

Abstract
In the title compound, C16H17N3OS, the dihedral angle between the planes of the benzene and pyridine rings is 71.33 (15)°. An intramolecular N—H⋯O hydrogen bond is present. In the crystal, weak aromatic C—H⋯O hydrogen bonds link the molecules into chains extending along a.
Keywords: crystal structure, hydrogen bonding, thiourea compounds, thiocarbonyl groups, benzamide
Related literature
For related structures, see: Saeed & Flörke (2007 ▶); Yusof et al. (2008 ▶, 2011 ▶); Shoukat et al. (2007 ▶); Hassan et al. (2008a
▶,b
▶,c
▶). For standard bond lengths, see: Allen et al. (1987 ▶). For graph-set analysis, see Bernstein et al. (1995 ▶).
Experimental
Crystal data
C16H17N3OS
M r = 299.39
Monoclinic,
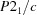
a = 16.0467 (12) Å
b = 4.8824 (4) Å
c = 23.0403 (18) Å
β = 124.997 (5)°
V = 1478.7 (2) Å3
Z = 4
Mo Kα radiation
μ = 0.22 mm−1
T = 100 K
0.47 × 0.20 × 0.14 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2005 ▶) T min = 0.903, T max = 0.980
12777 measured reflections
3409 independent reflections
2221 reflections with I > 2σ(I)
R int = 0.082
Refinement
R[F 2 > 2σ(F 2)] = 0.068
wR(F 2) = 0.182
S = 1.04
3409 reflections
199 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.66 e Å−3
Δρmin = −0.44 e Å−3
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: SAINT (Bruker, 2005 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814016377/zs2306sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814016377/zs2306Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814016377/zs2306Isup3.cml
CCDC reference: 1014035
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H1N2⋯O2 | 0.87 (4) | 1.90 (3) | 2.645 (3) | 143 (3) |
| C14—H14A⋯O2i | 0.95 | 2.51 | 3.421 (4) | 161 |
Symmetry code: (i)  .
.
Acknowledgments
The authors thank Universiti Sains Malaysia for a research grant (No. PKIMIA846017) which partially supported this work.
supplementary crystallographic information
S1. Comment
In the title compound, C16H17N3SO (Fig. 1), the bond lengths and angles are generally normal compared to those in N-alkyl-N-benzoylthiourea compounds (Allen et al., 1987). The bond lengths of the carbonyl and thiocarbonyl groups [C7—O2 = 1.229 (5) Å and C8—S1 = 1.677 (4) Å, respectively] have typical C═O and C═S double-bond character (Yusof et al. 2011). However, the thiocarbonyl is longer compared to the typical C═S bond which is 1.660 (2) Å. The C—N bond lengths for the title compound [C7—N1 = 1.375 (4) Å, C8—N1 = 1.397 (4) Å, C9—N2 = 1.460 (4) Å, C11—N3 = 1.335 (4), C15—N3 = 1.351 (4) Å] are all shorter than the average C—N single bond length [1.472 (5) Å], thus showing varying degrees of single bond character (Yusof et al. 2008). These bond features in the structure are presumed as a result of the intramolecular H-bonding interactions "locking" the molecule into a planar six-membered ring structure and are consistent with the expected delocalization in the title compound, confimed by the C9—N2—C8 and C8—N1—C7 bond angles [125.0 (3) and 128.8 (3)°, respectively], showing sp2 hybridization on the N2 and N1 atoms. The molecule maintains its cis–trans configuration with respect to the position of the methyl benzene and ethyl pyridine groups relative to the thiocarbonyl sulfur atom across the N1—C7 and N2—C8 bonds, respectively (Hassan et al. (2008b,2008c)). The conformation of the molecule with respect to the thiocarbonyl and carbonyl moieties is twisted, as refleced by the torsion angles [C8–N1–C7–O2, C7–N1–C8–N2 and C7—N1—C8—S1: 2.1 (5), -4.4 (4) and 175.9 (2)°, respectively. The angle between the benzene and pyridine rings is 71.33 (15)°. The N2 H-atom forms bifurcated intramolecular interactions with both a carbonyl O-atom and the pyridine N-atom (Table 1): a hydrogen bond with O2 (N2—H···O2) and an interaction with N3 (N2—H···N3), giving cyclic motifs [graph sets S6 (Bernstein et al., 1995)]. Present also are weak intramolecular C1—H···O2 and C9—H···S1 interactions [graph set S(5)]. In the crystal, molecules are connected through weak intermolecular C14—H···O2 hydrogen-bonding interactions, giving one-dimensional chain structures extending along x (Fig. 2). The N1—H1N1 group has no acceptor in the crystal.
S2. Experimental
S2.1. Synthesis and crystallization
Freshly prepared substituted p-benzoyl chloride (13 mmol) was added dropwise to a stirred acetone solution (30 ml) of ammonium thiocyanate (13 mmol). The mixture was stirred for 10 min. A solution of 2-(2-aminethylpyridine) in acetone was added and the reaction mixture was refluxed for 3 h., after which the solution was poured into a beaker containing some ice cubes. The resulting precipitate was collected by filtration, washed several times with a cold ethanol/water mixture and purified by recrystallization from ethanol (Hassan et al., 2008a). Yield 65%; white transparent crystals, m.p. 126.3 °C. Anal Calc. for C16 H17 N3 O S: C, 64.9; H, 5.6; N, 15.9; S, 8.2%. Found: C, 64.8; H, 5.7; N, 14.8; S, 8.7%.
S2.2. Refinement
The H-atoms on the N atoms were located in a difference-Fourier and were fully refined. All other H-atoms were positioned geometrically and refined using a riding model with C—H = 0.95–0.99 Å and Uiso(H) = 1.2Ueq(aromatic C), 1.5Ueq(methyl C) and 1.2Ueq(methylene C).
Figures
Fig. 1.

The molecular structure of the title compound, showing 50% probability displacement ellipsoids and the atom-numbering scheme. Intramolecular interactions are shown as dashed lines.
Fig. 2.

The crystal packing of the title compound viewed down the c axis. Hydrogen bonds are shown as dashed lines.
Crystal data
| C16H17N3OS | F(000) = 632 |
| Mr = 299.39 | Dx = 1.345 Mg m−3 |
| Monoclinic, P21/c | Melting point: 399.3 K |
| Hall symbol: -P 2ybc | Mo Kα radiation, λ = 0.71073 Å |
| a = 16.0467 (12) Å | θ = 2.6–25.5° |
| b = 4.8824 (4) Å | µ = 0.22 mm−1 |
| c = 23.0403 (18) Å | T = 100 K |
| β = 124.997 (5)° | Block, colourless |
| V = 1478.7 (2) Å3 | 0.47 × 0.20 × 0.14 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD diffractometer | 3409 independent reflections |
| Radiation source: fine-focus sealed tube | 2221 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.082 |
| φ and ω scans | θmax = 27.6°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2005) | h = −17→20 |
| Tmin = 0.903, Tmax = 0.980 | k = −6→6 |
| 12777 measured reflections | l = −30→24 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.068 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.182 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0899P)2] where P = (Fo2 + 2Fc2)/3 |
| 3409 reflections | (Δ/σ)max < 0.001 |
| 199 parameters | Δρmax = 0.66 e Å−3 |
| 0 restraints | Δρmin = −0.44 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.72652 (6) | 0.04528 (17) | 0.87044 (4) | 0.0286 (3) | |
| O2 | 0.78657 (14) | 0.5226 (4) | 0.72802 (11) | 0.0272 (5) | |
| N1 | 0.69712 (19) | 0.3898 (5) | 0.77218 (13) | 0.0227 (6) | |
| N2 | 0.84392 (17) | 0.1337 (5) | 0.82384 (13) | 0.0211 (5) | |
| N3 | 1.02858 (17) | 0.3126 (5) | 0.84401 (12) | 0.0229 (6) | |
| C1 | 0.6442 (2) | 0.9043 (6) | 0.63777 (15) | 0.0236 (7) | |
| H1A | 0.7034 | 0.8759 | 0.6386 | 0.028* | |
| C2 | 0.5737 (2) | 1.0965 (6) | 0.59246 (15) | 0.0250 (7) | |
| H2A | 0.5853 | 1.1996 | 0.5627 | 0.030* | |
| C3 | 0.4852 (2) | 1.1422 (6) | 0.58956 (15) | 0.0227 (6) | |
| C4 | 0.4718 (2) | 0.9901 (6) | 0.63468 (16) | 0.0246 (7) | |
| H4A | 0.4128 | 1.0199 | 0.6341 | 0.030* | |
| C5 | 0.5426 (2) | 0.7954 (6) | 0.68073 (15) | 0.0245 (7) | |
| H5A | 0.5315 | 0.6933 | 0.7108 | 0.029* | |
| C6 | 0.6301 (2) | 0.7508 (6) | 0.68249 (14) | 0.0200 (6) | |
| C7 | 0.7113 (2) | 0.5479 (6) | 0.72911 (14) | 0.0203 (6) | |
| C8 | 0.7611 (2) | 0.1903 (6) | 0.82127 (15) | 0.0218 (6) | |
| C9 | 0.9261 (2) | −0.0462 (6) | 0.87572 (15) | 0.0240 (7) | |
| H9A | 0.9508 | −0.1556 | 0.8523 | 0.029* | |
| H9B | 0.9001 | −0.1743 | 0.8952 | 0.029* | |
| C10 | 1.0135 (2) | 0.1207 (6) | 0.93572 (15) | 0.0242 (7) | |
| H10A | 0.9874 | 0.2330 | 0.9578 | 0.029* | |
| H10B | 1.0650 | −0.0066 | 0.9723 | 0.029* | |
| C11 | 1.0649 (2) | 0.3085 (6) | 0.91261 (15) | 0.0224 (6) | |
| C12 | 1.1454 (2) | 0.4739 (6) | 0.96253 (16) | 0.0282 (7) | |
| H12A | 1.1707 | 0.4633 | 1.0112 | 0.034* | |
| C13 | 1.1880 (2) | 0.6550 (7) | 0.93981 (18) | 0.0315 (8) | |
| H13A | 1.2426 | 0.7710 | 0.9728 | 0.038* | |
| C14 | 1.1503 (2) | 0.6643 (6) | 0.86918 (18) | 0.0323 (8) | |
| H14A | 1.1775 | 0.7882 | 0.8523 | 0.039* | |
| C15 | 1.0716 (2) | 0.4885 (6) | 0.82314 (17) | 0.0269 (7) | |
| H15A | 1.0466 | 0.4919 | 0.7745 | 0.032* | |
| C16 | 0.4073 (2) | 1.3480 (6) | 0.53901 (16) | 0.0279 (7) | |
| H16C | 0.3579 | 1.3783 | 0.5505 | 0.042* | |
| H16D | 0.3721 | 1.2787 | 0.4904 | 0.042* | |
| H16A | 0.4412 | 1.5211 | 0.5431 | 0.042* | |
| H1N2 | 0.852 (2) | 0.242 (7) | 0.7974 (17) | 0.039 (10)* | |
| H1N1 | 0.651 (3) | 0.424 (9) | 0.775 (2) | 0.078 (15)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0251 (4) | 0.0384 (5) | 0.0310 (5) | 0.0068 (3) | 0.0211 (4) | 0.0085 (3) |
| O2 | 0.0175 (11) | 0.0343 (12) | 0.0359 (12) | 0.0052 (9) | 0.0189 (10) | 0.0070 (9) |
| N1 | 0.0166 (13) | 0.0302 (14) | 0.0268 (14) | 0.0042 (10) | 0.0157 (12) | 0.0023 (10) |
| N2 | 0.0151 (12) | 0.0249 (14) | 0.0253 (14) | 0.0030 (10) | 0.0127 (11) | 0.0040 (10) |
| N3 | 0.0185 (13) | 0.0269 (14) | 0.0266 (14) | 0.0036 (10) | 0.0149 (11) | 0.0030 (10) |
| C1 | 0.0173 (14) | 0.0322 (17) | 0.0246 (16) | −0.0003 (12) | 0.0140 (13) | 0.0001 (12) |
| C2 | 0.0212 (15) | 0.0317 (17) | 0.0258 (16) | 0.0013 (12) | 0.0156 (13) | 0.0055 (13) |
| C3 | 0.0184 (15) | 0.0252 (16) | 0.0234 (16) | 0.0012 (12) | 0.0114 (13) | −0.0001 (12) |
| C4 | 0.0180 (15) | 0.0286 (16) | 0.0311 (17) | 0.0010 (12) | 0.0163 (13) | 0.0005 (13) |
| C5 | 0.0235 (16) | 0.0284 (17) | 0.0273 (16) | 0.0017 (12) | 0.0180 (14) | 0.0038 (13) |
| C6 | 0.0154 (14) | 0.0229 (15) | 0.0214 (15) | −0.0028 (11) | 0.0104 (12) | −0.0031 (11) |
| C7 | 0.0165 (14) | 0.0234 (15) | 0.0212 (15) | −0.0030 (11) | 0.0109 (12) | −0.0029 (12) |
| C8 | 0.0173 (14) | 0.0265 (16) | 0.0222 (15) | 0.0005 (12) | 0.0116 (12) | 0.0002 (12) |
| C9 | 0.0208 (15) | 0.0247 (16) | 0.0303 (17) | 0.0046 (12) | 0.0169 (14) | 0.0056 (12) |
| C10 | 0.0180 (15) | 0.0315 (17) | 0.0238 (16) | 0.0037 (12) | 0.0124 (13) | 0.0037 (12) |
| C11 | 0.0156 (15) | 0.0248 (16) | 0.0285 (16) | 0.0051 (12) | 0.0137 (13) | 0.0030 (12) |
| C12 | 0.0216 (16) | 0.0360 (18) | 0.0268 (17) | −0.0014 (13) | 0.0137 (14) | −0.0002 (13) |
| C13 | 0.0214 (16) | 0.0293 (17) | 0.044 (2) | −0.0020 (13) | 0.0185 (15) | −0.0035 (14) |
| C14 | 0.0265 (17) | 0.0298 (18) | 0.052 (2) | 0.0030 (14) | 0.0292 (17) | 0.0059 (15) |
| C15 | 0.0246 (16) | 0.0330 (18) | 0.0327 (17) | 0.0063 (13) | 0.0220 (14) | 0.0052 (13) |
| C16 | 0.0229 (16) | 0.0327 (18) | 0.0286 (17) | 0.0048 (13) | 0.0150 (14) | 0.0027 (13) |
Geometric parameters (Å, º)
| S1—C8 | 1.678 (3) | C5—H5A | 0.9500 |
| O2—C7 | 1.228 (3) | C6—C7 | 1.493 (4) |
| N1—C7 | 1.376 (4) | C9—C10 | 1.523 (4) |
| N1—C8 | 1.397 (4) | C9—H9A | 0.9900 |
| N1—H1N1 | 0.80 (5) | C9—H9B | 0.9900 |
| N2—C8 | 1.325 (3) | C10—C11 | 1.519 (4) |
| N2—C9 | 1.460 (3) | C10—H10A | 0.9900 |
| N2—H1N2 | 0.87 (3) | C10—H10B | 0.9900 |
| N3—C11 | 1.335 (4) | C11—C12 | 1.392 (4) |
| N3—C15 | 1.351 (4) | C12—C13 | 1.391 (4) |
| C1—C2 | 1.376 (4) | C12—H12A | 0.9500 |
| C1—C6 | 1.393 (4) | C13—C14 | 1.374 (4) |
| C1—H1A | 0.9500 | C13—H13A | 0.9500 |
| C2—C3 | 1.402 (4) | C14—C15 | 1.386 (4) |
| C2—H2A | 0.9500 | C14—H14A | 0.9500 |
| C3—C4 | 1.390 (4) | C15—H15A | 0.9500 |
| C3—C16 | 1.502 (4) | C16—H16C | 0.9800 |
| C4—C5 | 1.391 (4) | C16—H16D | 0.9800 |
| C4—H4A | 0.9500 | C16—H16A | 0.9800 |
| C5—C6 | 1.399 (4) | ||
| C7—N1—C8 | 128.8 (2) | N2—C9—H9A | 109.5 |
| C7—N1—H1N1 | 119 (3) | C10—C9—H9A | 109.5 |
| C8—N1—H1N1 | 111 (3) | N2—C9—H9B | 109.5 |
| C8—N2—C9 | 125.0 (2) | C10—C9—H9B | 109.5 |
| C8—N2—H1N2 | 113 (2) | H9A—C9—H9B | 108.1 |
| C9—N2—H1N2 | 121 (2) | C11—C10—C9 | 114.1 (2) |
| C11—N3—C15 | 117.7 (3) | C11—C10—H10A | 108.7 |
| C2—C1—C6 | 121.1 (3) | C9—C10—H10A | 108.7 |
| C2—C1—H1A | 119.4 | C11—C10—H10B | 108.7 |
| C6—C1—H1A | 119.4 | C9—C10—H10B | 108.7 |
| C1—C2—C3 | 120.9 (3) | H10A—C10—H10B | 107.6 |
| C1—C2—H2A | 119.5 | N3—C11—C12 | 122.6 (3) |
| C3—C2—H2A | 119.5 | N3—C11—C10 | 117.9 (3) |
| C4—C3—C2 | 117.8 (3) | C12—C11—C10 | 119.5 (3) |
| C4—C3—C16 | 121.4 (3) | C13—C12—C11 | 118.7 (3) |
| C2—C3—C16 | 120.8 (3) | C13—C12—H12A | 120.6 |
| C3—C4—C5 | 121.8 (3) | C11—C12—H12A | 120.6 |
| C3—C4—H4A | 119.1 | C14—C13—C12 | 119.3 (3) |
| C5—C4—H4A | 119.1 | C14—C13—H13A | 120.3 |
| C4—C5—C6 | 119.7 (3) | C12—C13—H13A | 120.3 |
| C4—C5—H5A | 120.2 | C13—C14—C15 | 118.4 (3) |
| C6—C5—H5A | 120.2 | C13—C14—H14A | 120.8 |
| C1—C6—C5 | 118.7 (3) | C15—C14—H14A | 120.8 |
| C1—C6—C7 | 116.4 (2) | N3—C15—C14 | 123.3 (3) |
| C5—C6—C7 | 124.9 (2) | N3—C15—H15A | 118.4 |
| O2—C7—N1 | 122.0 (3) | C14—C15—H15A | 118.4 |
| O2—C7—C6 | 121.0 (3) | C3—C16—H16C | 109.5 |
| N1—C7—C6 | 117.0 (2) | C3—C16—H16D | 109.5 |
| N2—C8—N1 | 115.7 (2) | H16C—C16—H16D | 109.5 |
| N2—C8—S1 | 126.4 (2) | C3—C16—H16A | 109.5 |
| N1—C8—S1 | 117.8 (2) | H16C—C16—H16A | 109.5 |
| N2—C9—C10 | 110.5 (2) | H16D—C16—H16A | 109.5 |
| C6—C1—C2—C3 | −0.5 (5) | C9—N2—C8—N1 | 173.5 (3) |
| C1—C2—C3—C4 | 1.0 (4) | C9—N2—C8—S1 | −6.9 (4) |
| C1—C2—C3—C16 | −178.7 (3) | C7—N1—C8—N2 | −4.4 (4) |
| C2—C3—C4—C5 | −0.9 (4) | C7—N1—C8—S1 | 175.9 (2) |
| C16—C3—C4—C5 | 178.8 (3) | C8—N2—C9—C10 | −98.0 (3) |
| C3—C4—C5—C6 | 0.4 (4) | N2—C9—C10—C11 | −63.9 (3) |
| C2—C1—C6—C5 | −0.1 (4) | C15—N3—C11—C12 | −1.0 (4) |
| C2—C1—C6—C7 | 179.8 (3) | C15—N3—C11—C10 | 177.2 (3) |
| C4—C5—C6—C1 | 0.1 (4) | C9—C10—C11—N3 | 1.1 (4) |
| C4—C5—C6—C7 | −179.7 (3) | C9—C10—C11—C12 | 179.4 (3) |
| C8—N1—C7—O2 | 2.1 (5) | N3—C11—C12—C13 | 1.5 (4) |
| C8—N1—C7—C6 | −178.7 (3) | C10—C11—C12—C13 | −176.6 (3) |
| C1—C6—C7—O2 | 0.8 (4) | C11—C12—C13—C14 | −0.5 (4) |
| C5—C6—C7—O2 | −179.3 (3) | C12—C13—C14—C15 | −0.9 (4) |
| C1—C6—C7—N1 | −178.3 (3) | C11—N3—C15—C14 | −0.5 (4) |
| C5—C6—C7—N1 | 1.5 (4) | C13—C14—C15—N3 | 1.5 (4) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H1N2···O2 | 0.87 (4) | 1.90 (3) | 2.645 (3) | 143 (3) |
| N2—H1N2···N3 | 0.87 (4) | 2.41 (4) | 2.860 (4) | 113 (3) |
| C1—H1A···O2 | 0.95 | 2.42 | 2.751 (4) | 100 |
| C9—H9B···S1 | 0.99 | 2.72 | 3.166 (4) | 108 |
| C14—H14A···O2i | 0.95 | 2.51 | 3.421 (4) | 161 |
Symmetry code: (i) −x+2, y+1/2, −z+3/2.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: ZS2306).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Bruker (2005). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Hassan, I. N., Yamin, B. M. & Kassim, M. B. (2008a). Acta Cryst. E64, o1727. [DOI] [PMC free article] [PubMed]
- Hassan, I. N., Yamin, B. M. & Kassim, M. B. (2008b). Acta Cryst. E64, o2083. [DOI] [PMC free article] [PubMed]
- Hassan, I. N., Yamin, B. M. & Kassim, M. B. (2008c). Acta Cryst. E64, o2167. [DOI] [PMC free article] [PubMed]
- Saeed, A. & Flörke, U. (2007). Acta Cryst. E63, o4259.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shoukat, N., Rauf, M. K., Bolte, M. & Badshah, A. (2007). Acta Cryst. E63, o3207.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Yusof, M. S. M., Muharam, S. H., Kassim, M. B. & Yamin, B. M. (2008). Acta Cryst. E64, o1137. [DOI] [PMC free article] [PubMed]
- Yusof, M. S. M., Wong, S. T. & Yamin, B. M. (2011). Acta Cryst. E67, o2483. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814016377/zs2306sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814016377/zs2306Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814016377/zs2306Isup3.cml
CCDC reference: 1014035
Additional supporting information: crystallographic information; 3D view; checkCIF report