

Abstract
Genome-wide association studies, DNA sequencing studies, and other genomic studies are finding an increasing number of genetic variants associated with clinical phenotypes that may be useful in developing diagnostic, preventive, and treatment strategies for individual patients. However, few common variants have been integrated into routine clinical practice. The reasons for this are several, but two of the most significant are limited evidence about the clinical implications of the variants and a lack of a comprehensive knowledge base that captures genetic variants, their phenotypic associations, and other pertinent phenotypic information that is openly accessible to clinical groups attempting to interpret sequencing data. As the field of medicine begins to incorporate genome-scale analysis into clinical care, approaches need to be developed for collecting and characterizing data on the clinical implications of variants, developing consensus on their actionability, and making this information available for clinical use. The National Human Genome Research Institute (NHGRI) and the Wellcome Trust thus convened a workshop to consider the processes and resources needed to: 1) identify clinically valid genetic variants; 2) decide whether they are actionable and what the action should be; and 3) provide this information for clinical use. This commentary outlines the key discussion points and recommendations from the workshop.
Keywords: genomic medicine, clinical actionability, database, electronic health records (EHR), pharmacogenomics, DNA sequencing
Introduction
Genome-wide association studies (GWAS), DNA sequencing studies, and other genomic studies are finding an increasing number of genetic variants associated with clinical phenotypes that may be useful in developing diagnostic, preventive, and treatment strategies for individual patients. Conceptually, one can view these genetic variants along a continuum between common polymorphisms and rare or private mutations [Manolio et al., 2009]. This range of allele frequencies has implications for the discovery of such variants in different populations, the study of such variants with regard to their clinical implications, and the detection and interpretation of specific variants in a given individual. The vast amount of human medical genetics research conducted over the last few decades began primarily with the discovery of mutations in genes responsible for Mendelian diseases. More recently this research expanded to include the common variation associated with multifactorial phenotypes discovered through GWAS [Hindorff et al., 2009]. Clinical genetic testing has been used in a specialized clinical genetics setting for over fifty years, providing specific molecular diagnoses for thousands of individuals with rare single gene and chromosomal disorders. Somatic tumor variants are now increasingly assayed in order to target chemotherapy [McLeod 2013] and germline pharmacogenomic variants are beginning to be incorporated into routine clinical practice in certain scenarios [Relling and Klein, 2011]. However, despite an increasing number of characterized variants from GWAS studies, few common variants have been integrated into routine clinical practice. The reasons for this are several, but two of the most significant are limited evidence about the clinical implications of the variants and a lack of a comprehensive knowledge base that captures genetic variants, their phenotypic associations, and other pertinent clinically relevant information that is openly accessible to clinical groups attempting to interpret sequencing data [Evans et al. 2011; Manolio et al., 2013].
Data on the clinical implications of genetic variants are currently contained in a patchwork of non-standardized repositories maintained by individual scientists, academic institutions, laboratories, government entities, and industry and are not easily accessible to health care providers and health care systems. Making this information accessible and useful for clinical care will require systematic collection, extraction, evaluation, and synthesis of these findings followed by standardized representation of the information in queryable databases, along with tools permitting user-defined filtering. It will be important to compile the available evidence and develop consensus views from the clinical, genetics, and laboratory communities on what variants are actionable and the clinical actions to be taken. This information must then be made available to clinicians through clinical decision support tools embedded in electronic health records (EHR) [Starren et al. 2013].
As the field of medicine begins to incorporate genome-scale analysis into clinical care, approaches need to be developed for collecting and characterizing data on the clinical implications of variants, developing consensus on their actionability, and making this information available for clinical use. The National Human Genome Research Institute (NHGRI) and the Wellcome Trust thus convened a workshop to consider the processes and resources needed to: 1) identify clinically valid genetic variants; 2) decide whether they are actionable and what the action should be; and 3) provide this information for clinical use. The following sections summarize the discussion and the workshop recommendations (see Box 1).
Box 1. Characterizing and Displaying Genetic Variants for Clinical Action Workshop Recommendations.
Existing Genetic Variation Resources
Hold a workshop or convene a working group to identify reasonable technical standards for exchange of genetic variant and clinical data to maximize exchange of among existing databases.
Coordinate with US and UK agencies, such as Agency for Healthcare Research and Quality (AHRQ), the Office of the National Coordinator for Health Information Technology (ONC), Department of Veterans Affairs (DVA), National Health Service (NHS), commercial electronic health record (EHR) vendors, and other relevant organizations to address data interoperability and viable approaches for integration of genomic information and actionable variants into a variety of EHR systems.
Facilitate the clinical annotation of genes and germline and somatic variants in relation to specific traits (including specificity, sensitivity, prevalence, positive and negative predictive values, and penetrance). Capture of penetrance data from exome chip studies may be a good opportunity to capture information on the “most common of the rare” variants.
Ensure that 1) ClinVar and similar resources capture genetic variants of unknown significance (VUS) and isolated reports of variant – condition associations identified through clinical sequencing projects, and 2) computer programs are developed to enable clinical sequencing labs to efficiently transmit data to such resources.
Identifying Genetic Variants for Potential Clinical Action
Support and expand research to determine clinical validity and utility/actionability of SNP and structural genetic variants.
Design studies to ensure that clinically valid variants for which actionability is unknown are appropriately stratified and have identified research pathways for determining actionability.
Ensure that studies of gene-disease and gene-drug associations in diverse populations are funded.
Support functional and other follow-up studies on novel variants found in specific genes with known utility (e.g., determine the consequence of every BRCA1 variant) to generate data to support better interpretation of variants of uncertain significance.
Explore mechanisms to facilitate communication between labs studying specific genes with potentially clinically relevant variants and researchers and clinicians with family, phenotype and other clinical information willing to partner to study the function of the genes and variants.
Maximize interactions among epidemiologists, bioinformaticians, and genomic scientists to facilitate obtaining needed information on clinical validity and utility. For example, develop training programs that bring these three disciplines together to tackle specific aspects of the pipeline needed to identify actionable variants and move them into the clinic.
Serve as a “convener” in conjunction with other NIH Institutes and Centers, professional organizations, and other groups to build consensus, prioritize and publicize recommendations regarding clinical validity and utility/actionability.
Creating a Translation Loop for Genomic Medicine
Create and support a coordinated resource to extend Ensembl, ClinVar, and other databases for use in clinical care by providing relevant phenotype information, other clinical annotation, and suggestions regarding clinical utility/actionability. Such a resource could help bridge the gap between researchers and primary care clinicians, who will need user-friendly clinical support tools and/or an EHR integration layer to readily utilize these data in clinical care.
Collaborate with data warehouses (e.g. Medco) on large-scale studies to better evaluate outcomes of specific uses of genomic variants in clinical care. Develop a process to identify research questions that could be answered using data warehouses.
Consider supporting competitions that promote development of algorithms for interpreting genomic variants and compare algorithm performance, such as the Critical Assessment of Genome Interpretation (CAGI; http://genomeinterpretation.org/) effort.
Encourage the dissemination of decision support logic and interpretive tools, including making a publicly available library, to enable diverse EHR systems to use the same logic and tools when developing clinical decision support tools.
Develop and test innovative genetic education tools for providers specifically focused around the appropriate use of genomic variants.
Develop approaches for long-term follow-up of patients with rare variants of interest to better understand the relationship of these variants to disease and other phenotypes, leveraging existing resources with healthcare systems (e.g., payer information, NHS records).
Catalyze discussion with the US Office for Human Research Protections (OHRP) and the UK National Research Ethics Service (NRES) regarding institutional review board guidance on boundaries and synergy between clinical care and research. Conduct policy analyses to better understand the perspectives of relevant organizations (e.g., FDA, CMS, NICE, UKGTN and professional organizations such as ACMG, CAP, and AMP) regarding using genetic variant information to inform clinical care.
Existing Genetic Variation Resources
Several existing resources provide some of the underlying data required to characterize genetic variants along the evidence continuum (Table I). For example, dbSNP is a catalog of short genetic variation, OMIM contains clinical descriptions and genetic variation primarily related to Mendelian diseases, and the NHGRI Genome Wide Association Catalog curates significant SNP-trait associations from genome-wide association studies. There are also approximately 1,600 locus-specific databases that curate clinical information on specific genes or diseases, although they vary in the types of data included and nomenclature followed (http://www.centralmutations.org/Lsdb.php). The National Center for Biotechnology Information (NCBI) has developed a new database, ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), to provide a public archive of reports about the relationships between human variants and phenotypes along with the supporting evidence from attributed sources such as clinical research studies and case reports, specialized databases, clinical practice guidelines, and peer-reviewed literature.
Table I. Commonly used databases and resources that relate genotype to human phenotypes and disease.
| Database/Resource | Brief Description | Main Purpose | URL |
|---|---|---|---|
| ClinVar | Aggregates information about sequence variation and its relationship to human health | Provide assertions of variation-phenotype relationships | http://www.ncbi.nlm.nih.gov/clinvar/ |
|
| |||
| dbSNP | Database of short genetic variation | Archive germline variation (both polymorphism and rare mutation). Provides alleles, genotypes and their respective frequencies by population | http://www.ncbi.nlm.nih.gov/snp |
|
| |||
| dbVaR | Database of genomic structural variation | Archive studies of structural variation and their interpretation | http://www.ncbi.nlm.nih.gov/dbvar/ |
|
| |||
| Ensembl | Genome databases for vertebrates and other eukaryotic species, | Develop a software system which produces and maintains automatic annotation on selected eukaryotic genomes | www.ensembl.org |
|
| |||
| Human Gene Mutation Database | Database of the first example of all mutations causing or associated with human inherited disease, plus disease-associated/functional polymorphisms reported in the literature | Collate published gene lesions responsible for human inherited disease and provide information of practical diagnostic importance to genetics professionals | http://www.hgmd.cf.ac.uk/ac/index.php |
|
| |||
| The International Standards for Cytogenomic Arrays (ISCA) Consortium | Central repository for cytogenomic array data generated in clinical testing laboratories. | Useful for classifying copy number variants of uncertain clinical significance | https://www.iscaconsortium.org/ |
|
| |||
| NHGRI GWAS Catalog | Compendium of SNP-trait associations gleaned from published GWAS studies | Provide a catalog of significant findings from all published GWAS studies to facilitate prioritization, replication, and follow-up | http://www.genome.gov/GWAStudies/ |
|
| |||
| NIH Genetic Testing Registry (GTR) | Uses laboratory-reported data to provide information about genetic tests for inherited genetic variations. Also reports disease- and gene-specific information integrated from NCBI's databases | Provide a catalogue of genetic tests in clinical use for clinicians. While most information will be at the gene level, tests for single variants will be included. Assertions of AV, CV, and CU are made by test submitters. NCBI assembles practice guidelines | http://www.ncbi.nlm.nih.gov/gtr/ |
|
| |||
| OMIM | Compendium of human genes and genetic phenotypes | Provide physicians and other professionals with full-text, referenced overviews for Mendelian disorders and > 12,000 genes | http://www.omim.org/ |
|
| |||
| PharmGKB | Pharmacogenomics knowledge resource | Provide information about the impact of genetic variation on drug response for clinicians and researchers | http://www.pharmgkb.org/ |
|
| |||
| PheGenI | Merges NHGRI GWAS catalog data with data-bases housed at the NCBI including Gene, dbGaP, OMIM, GTEx and dbSNP | Facilitate prioritization of GWAS variants to follow up, study design considerations, and generation of biological hypotheses | http://www.ncbi.nlm.nih.gov/gap/PheGenI |
Challenges in synthesizing these existing data resources include incomplete or inaccurate phenotypic data and a lack of standard terminologies, thus limiting interoperability. Representatives from the relevant databases and resources, along with bioinformatics and clinical experts, should work together to identify reasonable technical standards for exchange and reporting of genetic variant and associated phenotypic and clinical data. Interactions among existing databases should be maximized to ensure comparability and avoid duplication. In addition, none of these resources are currently designed to interact with EHR systems, meaning clinicians would have to access the information by interrupting their clinical workflow—a known barrier to use [Ross, 2009]. Consideration needs to be given to how such a resource would be utilized by clinicians and how to optimize that usage under conditions when a patient's individual sequence (or relevant extracts from) is or is not directly available to the clinician. EHR vendors should be convened with other relevant organizations such as major health payers and regulatory agencies to address data interoperability and viable approaches for integrating genomic information and actionable variants into EHR systems. Some possible technical characteristics of such a system were recently outlined and include maintaining separation of primary molecular observations from the clinical interpretations of those data, support data compression of sequence data to clinically manageable subsets, without losing the ability to produce a fully accurate copy of the original sequence, and support both human-viewable and machine-readable formats to facilitate implementation of clinical decision support rules [Masys et al., 2012]
Identifying Genetic Variants for Potential Clinical Action
In characterizing evidence about genes and specific genetic variants for clinical use, the concepts of analytic validity (AV), clinical validity (CV), and clinical utility (CU) are in broad use (Box 2). Out of the three concepts, defining CU and reaching consensus on what constitutes sufficient evidence for CU is the most challenging. Perspectives on what patient outcomes are significant can be highly variable, ranging from outcomes associated with a clear opportunity for medical intervention and medical benefit to opportunity for behavioral change to information that can be used in reproductive decision making and life planning. Evidence thresholds may need to be tailored to the cost, burden, and/or risk of proposed interventions. This means that different groups, such as patients, clinicians, payers, hospital systems, and government agencies may reach different conclusions about CU even after reviewing the same evidence. In the case of rare diseases in which formal CU may be difficult to assess, the diagnostic information can still guide management of the patient, which is a type of clinical utility [Grosse et al., 2010]. The discussion of what evidence is needed for CU frequently centers on whether a clinician should order a particular genetic assay to make a diagnostic or therapeutic decision, and does not address the question of what a clinician could or should do if that information were already available. The latter is important to consider under several possible future scenarios; people may order their own sequence from commercial vendors and then present their clinician with the findings for interpretation or participation in a research orientated biobank may lead to extensive genetic variants including sequence data being available and potentially actionable. The Clinical Pharmacogenetic Implementation Consortium (http://www.pharmgkb.org/page/cpicGeneDrugPairs) has created a set of pharmacogenetic guidelines based on this principle. The concept of “personal utility” has been put forward as another facet of medical decision-making regarding the use of genetic testing [Foster et al. 2009]. For example, the specific genetic etiology of a rare disorder can have immense value to families regardless of the ability to intervene in a particular condition. Similar “personal utility” might be derived from more common genetic variation, such as APOE status and Alzheimer's disease risk [Roberts et al. 2011] and other profiles of variants with small contributions to multifactorial disease [Gollust et al., 2012], but this type of information is by definition highly context-dependent and requires input from the patient regarding whether such information is desired.
Box 2. Definitions*.
Analytic Validity (AV) How accurately and reliably the test measures the genotype of interest.
Clinical Validity (CV) How consistently and accurately the test detects or predicts the intermediate or final outcomes of interest.
Clinical Utility (CU) How likely the test is to significantly improve patient outcomes.
*as defined by the CDC's Office of Public Health Genomics (OPHG) ACCE Model Process for Evaluating Genetic Tests (http://www.cdc.gov/genomics/gtesting/ACCE/index.htm).
An intermediate category between CV and CU has been termed clinical actionability (see Figure 1). This can be applied to variants having either proven CU or evidence sufficient to indicate how existing variant information could or should be used clinically though insufficient to establish CU unequivocally [Berg et al,. 2011]. This concept of actionability also allows flexibility for clinicians or even institutions to tailor their use of variant information to a particular patient, clinical setting, or local standard of practice. However, lack of clear clinical utility also means that an action taken in response to the finding of a genetic variant may in fact have detrimental outcomes. Furthermore, the definition of “actionable” can range from a broad inclusion of variants with possible “personal utility” to a more narrow definition of variants that have well-established implications for the management of the individual patient.
Figure 1.
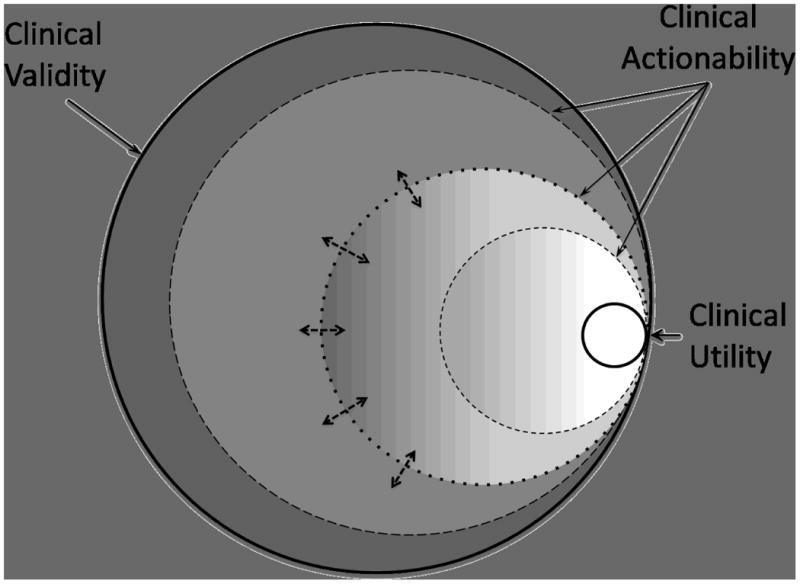
Schematic diagram showing overlap of clinical validity (outer boundary, containing all genetic variants with a valid clinical association), clinical utility (inner white circle), and the boundaries of clinical actionability (shaded circles). Depending on the criteria used to define “actionability” (depicted by double arrows) the boundaries could be more or less inclusive (dashed circles). In this scheme, variants with clinical utility are a subset of all clinically actionable variants.
As a practical matter, the variants identified in an individual patient will fall into two broad categories: “known” genetic variants that have been observed previously and “novel” genetic variants that have not been observed before and may be unique to that individual or private to his or her family members. With regard to “known” genetic variants, the existence of prior information about these variants should in theory facilitate the annotation of their clinical significance or lack thereof, whereas “novel” genetic variants may require quite different methods for determining their clinical relevance. Many of the databases of known genetic variants listed in Table I are primary data archives, and thus maintain experimental results regarding genetic variants and their phenotypic correlations rather than curators' interpretation of these data. However, numerous medical centers and research programs are beginning to evaluate these databases and other pieces of information to explore the use of such variants in clinical care, and many are developing approaches for identifying variants to be assayed and the actions to be recommended when they are detected (see Table II for a brief description of some of these programs and approaches). These groups are often evaluating the same assays, reviewing the same literature, and assessing the same evidence. In addition, efforts to identify variants relevant to drug response have been ongoing through the National Institute of General Medical Sciences (NIGMS)-led Pharmacogenomics Knowledge Base (PharmGKB) and Pharmacogenomics Research Network (PGRN) and independent research groups such as Coriell's Pharmacogenomics Advisory Group. Unifying these often isolated efforts and developing a consensus framework for evaluating existing data might reduce duplication of effort and eventually speed adoption of actionable genetic variants into clinical practice.
Table II. Current approaches for identifying genomic variants for potential clinical use.
| Program | Brief Description of Approach |
|---|---|
| Coriell Personalized Medicine Collaborative, CPMC® | Longitudinal study examining impact of potentially actionable genomic results for common complex diseases and drug response. Uses two external oversight bodies, Informed Cohort Oversight Board (Complex Diseases) and Pharmacogenomics Advisory Group (drug response) to determine what variants will be returned to study participants (Stack, Gharani et al. 2011) |
|
| |
| Clinical Genome Resource (ClinGen) | Developing a pipeline for the submission by clinical laboratories of sequence variants and related data to ClinVar, curating these variants with clinical and functional data, developing a consensus process to bin genes and variants into categories of clinical actionability, and systematically disseminating this information. |
|
| |
| Electronic Medical Records and Genomics Network (eMERGE) Return of Results Working Group | Recommended that Klinefelter, Turner, and homozygosity for Factor V Leiden be considered for return to research participants.(Fullerton, Wolf et al. 2012) The current focus is to define an initial set of variants that are potentially useful in clinical practice for purposes such as assessment of genetic risk for complex disorders or selection or dosing of drugs. This initial set will focus on common disease risk variants and pharmacogenomic variants for which eMERGE sites expect to have data. |
|
| |
| International Collaboration for Clinical Genomics (ICCG) | Participating in the ClinGen program. Curation and evidence-based review of structural and sequence-level variant data deposited within ClinVar to assign consensus annotation of variants with regard to their clinical significance (e.g., “pathogenic”, “benign”, etc.) and standardize clinical interpretations. |
|
| |
| Clinical Pharmacogenetics Implementation Consortium, CPIC | Expert consensus of CPIC members. Consensus results in a clinical algorithm that defines the clinical context and defines the information required and provides a specific clinical recommendation. Published in peer-review literature. Focus is on how available genetic test results should be used to optimize drug therapy, rather than whether tests should be ordered. (Relling and Klein 2011) Recommendations include CYP2C9, VKORC1 for warfarin dosing; CYP2C19 for clopidogrel therapy; TPMT and thiopurine dosing; SLCO1B1 and simvastatin; CYP2D6 and codeine. |
|
| |
| Evaluation of Genomic Applications in Practice and Prevention, EGAPP Working Group | Systematic evidence review with a focus on clinical utility followed by synthesis of evidence and development of recommendation statement (Teutsch, Bradley et al. 2009) Evidence reviews and recommendation statements do provide information about the clinical context and how the variant information is proposed to be used that could inform clinical action to be taken. |
|
| |
| NHGRI Clinical Sequencing Program (CSER)'s Actionable Variants and Return of Results Working Groups | The Actionable Variants WG coordinate approaches to defining and binning genetic variants potentially useful for clinical purposes; share and review external resources for similar purposes. Discuss emerging issues and develop standards related to returning results to study participants (including incidental findings, where determined to be appropriate).<break/>The Return of Results WG Analyze the relevant normative and clinical issues, including such issues as whether or when there exists an ethical duty to return results, what are the appropriate normative and clinical criteria for determining whether results should be returned, the meaning of “actionability” and whether “actionability,” should be the relevant standard for determining which results are returnable. |
|
| |
| FDA Table of Pharmacogenomic Biomarkers in Drug Labels | FDA-approved drugs with pharmacogenomic information in their labels. Some, but not all, of the labels include specific actions to be taken based on genetic information (http://www.fda.gov/drugs/scienceresearch/%20researchareas/pharmacogenetics/ucm083378.htm) |
Although significant progress has been made identifying disease variant associations, a key barrier to the identification of variants for clinical use is the lack of clinical translational research beyond this initial identification of an association to assess the risks, costs, and health benefits of utilizing genomic information in the practice of clinical medicine. The importance of this problem was evident from an informal poll of our workshop attendees showing little consensus on the appropriateness of using genetic variant information in a variety of clinical scenarios and from the discordance among genetics experts asked to assess the importance of returning specific secondary findings after clinical sequencing [Green et al., 2012]. The 2011 NHGRI strategic plan [Green and Guyer, 2011] identifies the need for funding clinical research to accelerate the pace of knowledge generation needed to increase the clinical use of genetic information. Given the large number of variants that currently have limited evidence of actionability, despite their clinical validity [Berg et al, 2011], clear pathways will need to be developed for providing the evidence to move them into the actionable range or demonstrate definitively their lack of clinical utility. Again, a critical component of that pathway is deposition of primary data in well-defined, standardized formats to public databases. To support standardized assessment of information about human variation, it is critical that both the primary data and the current interpretation of those data be freely accessible. Processes and decision support tools can then be applied to the existing data to identify which variants are clinically actionable and provide these variants and supporting information for clinical use as appropriate.
In addition to devising a plan to accumulate evidence of actionability for known clinically valid variants, additional effort must be dedicated to expand the set of current associations beyond populations of European ancestry. There is a paucity of data on associations in non-European ancestry populations. Differences in allele frequencies and linkage disequilibrium patterns across ancestry groups (The International HapMap Consortium, 2005; Abecasis et al., 2010; Altshuler et al., 2010], as well as major differences in disease burden and severity [Ramos and Rotimi, 2009], make research in these under-studied populations a critical need. Further, determining the effect of rare variants on gene function is essential to determining the clinical impact of these variants [Marian 2012]. This might be addressed by enhancing communications between researchers who identify rare or unique variants in well-phenotyped individuals and families and researchers with interests in the specific gene(s) implicated in these conditions. Another critical group of stakeholders are the diagnostic laboratories performing clinical molecular tests for rare disorders. These clinical laboratories often hold a wealth of information about variants detected in specific genes and their interpretation of the pathogenicity of those variants. Thus, development of data-sharing models that protect patient privacy could enhance communication regarding the clinical significance of genetic variants. This model was used to create the International Standards for Cytogenomic Arrays (ISCA) Consortium database where multiple academic and commercial laboratories contribute data on copy number variants (CNVs) and associated phenotypes. To date data on over 32,000 patients has been collected allowing rapid identification of novel CNVs associated with a range of disorders. Together, these efforts would create a so-called ‘translational loop’, from bench to bedside and back again, as described below.
Creating a Translation Loop for Genomic Medicine
To achieve more effective translation of genomic variant information into clinical practice requires not only a ‘push’ to move evidence-based information generated in the laboratory into the clinic, but the ability to ‘pull’ or extract information from clinical data systems to assess the impact of implementation of that research on real world clinical outcomes and effectiveness (see Figure 2). This assessment should lead to further research to validate or expand evidence and, where appropriate, modify clinical practice, creating the translation loop. To ensure this translation loop is successful requires the identification of novel research approaches that may be purposed to answer specific clinical questions. As noted above, the spectrum of genetic variation from common to rare (or private) variants will dictate the approaches that can be used to determine the clinical significance and actionability of different types of genetic variants. For example, pharmacogenomic alleles that are part of the natural human population variation should be amenable to large prospective studies that can formally assess the evidence for clinical utility such as that being done by the Electronic Medical Records and Genomics pharmacogenomics project (eMERGE-PGX) [Gottesman O et al., 2013]. Although randomized control trials have been the standard in evaluating clinical effectiveness and should continue to be utilized to better understand the impact of various aspects of genomic medicine, they can be time consuming and expensive. In addition, the application of clinical trial results with their tight controls to the complicated milieu of routine patient care is difficult [Hoes, 2009]. Other methodologies that produce useful observational data need to be considered. The use of extant publicly accessible data warehouses (such as was done for the Medco-Mayo warfarin effectiveness study [Epstein, 2011] or data from consumer genomics companies are such approaches. In order to take advantage of these data warehouses and other clinical data repositories, it is important to develop systems that can retrieve information about defined outcomes of interest from these sources and other transactional data warehouses (e.g. payer databases, pharmacy databases). Information from these sources can be used to assess the use of a given variant by clinician; identify individual and organizational factors that facilitate or impede the actual use of the variant; assess the effect on health behaviors, compliance, and other medical outcomes from the patient perspective; estimate the effect on cost of care; and compare decision-making algorithms to determine which are best for clinical use.
Figure 2.
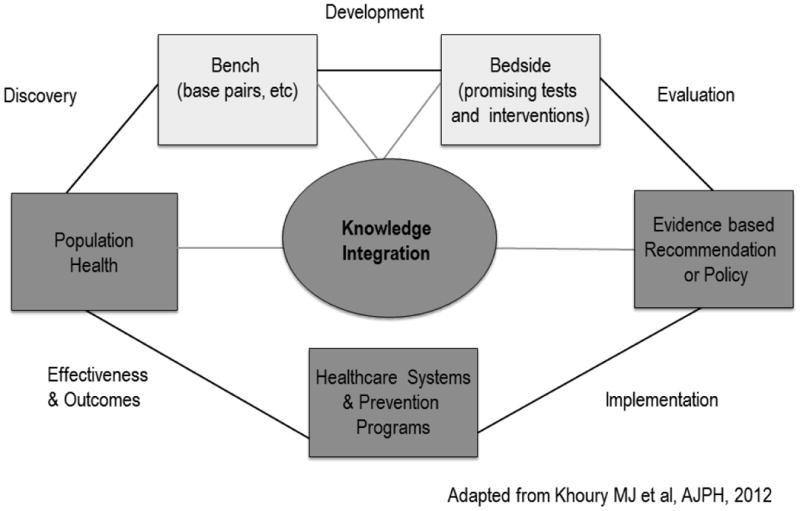
A framework for a translational loop in genomic medicine, with green representing the first phase of translation and blue representing the second phase of translation), with a feedback loop to basic science discoveries. Adapted with permission from M. Khoury [Khoury et al., 2012].
Often clinical implementation does not occur until practice guidelines have been developed by leading professional organizations; therefore, movement through the translational loop may be prompted by observational studies which facilitate the use of novel data in a real world setting. Data from observational studies, rather than clinical trials, may then provide both the data and impetus for large scale clinical implementation. Alternatively, these approaches could be used as preliminary, low cost methods to identify potential signals that could be prioritized for examination in more traditional research designs. Dissemination of these novel research approaches would avoid each health care system having to create its own specific algorithm or educational resources for the use of common genetic variants in clinical care, allowing the organizations to focus on the processes needed to implement this in clinical care. This approach could also allow the testing of various types of implementation approaches to see which are more effective in the clinic, provided that such studies are performed systematically and outcomes are rigorously measured. These types of novel approaches are probably most applicable to common variants responsible for pharmacogenomic traits and risk for multifactorial diseases.
Another benefit of a translation loop is the ability to more rapidly understand the prevalence and clinical impact of rare variants that are expected to be identified in genes of interest as next generation sequencing is increasingly deployed in clinical and research settings. This is of particular importance if rare variants with high clinical impact are to be identified and used to improve patient care. However, the assessment of rare variants in clinical medicine may require different methods than those used to assess pharmacogenomic variants or common risk factors for multifactorial diseases. There are two contexts in which rare variants might be evaluated: 1) in the diagnostic evaluation of a person with a suspected single gene disorder, and 2) as part of a collection of “incidental” or “secondary” findings from a genome-scale sequencing assay. In the molecular diagnostic setting, the goal is to identify the single variant or combination of variants that explains the patient's presenting phenotype. In contrast to the diagnostic setting, where there is a substantial a priori probability of a genetic etiology, rare variants identified as genomic incidental or secondary findings are problematic because in the absence of a family history or phenotypic features, the a priori probability that the individual is affected with a particular single gene disorder caused by a given variant is very small. Thus, evidentiary standards may be different for rare variants depending on the clinical context.
Disparate types of evidence are used to define whether a given rare variant is likely to provide such an explanation for a patient's diagnosis, including the allele frequency in control populations, the effect of the variant on the translated protein compared to the types of changes that typically cause the disorder, in silico predictions that take into account evolutionary conservation and protein structure, and family segregation studies. The final element needed to close the translation loop is the capability to aggregate assertions made by researchers and molecular diagnostic laboratories regarding the pathogenicity (or lack thereof) for particular variants in order to provide updated knowledge in an efficient way to the central databases that store information on genotypes and phenotypes (such as those identified in Table I.) A curated centralized database could then be accessed by molecular diagnostic laboratories or electronic health records in order to annotate the clinical significance of variants identified through genome-scale sequencing. Health systems could then implement automated mechanisms for updating clinically actionable information such that messages could be passed into EHRs for patients who carry this variant, including notifying their clinician in specific situations based on context-sensitive rules [Aronson et al., 2012].
Inherent in this translation loop is a blurring of boundaries between research and clinical care. Current rules and regulations are not adequately explicit about this distinction, leading to variable interpretations by institutional IRBs. This impedes the collaboration needed among institutions to generate sufficient patient numbers to be confident of the impact of variants on clinical care. The advanced notice of proposed rule-making for revision of the Common Rule [http://www.hhs.gov/ohrp/humansubjects/anprm2011page.html] may impact the ability to pursue this type of research. One aspect of this proposal is that written consent for research use of any biospecimens collected for clinical purposes would be required. The impact of current and proposed new policies on the use of genetic variants in clinical practice will need to be studied. Exploration of these and other policy issues with institutions responsible for the oversight of human subjects research such as the Office for Human Research Protections (OHRP) / National Research Ethics Service (NRES) and their international counterparts is needed to try to harmonize approaches to these types of studies.
Conclusions
GWAS, DNA sequencing studies, and other genomic studies are producing a profusion of genetic variants that are or may be associated with clinical phenotypes. For each of these variants it is important to determine population-specific frequencies; identify which variants have clinical effects and characterize those effects; and follow variant carriers over time to study natural history and measure the impact of clinical intervention that result from knowledge of these variants. To minimize the risk of being overwhelmed by the number of variants that need to be evaluated for potential clinical actionability, strategies to prioritize investigation are needed. This prioritization should include an upfront assessment of the likelihood of clinical value based on standardized frameworks such as the method proposed by Berg et al [Berg et al., 2011]. The scalability of such prioritization will be challenging because each variant will most likely need a detailed review of available evidence for multiple clinical scenarios. [Fullerton, Wolf et al. 2012]. Classification of genes or variants into such schema should be vetted by larger groups of experts and, ultimately, professional societies can review these evidence syntheses to generate clinical practice guidelines on which clinical decision support tools can be built. Speeding the adoption of actionable genetic findings for use in clinical care first requires the development of a dynamic and comprehensive resource of clinically relevant genetic variants. To meet this need, the NHGRI issued a funding opportunity titled ‘Clinically Relevant Genetic Variants Resource (CRVR): A Unified Approach for Identifying Genetic Variants for Clinical Use (U01) (http://grants.nih.gov/grants/guide/rfa-files/rfa-hg-12-016.html). CRVR has since evolved into the Clinical Genome Resource - a collaboration between CRVR grantees and the International Collaboration for Clinical Genomics (ICCG formerly the ISCA referenced above) to develop a pipeline for the submission by clinical laboratories of sequence variants and related data to a central database (ClinVar), curate these variants with clinical and functional data, develop a consensus process to bin genes and variants into categories of clinical actionability, identify clinically relevant variants for consideration for clinical use, and systematically disseminate this information including creating a resource that is compatible with standards based electronic health record systems. Engaging the numerous individual research efforts and relevant stakeholders including genetics researchers, bioinformaticians, clinicians and medical institutions, professional societies, and regulatory agencies, is paramount for shaping such a comprehensive and useful resource.
Acknowledgments
Dr. David Mrazek passed away May 6, 2013. He was an important contributor to the ClinAction Workshop and a trailblazer in the field of psychiatric pharmacogenomices. David was the chairman of the Department of Psychiatry and Psychology at the Mayo Clinic and a professor of psychiatry at the Mayo Clinic College of Medicine.
Biography
Author Biographies: Dr. Erin Ramos is a genetic epidemiologist in the Division of Genomic Medicine, National Human Genome Research Institute (NHGRI). She manages a portfolio of research in genomic medicine and population genomes, including the Clinical Genome Resource (ClinGen), and was the lead organizer for the Characterizing and Displaying Genetic Variants for Clinical Action (ClinAction) Workshop.
Corina Din-Lovinescu was a program analyst for the Genetic Association Information Network Data Access Committee and the Genomics and Randomized Trials Network (GARNET), two programs of the NHGRI. She was instrumental in the planning of the ClinAction Workshop and is now a medical student at the Touro College of Osteopathic Medicine.
Dr. Jonathan Berg is an Assistant Professor in the Department of Genetics at UNC Chapel Hill, where he maintains an active clinical practice in Cancer and Adult Genetics and pursues research related to the application of genomic sequencing in different aspects of clinical medicine. He is a co-PI or investigator on grants that explore the use of next-generation sequencing as a diagnostic tool, for augmented newborn screening, and for population screening of healthy adults.
Dr. Lisa D. Brooks manages the Genetic Variation Program in the Division of Genome Sciences at the NHGRI. She manages computational studies of genetic variation and its relation to phenotype, as well as the 1000 Genomes Project, ClinGen, and BD2K.
Dr. Audrey Duncanson is a Senior Portfolio Developer, Wellcome Trust. She helps develop and implement strategic funding initiatives in genetics, genomics and the molecular sciences, and oversees the portfolio in these areas. Her current responsibilities include the Wellcome Trust Case Control Consortium, the UK10K sequencing project and H3 Africa.
Dr. Michael Dunn is Head of the Genetic and Molecular Sciences team in Science Funding. Having obtained a PhD in biochemistry at the University of Cambridge, he went on to work on the genetics of type 1 diabetes at the Wellcome Trust Centre for Human Genetics in Oxford. Michael joined the Trust in 2000 where he manages and develops its scientific portfolio and community of investigators to enable the Trust to achieve its mission.
Dr. Peter Good is the deputy director for the Division of Genomic Sciences, NHGRI where he advises NHGRI leaders on bioinformatics as a key component of genomic research. Dr. Good oversaw development of UniProt, a database of protein sequence and functional information derived from genome sequencing projects and guided other NHGRI projects including The Cancer Genome Atlas, the Knockout Mouse Project, and the ENCyclopedia of DNA Elements (ENCODE).
Dr. Tim Hubbard is a Professor of Bioinformatics, Head of Department of Medical and Molecular Genetics at King's College London and overall Director of Bioinformatics for King's Health Partners/King's College London. He also has a part-time appointment as Head of Bioinformatics at Genomics England, the company set up by the Government to help deliver the 100k Genome Project.
Gail Jarvik, M.D., Ph.D. is an Internist, Medical Geneticist, Head of Medical Genetics, the Arno G.
Motulsky Endowed Chair of Medicine, Professor of Genome Sciences at the University of Washington, and the Director of the Northwest Institute for Genetic Medicine. She is a practicing physician in both Internal Medicine and Medical Genetics with research focuses on the genetics of complex disease and the utility of high-throughput genomic data in clinical medicine.
Dr. Christopher O'Donnell, a cardiologist and genetic epidemiologist, serves as the Senior Advisor for Genome Research to the Director of the National Heart, Lung and Blood Institute (NHLBI). He is a Tenured Senior Investigator in the NHLBI Division of Intramural Research, where he serves as Chief of the Cardiovascular Epidemiology and Human Genomics Branch and Associate Director of the Framingham Heart Study.
Dr. Steve Sherry serves as the Chief of the Reference Collections Section, Information Engineering Branch at National Center for Biotechnology Information.
Dr. Naomi Aronson is the Executive Director of the Blue Cross and Blue Shield Association Technology Evaluation Center (TEC). Dr. Aronson has overseen TEC's development as a nationally recognized technology assessment program and an Evidence-based Practice Center (EPC) of the Agency for Healthcare Research and Quality (AHRQ).
Dr. Leslie Biesecker is a clinical and molecular geneticist with expertise in highly penetrant genetic and genomic disorders. He has been in the intramural research program of the NIH for 20 years and is a member of numerous editorial and advisory boards.
Dr. Bruce Blumberg is a clinical geneticist and Institutional Director of Graduate Medical Education at Kaiser Permanente Northern California. He is a Clinical Professor of Pediatrics at UCSF and an Adjunct Clinical Professor of Pediatrics at Stanford University School of Medicine.
Ned Calonge, MD, serves as President and CEO of The Colorado Trust and has worked closely with nonprofit organizations in every county across the state to improve health and well-being or all Coloradans. Dr. Calonge is a member and past President of the Colorado Medical Board, which licenses and regulates physicians. He teaches epidemiology, biostatistics and research methods at the University of Colorado Schools of Medicine and Public Health.
Dr. Helen Colhoun is a professor of public health at the University of Dundee, Scotland, UK. Her research includes studies that involve large scale linkage of electronic health record data to biospecimen resources used for genetic analyses of determinants of diabetes and diabetic complications.
Dr. Robert Epstein is an epidemiologist and faculty member at University of Maryland Medical System, Department of Epidemiology and Public Health. Prior to his current role Dr. Epstein was Medco's Chief Medical Officer for 13 years, where he led formulary development, clinical guideline development, drug information services, personalized medicine programs, and client analytics and reporting.
Dr. Paul Flicek leads the Vertebrate Genomics Team at the European Bioinformatics Institute and heads the cluster of resources focused on genes, genomes and variation. He is a senior scientist of the European Molecular Biology Laboratory.
Erynn S. Gordon, MS, LCGC is a board certified genetic counselor with over 12 years of clinical and research experience in neurology, oncology, and genetics. Before joining Invitae, Erynn managed the Coriell Personalized Medicine Collaborative, examining the utility of personalized medicine.
Eric D. Green, MD, PhD, became the third Director of the NHGRI in December 2009. He is responsible for providing overall leadership of the NHGRI's research portfolio and other initiatives. Immediately prior to this appointment, he was the Scientific Director of NHGRI, a position he had held since 2002, and Director of the NIH Intramural Sequencing Center (NISC).
Robert C. Green, MD, MPH is a medical geneticist at Brigham and Women's Hospital and Harvard Medical School. Dr. Green leads a research program in translational genomics and leads and co-leads respectively the NIH funded MedSeq and BabySeq Projects, research projects exploring the integration of genome sequencing into the practice of medicine.
Dr. Matthew Hurles earned his degree in biochemistry at Oxford University, UK, and PhD in Leicester, UK. He was a Research Fellow at the McDonald Institute for Archaeological Research at Cambridge University, UK, analyzing genetic variation with the aim of improving our understanding of the human past. He is now at the Wellcome Trust Sanger Institute near Cambridge, UK, investigating the unusual evolutionary dynamics of recently duplicated genomic regions.
Kensaku Kawamoto, MD, PhD, is Associate Chief Medical Information Officer for the University of Utah Health Sciences Center. He is also its Director of Knowledge Management and Mobilization and an Assistant Professor of Biomedical Informatics.
Dr. William A. Knaus is the Director of the Applied Genomics Research & Health Heritage Family Medical History Project, Center for Biomedical Research Informatics NorthShore University Health Systems, Evanston, Illinois.
Dr. Ledbetter is a board-certified Clinical Cytogeneticist whose research interest is in the development and application of new genetic/genomic technologies to improve early diagnosis for neurodevelopmental disorders such as autism and intellectual disabilities. He currently serves as Executive Vice President and Chief Scientific Officer at Geisinger Health Systems where he is investigating the clinical utility of genomic medicine in an integrated healthcare “laboratory”.
Howard P. Levy, MD, PhD is an Assistant Professor at Johns Hopkins University. He is board certified in Internal Medicine and Clinical Genetics, and specializes in general primary care medicine, genetics of common complex disease, and the care of adults with Mendelian genetic disease.
Dr. Elaine Lyon is an associate professor of Pathology at the University of Utah School of Medicine. In addition, she is a medical director of Genetics/Genomics at ARUP Laboratories, a national reference laboratory owned by the University of Utah.
Dr. Donna Maglott has been a staff scientist at NCBI since 1998 where she contributed to diverse projects such as RefSeq, Gene, OMIM, Map Viewer, Eukaryotic Genome Annotation, UniGene, UniSTS, dbSNP, GTR, ClinVar, and MedGen.
Dr. Howard McLeod is the Medical Director of the DeBartolo Family Personalized Medicine Institute at the Moffitt Cancer Center in Tampa, Florida. He is also a Senior Member of the Division of Population Sciences and an Endowed Research Chair of the state of Florida.
Dr. Nazneen Rahman, MD, PhD, is Professor of Human Genetics at Institute of Cancer Research, London and Head of Clinical Cancer Genetics Unit at the Royal Marsden Hospital. Her research is focussed on the identification and clinical translation of disease predisposition genes.
Gurvaneet S. Randhawa, MD, MPH, is a past director of the U.S. Preventive Services Task Force program. He provides scientific direction to AHRQ grants that are building an clinical electronic infrastructure to collect prospective, patient-centered outcomes data used for comparative effectiveness research, patient-centered outcomes research and quality improvement of clinical care. He is also a senior AHRQ adviser on clinical genomics and personalized medicine.
Catherine Wicklund is an Associate Professor and Director of the Graduate Program in Genetic Counseling at Northwestern University Feinberg School of Medicine, Center for Genetic Medicine. She is the National Society of Genetic Counselors representative on the IOM Roundtable on Translating Genomic-Based Research for Health and is an appointed member of the Discretionary Advisory Committee on Heritable Disorders in Newborns and Children.
Dr. Teri Manolio, a physician and epidemiologist, serves as the Director of the Division of Genomic Medicine, NHGRI. Dr. Manolio is leading efforts to support research translating genomic discoveries into more effective diagnoses, preventive measures, and treatments.
Dr. Rex L. Chisholm is the Adam and Richard Lind Professor of Human Genetics at Northwestern University Feinberg School of Medicine. His research focuses on biomedical informatics and the contributions of genetic variation to human disease and therapeutic outcomes.
Dr. Williams is the director of the Genomic Medicine Institute (GMI) for Geisinger Health System. The GMI is responsible for implementation of genomic medicine for the system. Dr. Williams was co-chair of the Characterizing and Displaying Genetic Variants for Clinical Action Workshop.
References
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Chang K, Hawes A, Lewis LR, Ren Y, Wheeler D, Muzny DM, Barnes C, Darvishi K, Hurles M, Korn JM, Kristiansson K, Lee C, McCarrol SA, Nemesh J, Keinan A, Montgomery SB, Pollack S, Price AL, Soranzo N, Gonzaga-Jauregui C, Anttila V, Brodeur W, Daly MJ, Leslie S, McVean G, Moutsianas L, Nguyen H, Zhang Q, Ghori MJ, McGinnis R, McLaren W, Takeuchi F, Grossman SR, Shlyakhter I, Hostetter EB, Sabeti PC, Adebamowo CA, Foster MW, Gordon DR, Licinio J, Manca MC, Marshall PA, Matsuda I, Ngare D, Wang VO, Reddy D, Rotimi CN, Royal CD, Sharp RR, Zeng C, Brooks LD, McEwen JE. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson SJ, Clark EH, Varugheese M, Baxter S, Babb LJ, Rehm HL. Communicating new knowledge on previously reported genetic variants. Genetics in medicine : official journal of the American College of Medical Genetics. 2012 doi: 10.1038/gim.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Khoury MJ, Evans JP. Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genetics in medicine : official journal of the American College of Medical Genetics. 2011;13(6):499–504. doi: 10.1097/GIM.0b013e318220aaba. [DOI] [PubMed] [Google Scholar]
- Epstein RS, Moyer TP, Aubert RE, O Kane DJ, Xia F, Verbrugge RR, Gage BF, Teagarden JR. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study) J Am Coll Cardiol. 2010;55(25):2804–12. doi: 10.1016/j.jacc.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Evans JP, Meslin EM, Marteau TM, Caulfield T. Genomics. Deflating the genomic bubble. Science. 2011;331(6019):861–862. doi: 10.1126/science.1198039. [DOI] [PubMed] [Google Scholar]
- Foster MW, Mulvihill JJ, Sharp RR. Evaluating the utility of personal genomic information. Genetics in medicine : official journal of the American College of Medical Genetics. 2009;11(8):570–574. doi: 10.1097/GIM.0b013e3181a2743e. [DOI] [PubMed] [Google Scholar]
- Fullerton SM, Wolf WA, Brothers KB, Clayton EW, Crawford DC, Denny JC, Greenland P, Koenig BA, Leppig KA, Lindor NM, McCarty CA, McGuire AL, McPeek Hinz ER, Mirel DB, Ramos EM, Ritchie MD, Smith ME, Waudby CJ, Burke W, Jarvik GP. Return of individual research results from genome-wide association studies: experience of the Electronic Medical Records and Genomics (eMERGE) Network. Genetics in medicine : official journal of the American College of Medical Genetics. 2012;14(4):424–431. doi: 10.1038/gim.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollust SE, Gordon ES, Zayac C, Griffin G, Christman MF, Pyeritz RE, Wawak L, Bernhardt BA. Motivations and perceptions of early adopters of personalized genomics: perspectives from research participants. Public health genomics. 2012;15(1):22–30. doi: 10.1159/000327296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, Manolio TA, Sanderson SC, Kannry J, Zinberg R, Basford MA, Brilliant M, Carey DJ, Chisolm RL, Chute CG, Connolly JJ, Crosslin D, Denny JC, Gallego CJ, Haines JL, Hakonarson H, Harley J, Jarvik GP, Kohane I, Kullo IJ, Larson EB, McCarty C, Ritchie MD, Roden D, Smith ME, Bottinger EP, Williams MS. The Electronic Medical Records and Genomics (eMERGE) Network: Past, Present and Future. Genet Med advance online publication. 2013 Jun 6; doi: 10.1038/gim.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ED, Guyer MS. Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470(7333):204–213. doi: 10.1038/nature09764. [DOI] [PubMed] [Google Scholar]
- Green RC, Berg JS, Berry GT, Biesecker LG, Dimmock DP, Evans JP, Grody WW, Hegde MR, Kalia S, Korf BR, Krantz I, McGuire AL, Miller DT, Murray MF, Nussbaum RL, Plon SE, Rehm HL, Jacob HJ. Exploring concordance and discordance for return of incidental findings from clinical sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2012;14(4):405–410. doi: 10.1038/gim.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse SD, Kalman L, Khoury MJ. Evaluation of the validity and utility of genetic testing for rare diseases. Adv Exp Med Biol. 2010;686:115–131. doi: 10.1007/978-90-481-9485-8_8. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(23):9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoes AW. Clinical epidemiology: principles, methods, and applications for clinical research. Sudbury, MA: Jones and Bartlett Publishers; 2009. [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437(7063):1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury MJ, Gwinn M, Bowen MS, Dotson WD. Beyond Base Pairs to Bedside: A population Perspective on How Genomics Can Improve Health.”. American Journal of Public Health. 2012;102(1):34–7. doi: 10.2105/AJPH.2011.300299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Chisholm RL, Ozenberger B, Roden DM, Williams MS, Wilson R, Bick D, Bottinger EP, Brilliant MH, Eng C, Frazer KA, Korf B, Ledbetter DH, Lupski JR, Marsh C, Mrazek D, Murray MF, O'Donnell PH, Rader DJ, Relling MV, Shuldiner AR, Valle D, Weinshilboum R, Green ED, Ginsburg GS. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013;15(4):258–267. doi: 10.1038/gim.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian AJ. Molecular genetic studies of complex phenotypes. Translational research : the journal of laboratory and clinical medicine. 2012;159(2):64–79. doi: 10.1016/j.trsl.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masys DR, Jarvik GP, Abernethy NF, Anderson NR, Papanicolaou GJ, Paltoo DN, Hoffman MA, Kohane IS, Levy HP. Technical desiderata for the integration of genomic data into Electronic Health Records. J Biomed Inform. 2012;45(3):419–422. doi: 10.1016/j.jbi.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod HL. Cancer pharmacogenomics: early promise, but concerted effort needed. Science. 2013;339(6127):1563–1566. doi: 10.1126/science.1234139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos E, Rotimi C. The A's, G's, C's, and T's of health disparities. BMC medical genomics. 2009;2:29. doi: 10.1186/1755-8794-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clinical pharmacology and therapeutics. 2011;89(3):464–467. doi: 10.1038/clpt.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JS, Christensen KD, Green RC. Using Alzheimer's disease as a model for genetic risk disclosure: implications for personal genomics. Clinical genetics. 2011;80(5):407–414. doi: 10.1111/j.1399-0004.2011.01739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S. Results of a survey of an online physician community regarding use of electronic medical records in office practices. J Med Pract Manage. 2009;24(4):254–256. [PubMed] [Google Scholar]
- Stack CB, Gharani N, Gordon ES, Schmidlen T, Christman MF, Keller MA. Genetic risk estimation in the Coriell Personalized Medicine Collaborative. Genetics in medicine : official journal of the American College of Medical Genetics. 2011;13(2):131–139. doi: 10.1097/GIM.0b013e318201164c. [DOI] [PubMed] [Google Scholar]
- Starren J, Williams MS, Bottinger EP. Crossing the omic chasm: a time for omic ancillary systems. JAMA. 2013;309(12):1237–1238. doi: 10.1001/jama.2013.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teutsch SM, Bradley LA, Palomaki GE, Haddow JE, Piper M, Calonge N, Dotson WD, Douglas MP, Berg AO. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genetics in medicine : official journal of the American College of Medical Genetics. 2009;11(1):3–14. doi: 10.1097/GIM.0b013e318184137c. [DOI] [PMC free article] [PubMed] [Google Scholar]