doi: 10.1136/bmj.313.7066.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson 2000 .Anderson IM, Nutt DJ, Deakin JF. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 1993 British Association for Psychopharmacology guidelines. Journal of Psychopharmacology. 2000;14(1):3–20. doi: 10.1177/026988110001400101. [DOI] [PubMed] [Google Scholar]
- Anttila 2001 .Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Reviews. 2001;7(3):249–64. doi: 10.1111/j.1527-3458.2001.tb00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APA 1980 .American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders (DSM-III) 3rd Edition American Psychiatric Association; Washington, DC: 1980. [Google Scholar]
- APA 1987 .American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders (DSM-III-R) 3rd Edition American Psychiatric Association; Washington, DC: 1987. [Google Scholar]
- APA 1994 .American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) 4th Edition American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- APA 2000 .American Psychiatric Association Practice guideline for the treatment of patients with major depressive disorder (revision) American Journal of Psychiatry. 2000;157(Suppl (4)):1–45. [PubMed] [Google Scholar]
- Arroll 2005 .Arroll B, Macgillivray S, Ogston S, Reid I, Sullivan F, Williams B, et al. Efficacy and tolerability of tricyclic antidepressants and SSRIs compared with placebo for treatment of depression in primary care: a meta-analysis. Annals of Family Medicine. 2005;3(5):449–56. doi: 10.1370/afm.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbui 2000 .Barbui C, Hotopf M, Freemantle N, Boynton J, Churchill R, Eccles MP, et al. Treatment discontinuation with selective serotonin reuptake inhibitors (SSRIs) versus tricyclic antidepressants (TCAs) Cochrane Database of Systematic Reviews. 2000;(4) doi: 10.1002/14651858.CD002791.pub2. DOI: 10.1002/14651858.CD002791. [DOI] [PubMed] [Google Scholar]
- Bhandari 2004 .Bhandari M, Busse JW, Jackowski D, Montori VM, Schunemann H, Sprague S, et al. Association between industry funding and statistically significant pro-industry findings in medical and surgical randomized trials. CMAJ. 2004;170(4):477–80. [PMC free article] [PubMed] [Google Scholar]
- Bucher 1997 .Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. Journal of Clinical Epidemiology. 1997;50(6):683–91. doi: 10.1016/s0895-4356(97)00049-8. [DOI] [PubMed] [Google Scholar]
- Buchkowsky 2004 .Buchkowsky SS, Jewesson PJ. Industry sponsorship and authorship of clinical trials over 20 years. Annals of Pharmacotherapy. 2004;38(4):579–85. doi: 10.1345/aph.1D267. [DOI] [PubMed] [Google Scholar]
- Chan 2004 .Chan AW, Hrobjartsson A, Haahr MT, Gozsche PC, Altman DG. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA. 2004;291:2457–65. doi: 10.1001/jama.291.20.2457. [DOI] [PubMed] [Google Scholar]
- Churchill 2009 .Churchill R, Caldwell D, McGuire H, Barbui C, Cipriani A, Furukawa TA. Reboxetine versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews. 2009;(2) DOI: 10.1002/14651858.CD007852. [Google Scholar]
- Cipriani 2005 .Cipriani A, Brambilla P, Furukawa T, Geddes J, Gregis M, Hotopf M, et al. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews. 2005;(5) doi: 10.1002/14651858.CD004185.pub2. DOI: 10.1002/14651858.CD004185.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani 2007a .Cipriani A, Furukawa TA, Veronese A, Watanabe N, Churchill R, McGuire H, et al. Paroxetine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews. 2007;(2) doi: 10.1002/14651858.CD006531. DOI: 10.1002/14651858.CD006531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani 2007b .Cipriani A, Signoretti A, Furukawa TA, Churchill R, Tomelleri S, Omori I, et al. Venlafaxine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews. 2007;(2) doi: 10.1002/14651858.CD006530. DOI: 10.1002/14651858.CD006530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani 2009 .Cipriani A, La Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, et al. Sertraline versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews. 2009;(2) doi: 10.1002/14651858.CD006117.pub2. DOI: 10.1002/14651858.CD006117.pub4. [DOI] [PubMed] [Google Scholar]
- Cipriani 2009a .Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):756–8. doi: 10.1016/S0140-6736(09)60046-5. [DOI] [PubMed] [Google Scholar]
- Cipriani 2009b .Cipriani A, Santilli C, Furukawa TA, Signoretti A, Nakagawa A, McGuire H, et al. Escitalopram versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews. 2009;(2) doi: 10.1002/14651858.CD006532.pub2. DOI: 10.1002/14651858.CD006532.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuna 2004 .Ciuna A, Andretta M, Corbari L, Levi D, Mirandola M, Sorio A, et al. Are we going to increase the use of antidepressants up to that of benzodiazepines? European Journal of Clinical Pharmacology. 2004;60(9):629–34. doi: 10.1007/s00228-004-0810-8. [DOI] [PubMed] [Google Scholar]
- Clayton 2002 .Clayton AH, Pradko JF, Croft HA, Montano CB, Leadbetter RA, Bolden-Watson C, et al. Prevalence of sexual dysfunction among newer antidepressants. Journal of Clinical Psychiatry. 2002;63(4):357–66. doi: 10.4088/jcp.v63n0414. [DOI] [PubMed] [Google Scholar]
- de Boer 1995 .de Boer T. The effects of mirtazapine on central noradrenergic and serotonergic neurotransmission. International clinical psychopharmacology. 1995;10(Suppl 4):19–23. doi: 10.1097/00004850-199512004-00004. [DOI] [PubMed] [Google Scholar]
- de Boer 1996 .de Boer T. The pharmacologic profile of mirtazapine. Journal of Clinical Psychiatry. 1996;57(Suppl 4):19–25. [PubMed] [Google Scholar]
- Ellis 2004 .Ellis P. Australian and New Zealand clinical practice guidelines for the treatment of depression. Australian and New Zealand Journal of Psychiatry. 2004;38(6):389–407. doi: 10.1080/j.1440-1614.2004.01377.x. [DOI] [PubMed] [Google Scholar]
- Fawcett 1998 .Fawcett J, Barkin RL. A meta-analysis of eight randomized, double-blind, controlled clinical trials of mirtazapine for the treatment of patients with major depression and symptoms of anxiety. Journal of Clinical Psychiatry. 1998;59(3):123–7. [PubMed] [Google Scholar]
- Furukawa 2002 .Furukawa TA, Guyatt GH, Griffith LE. Can we individualize the ‘number needed to treat’? An empirical study of summary effect measures in meta-analyses. International Journal of Epidemiology. 2002;31(1):72–6. doi: 10.1093/ije/31.1.72. [DOI] [PubMed] [Google Scholar]
- Furukawa 2005 .Furukawa TA, Cipriani A, Barbui C, Brambilla P, Watanabe N. Imputing response rates from means and standard deviations in meta-analysis. International Clinical Psychopharmacology. 2005;20(1):49–52. doi: 10.1097/00004850-200501000-00010. [DOI] [PubMed] [Google Scholar]
- Furukawa 2006 .Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta-analyses can provide accurate results. Journal of Clinical Epidemiology. 2006;59(1):7–10. doi: 10.1016/j.jclinepi.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Furukawa 2007 .Furukawa TA, Watanabe N, Omori IM, Montori VM, Guyatt GH. Association between unreported outcomes and effect size estimates in Cochrane meta-analyses. JAMA. 2007;297(5):468–70. doi: 10.1001/jama.297.5.468-b. [DOI] [PubMed] [Google Scholar]
- Gartlehner 2008 .Gartlehner G, Gaynes BN, Hansen RA, Thieda P, DeVeaugh-Geiss A, Krebs EE, et al. Comparative benefits and harms of second-generation antidepressants: background paper for the American College of Physicians. Annals of Internal Medicine. 2008;149(10):734–50. doi: 10.7326/0003-4819-149-10-200811180-00008. [DOI] [PubMed] [Google Scholar]
- Guaiana 2003 .Guaiana G, Barbui C, Hotopf M. Amitriptyline versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews. 2003;(2) doi: 10.1002/14651858.CD004186. DOI: 10.1002/14651858.CD004186.pub2. [DOI] [PubMed] [Google Scholar]
- Guaiana 2005 .Guaiana G, Andretta M, Corbari L, Mirandola M, Sorio A, D’Avanzo B, et al. Antidepressant drug consumption and public health indicators in Italy, 1955 to 2000. Journal of Clinical Psychiatry. 2005;66(6):750–5. doi: 10.4088/jcp.v66n0612. [DOI] [PubMed] [Google Scholar]
- Guy 1970 .Guy W, Bonato RR. Manual for the ECDEU Assessment Battery.2. National Institute of Mental Health; Chevy Chase, MD: 1970. [Google Scholar]
- Hamilton 1960 .Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen 2005 .Hansen RA, Gartlehner G, Lohr KN, Gaynes BN, Carey TS. Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Annals of Internal Medicine. 2005;143(6):415–26. doi: 10.7326/0003-4819-143-6-200509200-00006. [DOI] [PubMed] [Google Scholar]
- Higgins 2008 .Higgins JP, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1. The Cochrane Collaboration; 2008. updated September 2008. [Google Scholar]
- Hotopf 1997 .Hotopf M, Hardy R, Lewis G. Discontinuation rates of SSRIs and tricyclic antidepressants: a meta-analysis and investigation of heterogeneity. Br J Psychiatry. 1997;170:120–7. doi: 10.1192/bjp.170.2.120. [DOI] [PubMed] [Google Scholar]
- Hoyberg 1996 .Hoyberg OJ, Maragakis B, Mullin J, Norum D, Stordall E, Ekdahl P, et al. A double-blind multicentre comparison of mirtazapine and amitriptyline in elderly depressed patients. Acta Psychiatrica Scandinavica. 1996;93(3):184–90. doi: 10.1111/j.1600-0447.1996.tb10629.x. [DOI] [PubMed] [Google Scholar]
- Imperadore 2007 .Imperadore G, Cipriani A, Signoretti A, Furukawa TA, Watanabe N, Churchill R, et al. Citalopram versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews. 2007;(2) doi: 10.1002/14651858.CD006534.pub2. DOI: 10.1002/14651858.CD006534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis 2006 .Ioannidis JP. Indirect comparisons: the mesh and mess of clinical trials. Lancet. 2006;368(9546):1470–2. doi: 10.1016/S0140-6736(06)69615-3. [DOI] [PubMed] [Google Scholar]
- Kent 2000 .Kent JM. SNaRIs, NaSSAs, and NaRIs: new agents for the treatment of depression. Lancet. 2000;355(9207):911–8. doi: 10.1016/S0140-6736(99)11381-3. [DOI] [PubMed] [Google Scholar]
- Lexchin 2003 .Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ. 2003;326(7400):1167–70. doi: 10.1136/bmj.326.7400.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu 2006 .Lu G, Ades AE. Assessing evidence inconsistency in mixed treatment comparisons. Journal of the American Statistical Association. 2006;101:447–59. [Google Scholar]
- Luborsky 1962 .Luborsky L. Clinician’s judgments of mental health. Archives of General Psychiatry. 1962;7:407–17. doi: 10.1001/archpsyc.1962.01720060019002. [DOI] [PubMed] [Google Scholar]
- Lumley 2002 .Lumley T. Network meta-analysis for indirect treatment comparisons. Statistics in Medicine. 2002;21(16):2313–24. doi: 10.1002/sim.1201. [DOI] [PubMed] [Google Scholar]
- Montgomery 1979 .Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry. 1979;134:382–9. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- Montgomery 2004 .Montgomery JH, Byerly M, Carmody T, Li B, Miller DR, Varghese F, et al. An analysis of the effect of funding source in randomized clinical trials of second generation antipsychotics for the treatment of schizophrenia. Controlled Clinical Trials. 2004;25(6):598–612. doi: 10.1016/j.cct.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Mullin 1996 .Mullin J, Lodge A, Bennie E, McCreadie R, Bhatt GS, Fenton G. A multicentre, double-blind, amitriptyline-controlled study of mirtazapine in patients with major depression. Journal of Psychopharmacology. 1996;10(3):235–40. doi: 10.1177/026988119601000310. [DOI] [PubMed] [Google Scholar]
- Nakagawa 2009 .Nakagawa A, Watanabe N, Omori I, Cipriani A, Barbui C, McGuire H, et al. Milnacipran versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews. 2009;(3) doi: 10.1002/14651858.CD006529.pub2. DOI: 10.1002/14651858.CD006529.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICE 2004 .National Institute for Clinical Excellence . Depression: management of depression in primary and secondary care. National Institute for Clinical Excellence; London: 2004. [Google Scholar]
- Norman 1989 .Norman GR. Issues in the use of change scores in randomized trials. Journal of Clinical Epidemiology. 1989;42(11):1097–105. doi: 10.1016/0895-4356(89)90051-6. [DOI] [PubMed] [Google Scholar]
- Nose 2007 .Nose M, Cipriani A, Furukawa TA, Omori I, Churchill R, McGuire H, et al. Duloxetine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews. 2007;(2) doi: 10.1002/14651858.CD006530. DOI: 10.1002/14651858.CD006533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori 2010 .Omori I, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire C, et al. Fluvoxamine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews. 2010;(3) doi: 10.1002/14651858.CD006114.pub2. DOI: 10.1002/14651858.CD006114.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxman 1992 .Oxman AD, Guyatt GH. A consumer’s guide to subgroup analyses. Annals of Internal Medicine. 1992;116(1):78–84. doi: 10.7326/0003-4819-116-1-78. [DOI] [PubMed] [Google Scholar]
- Perlis 2005 .Perlis RH, Perlis CS, Wu Y, Hwang C, Joseph M, Nierenberg AA. Industry sponsorship and financial conflict of interest in the reporting of clinical trials in psychiatry. American Journal of Psychiatry. 2005;162(10):1957–60. doi: 10.1176/appi.ajp.162.10.1957. [DOI] [PubMed] [Google Scholar]
- Procyshyn 2004 .Procyshyn RM, Chau A, Fortin P, Jenkins W. Prevalence and outcomes of pharmaceutical industry-sponsored clinical trials involving clozapine, risperidone, or olanzapine. Canadian Journal of Psychiatry - Revue Canadienne De Psychiatrie. 2004;49(9):601–6. doi: 10.1177/070674370404900905. [DOI] [PubMed] [Google Scholar]
- Puech 1997 .Puech A, Montgomery SA, Prost JF, Solles A, Briley M. Milnacipran, a new serotonin and noradrenaline reuptake inhibitor: an overview of its antidepressant activity and clinical tolerability. International Clinical Psychopharmacology. 1997;12(2):99–108. doi: 10.1097/00004850-199703000-00005. [DOI] [PubMed] [Google Scholar]
- Quitkin 2001 .Quitkin FM, Taylor BP, Kremer C. Does mirtazapine have a more rapid onset than SSRIs? Journal of Clinical Psychiatry. 2001;62(5):358–61. doi: 10.4088/jcp.v62n0509. [DOI] [PubMed] [Google Scholar]
- Ramsay 2005 .Ramsay C, Gates S. Unit of analysis issues; UK Contributors meeting; Manchester: The Cochrane Collaboration. 2005. [Google Scholar]
- Rush 2007 .Rush AJ. STAR*D: what have we learned? American Journal of Psychiatry. 2007;164(2):201–4. doi: 10.1176/ajp.2007.164.2.201. [DOI] [PubMed] [Google Scholar]
- Salanti 2008 .Salanti G, Higgins JP, Ades AE, Ioannidis JP. Evaluation of networks of randomized trials. Statistical Methods in Medical Research. 2008;17(3):279–301. doi: 10.1177/0962280207080643. [DOI] [PubMed] [Google Scholar]
- Schatzberg 2002 .Schatzberg AF, Kremer C, Rodrigues HE, Murphy GM. Double-blind, randomized comparison of mirtazapine and paroxetine in elderly depressed patients. American Journal of Geriatric Psychiatry. 2002;10(5):541–50. [PubMed] [Google Scholar]
- Schulz 2010 .Schulz KF, Altman DG, Moher D, CONSORT Group CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c332. doi: 10.1136/bmj.c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith 2002 .Smith D, Dempster C, Glanville J, Freemantle N, Anderson I. Efficacy and tolerability of venlafaxine compared with selective serotonin reuptake inhibitors and other antidepressants: a meta-analysis. British Journal of Psychiatry. 2002;180:396–404. doi: 10.1192/bjp.180.5.396. [DOI] [PubMed] [Google Scholar]
- Song 2003 .Song F, Altman DG, Glenny AM, Deeks JJ. Validity of indirect comparison for estimating efficacy of competing interventions: empirical evidence from published meta-analyses. BMJ. 2003;326(7387):472. doi: 10.1136/bmj.326.7387.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens 1997 .Steffens DC, Krishnan KR, Helms MJ. Are SSRIs better than TCAs? Comparison of SSRIs and TCAs: a meta-analysis. Depression and Anxiety. 1997;6(1):10–18. doi: 10.1002/(sici)1520-6394(1997)6:1<10::aid-da2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Thase 2005 .Thase ME, Baker R, Simmons J, van Willigenburg AP, Heukels AJ, Janssens CJ, et al. More rapid onset of effect on the core symptoms of depression with mirtazapine than SSRIs; American Psychiatric Association 2005 Annual Meeting; Atlanta. 2005. [Google Scholar]
- Thase 2006 .Thase ME, Baker RA, Simmons JH, Van Willigenburg AP, Schutte AJ. Effect of mirtazapine versus SSRIs on core symptoms of depression. Journal of Affective Disorders. 2006;91(Suppl 1):S83. [Google Scholar]
- Thase 2010 .Thase ME, Nierenberg AA, Vrijland P, van Oers HJ, Schutte AJ, Simmons JH. Remission with mirtazapine and selective serotonin reuptake inhibitors: a meta-analysis of individual patient data from 15 controlled trials of acute phase treatment of major depression. International Clinical Psychopharmacology. 2010;25(4):189–98. doi: 10.1097/YIC.0b013e328330adb2. [DOI] [PubMed] [Google Scholar]
- Trivedi 2006 .Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. American Journal of Psychiatry. 2006;163(1):28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- Ware 1993 .Ware JE, Snow KK, Kosinski M, Gandek B. SF-36 Health Survey Manual and Interpretation Guide. New England Medical Centre; Boston, MA: 1993. [Google Scholar]
- WHO 1992 .World Health Organization . The Tenth Revision of the International Classification of Diseases and Related Health Problems (ICD-10) World Health Organization; Geneva: 1992. [Google Scholar]
- WHO 2006a .World Health Organization . Revised Global Burden of Disease 2002 estimates. World Health Organisation; Geneva: 2006. http://www.who.int/healthinfo/bodgbd2002/en/index [Google Scholar]
- WHO 2006b .World Health Organization . WHO Collaborative Centre for Drug Statistics Methodology. World Health Organisation; Geneva: 2006. http://www.whocc.no/atcddd/ [Google Scholar]
- WHOQOL Group 1998 .WHOQOL Group The World Health Organization quality of life assessment (WHOQOL): Development and general psychometric properties. Social Science and Medicine. 1998;46(12):1569–85. doi: 10.1016/s0277-9536(98)00009-4. [DOI] [PubMed] [Google Scholar]
- Wiebe 2003 .Wiebe S, Guyatt G, Weaver B, Matijevic S, Sidwell C. Comparative responsiveness of generic and specific quality-of-life instruments. Journal of Clinical Epidemiology. 2003;56(1):52–60. doi: 10.1016/s0895-4356(02)00537-1. [DOI] [PubMed] [Google Scholar]
- Williams 2000 .Williams JW, Jr, Mulrow CD, Chiquette E, Noel PH, Aguilar C, Cornell J. A systematic review of newer pharmacotherapies for depression in adults: evidence report summary. Annals of Internal Medicine. 2000;132(9):743–56. doi: 10.7326/0003-4819-132-9-200005020-00011. [DOI] [PubMed] [Google Scholar]
- Williams 2001 .Williams JB. Standardizing the Hamilton Depression Rating Scale: past, present, and future. European Archives of Psychiatry and Clinical Neuroscience. 2001;251(Suppl 2):II6–12. doi: 10.1007/BF03035120. [DOI] [PubMed] [Google Scholar]
- Wing 1994 .Wing J. Measuring mental health outcomes: a perspective from the Royal College of Psychiatrists. Outcomes into Clinical Practice. BMJ Publishing; London: 1994. [Google Scholar]
References to other published versions of this review
- Watanabe 2008 .Watanabe N, Omori IM, Nakagawa A, Cipriani A, Barbui C, McGuire H, et al. Mirtazapine versus other antidepressants in the acute-phase treatment of adults with major depression: systematic review and meta-analysis. Journal of Clinical Psychiatry. 2008;69(9):1404–15. doi: 10.4088/jcp.v69n0908. [DOI] [PubMed] [Google Scholar]
- Watanabe 2010 .Watanabe N, Omori IM, Nakagawa A, Cipriani A, Barbui C, McGuire H, Churchill R, Furukawa TA, MANGA (Meta-Analysis of New Generation Antidepressants) Study Group Safety reporting and adverse-event profile of mirtazapine described in randomized controlled trials in comparison with other classes of antidepressants in the acute-phase treatment of adults with depression: systematic review and meta-analysis. CNS Drugs. 2010;24(1):35–53. doi: 10.2165/11319480-000000000-00000. DOI: 10.2165/11319480-000000000-00000. [DOI] [PubMed] [Google Scholar]
- * Indicates the major publication for the study