

Despite their different compositions and space groups, the irregular KF8 coordination polyhedra of the potassium cations in these structures are almost identical. The layer stacking sequences are AAA… in the p-methoxy compound and ABAB… in the p-fluoro compound.
Keywords: crystal structure, boron, Lewis acid, layered structure
Abstract
The title compounds, K+·C7H7BF3O−, (I), and K+·C6H4BF4 −, (II), are molecular salts containing para-substituted phenyltrifluoridoborate anions. In each compound, the B atom adopts a distorted tetrahedral BCF3 geometry. Despite their different compositions and space groups, the irregular KF8 coordination polyhedra of the potassium cations in the structures are almost identical. These polyhedra share faces and edges, generating infinite (010) layers in (I) and infinite (001) layers in (II). In (I), adjacent layers are stacked in an AAA… fashion, whereas in (II), they are stacked in an ABAB… sequence.
Chemical context
The phenyltrifluoridoborate anion is an interesting intermediate species between the well-known tetrafluoridoborate (BF4
−) and tetraphenylborate [B(C6H5)4
−] ions (Conole et al., 1995 ▸) and may serve as a bulky charge-balancing anion (Quach et al., 2001 ▸; Fei et al., 2010 ▸). As part of our studies in this area, we now describe the syntheses and structures of the para-substituted phenyltrifluoridoborate salts K+C7H7BF3O− (I) and K+C6H4BF4
− (II).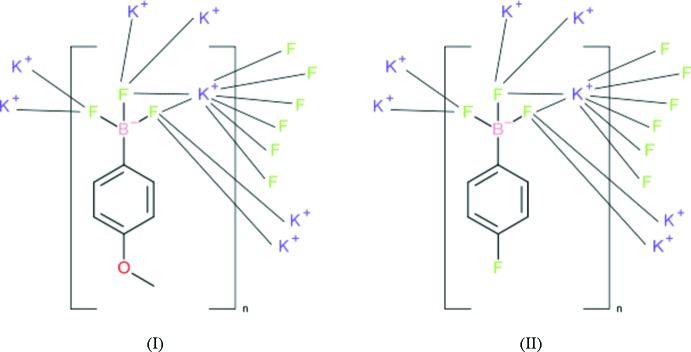
Structural commentary
Compound (I) comprises one cation and one anion in the asymmetric unit (Fig. 1 ▸). In the anion, the C7 atom of the methoxy group is close to coplanar with the benzene ring [displacement = 0.048 (2) Å]. The B atom adopts its expected tetrahedral BF3C geometry (Conole et al., 1995 ▸) and the C1—B1 bond length of 1.5987 (18) Å is consistent with previous data (Quach et al., 2001 ▸). One of the B—F bonds (to F1) in (I) is notably longer than the other two, which might reflect the different modes of coordination of the fluorine atoms to the potassium ions. The F—B—F bond angles (mean = 105.7°) are significantly smaller than the C—B—F angles (mean = 113.0°). F1 is displaced by −1.427 (2) Å from the plane of the benzene ring and F2 and F3 are displaced in the opposite sense, by 0.715 (2) and 0.252 (2) Å, respectively.
Figure 1.

The asymmetric unit of (I) showing 50% displacement ellipsoids.
The potassium ion in (I) is coordinated by eight fluorine atoms, with one of the K—F bonds substantially longer than the others (Table 1 ▸): the next-nearest F atom is over 4 Å distant. The coordination geometry of the K+ ion, which arises from one tridentate, one bidentate and three monodentate BF3 − groups, is irregular and highly asymmetric (Fig. 2 ▸), with five of the F atoms forming an approximate plane and the other three (arising from one BF3 group) lying to one side. The metal ion is displaced by 1.00 Å from the geometric centroid of the eight F atoms. In terms of the F atoms in (I), F1 bonds to three different metal ions (mean K—F = 2.734 Å), generating a distorted FBK3 tetrahedron, whereas F2 bonds to two K+ ions (mean K—F = 2.755 Å) in an FBK2 distorted T-shape. If the geometry around F3 is not merely deemed to be irregular, it could be described as an FBK3 trigonal-based pyramid, with the long K—F bond (Table 1 ▸) as the apex (mean K—F = 2.963 Å). The extended structure in (I) consists of (010) sheets in which the KF8 polyhedra share faces in the [100] direction and edges in [001]: the shortest K⋯K separation is 4.4523 (4) Å.
Table 1. Selected bond lengths (Å) for (I) .
| K1—F3i | 2.6156 (10) | K1—F2iv | 2.8885 (8) |
| K1—F2ii | 2.6211 (7) | K1—F3iv | 3.4886 (9) |
| K1—F1iii | 2.6550 (10) | B1—F3 | 1.4162 (19) |
| K1—F1iv | 2.7568 (10) | B1—F2 | 1.4196 (14) |
| K1—F3 | 2.7836 (8) | B1—F1 | 1.4403 (17) |
| K1—F1 | 2.7887 (8) |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  .
.
Figure 2.

The coordination of the potassium ion in (I). See Table 1 ▸ for symmetry codes.
The asymmetric unit of compound (II) also consists of an ion-pair (Fig. 3 ▸). The geometry of the anion in (II) is very similar to that of the equivalent species in (I): the C1—B1 bond length is 1.590 (2) Å and the mean F—B—F and C—B—F bond angles are 105.5 and 113.2°, respectively. The displacements of F1, F2 and F3 from the benzene-ring plane are −1.386 (2), 0.813 (3) and 0.131 (3) Å, respectively. As seen for (I), the B1—F1 bond in (II) is noticeably longer than the B1—F2 and B1—F3 bonds.
Figure 3.

The asymmetric unit of (II) showing 50% displacement ellipsoids.
It is notable that the K+ ion in (II) adopts a very similar coordination geometry (Table 2 ▸) to the equivalent species in (I), despite the different space groups. Again, a very asymmetric KF8 coordination polyhedron (Fig. 4 ▸) arises from one tridentate, one bidentate and three monodentate anions; one K—F bond is much longer than the others and the potassium ion is displaced by 0.98 Å from the geometric centroid of the fluorine atoms.
Table 2. Selected bond lengths (Å) for (II) .
| K1—F3i | 2.6116 (10) | K1—F2iv | 2.8853 (10) |
| K1—F2ii | 2.6159 (9) | K1—F3iv | 3.3927 (10) |
| K1—F1iii | 2.6527 (9) | B1—F2 | 1.4166 (17) |
| K1—F1iv | 2.7612 (10) | B1—F3 | 1.4182 (19) |
| K1—F3 | 2.7732 (9) | B1—F1 | 1.4393 (18) |
| K1—F1 | 2.8050 (9) |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  .
.
Figure 4.

The coordination of the potassium ion in (II). See Table 2 ▸ for symmetry codes.
The extended structure of (II) consists of (001) sheets [rather than (010) sheets, as seen in (I)] of face- and edge-sharing KF8 groups with the same topology as in (I): the shortest K⋯K separation is 4.4255 (5) Å.
Supramolecular features
In (I) the methoxyphenyl groups lie roughly normal to (010). When the packing is viewed along [101] (Fig. 5 ▸), it may be seen that adjacent benzene ring planes are rotated by 90°, which facilitates the formation of a weak edge-to-face intra-sheet C—H⋯π interaction (Table 3 ▸). An intra-sheet C2—H2⋯F2 hydrogen bond also occurs. The only possible inter-sheet interaction in (I) is an extremely weak C—H⋯O hydrogen bond with an H⋯O separation essentially the same as the van der Waals separation of these species. The layer-stacking sequence for (I) is AAA….
Figure 5.

The unit-cell packing in (I) viewed approximately down [101].
Table 3. Hydrogen-bond geometry (Å, °) for (I) .
Cg1 is the centroid of the C1–C6 benzene ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2⋯F2v | 0.95 | 2.50 | 3.3359 (17) | 147 |
| C7—H7a⋯O1vi | 0.98 | 2.72 | 3.496 (1) | 137 |
| C3—H3⋯Cg1v | 0.95 | 2.85 | 3.7171 (15) | 152 |
Symmetry codes: (v)  ; (vi)
; (vi)  .
.
When the crystal structure of (II) is viewed down [110] (Fig. 6 ▸), adjacent aromatic rings show the same 90° rotation as they do in (I), but the only directional interaction identified is an intralayer weak C—H⋯F hydrogen bond (Table 4 ▸) and there are no C—H⋯π interactions. There are no identified inter-layer interactions and the stacking sequence is ABAB….
Figure 6.

The unit-cell packing in (II) viewed approximately down [110].
Table 4. Hydrogen-bond geometry (Å, °) for (II) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C6—H6⋯F2v | 0.95 | 2.53 | 3.4099 (19) | 154 |
Symmetry code: (v)  .
.
Database survey
Compound (I) is closely related to K+.C6H5BF3 − (Conole et al., 1995 ▸), (III). Compounds (I) and (III) have the same space group and a similar unit cell, extended in the b-axis direction for (I) to accommodate the methoxy group. The potassium ion in (III) has almost the same KF8 coordination geometry as the equivalent species in (I) and (II) described above. In (III), weak edge-to-face C—H⋯π interactions occur between approximately perpendicular aromatic rings, as they do in (I). As already noted, the C6H5BF3 − anion has found use as a bulky charge-balancing species (Quach et al., 2001 ▸; Fei et al., 2010 ▸).
Synthesis and crystallization
(I) and (II) were received as commercial samples from Aldrich and recrystallized from ethanol solution, yielding colourless blocks.
Refinement
The H atoms were placed in idealized positions (C—H = 0.95–0.98 Å) and refined as riding atoms with U iso(H) = 1.2U eq(C) or 1.5U eq(methyl C). The methyl group in (I) was allowed to rotate, but not to tip, to best fit the electron density.. Experimental details are given in Table 5 ▸.
Table 5. Experimental details.
| (I) | (II) | |
|---|---|---|
| Crystal data | ||
| Chemical formula | K+·C7H7BF3O− | K+·C6H4BF4 − |
| M r | 214.04 | 202.00 |
| Crystal system, space group | Orthorhombic, P c a21 | Orthorhombic, P b c a |
| Temperature (K) | 120 | 100 |
| a, b, c (Å) | 7.1347 (2), 17.2819 (7), 7.3289 (3) | 7.1317 (5), 7.3757 (5), 29.129 (2) |
| V (Å3) | 903.66 (6) | 1532.22 (18) |
| Z | 4 | 8 |
| Radiation type | Mo Kα | Mo Kα |
| μ (mm−1) | 0.59 | 0.70 |
| Crystal size (mm) | 0.52 × 0.15 × 0.15 | 0.07 × 0.05 × 0.01 |
| Data collection | ||
| Diffractometer | Rigaku CCD | Rigaku CCD |
| Absorption correction | Multi-scan (SADABS; Bruker, 2012 ▸) | Multi-scan (SADABS; Bruker, 2012 ▸) |
| T min, T max | 0.750, 0.917 | 0.953, 0.993 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 5789, 1833, 1822 | 9537, 1726, 1435 |
| R int | 0.026 | 0.037 |
| (sin θ/λ)max (Å−1) | 0.649 | 0.649 |
| Refinement | ||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.021, 0.059, 1.10 | 0.027, 0.065, 1.06 |
| No. of reflections | 1833 | 1726 |
| No. of parameters | 120 | 109 |
| No. of restraints | 1 | 0 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.24, −0.20 | 0.27, −0.23 |
| Absolute structure | Flack (1983 ▸), 712 Friedel pairs | – |
| Absolute structure parameter | 0.02 (3) | – |
Supplementary Material
Crystal structure: contains datablock(s) I, II, global. DOI: 10.1107/S1600536814009684/wm0001sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814009684/wm0001Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S1600536814009684/wm0001IIsup3.hkl
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
We thank the National Crystallography Service (University of Southampton) for the data collections.
supplementary crystallographic information
(I) Potassium trifluorido(4-methoxyphenyl)borate. Crystal data
| K+·C7H7BF3O− | Dx = 1.573 Mg m−3 |
| Mr = 214.04 | Mo Kα radiation, λ = 0.71075 Å |
| Orthorhombic, Pca21 | Cell parameters from 4830 reflections |
| a = 7.1347 (2) Å | θ = 1.2–27.5° |
| b = 17.2819 (7) Å | µ = 0.59 mm−1 |
| c = 7.3289 (3) Å | T = 120 K |
| V = 903.66 (6) Å3 | Block, colourless |
| Z = 4 | 0.52 × 0.15 × 0.15 mm |
| F(000) = 432 |
(I) Potassium trifluorido(4-methoxyphenyl)borate. Data collection
| Rigaku CCD diffractometer | 1833 independent reflections |
| Radiation source: fine-focus sealed tube | 1822 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.026 |
| ω scans | θmax = 27.5°, θmin = 1.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2012) | h = −9→9 |
| Tmin = 0.750, Tmax = 0.917 | k = −22→20 |
| 5789 measured reflections | l = −8→9 |
(I) Potassium trifluorido(4-methoxyphenyl)borate. Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.021 | w = 1/[σ2(Fo2) + (0.035P)2 + 0.1245P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.059 | (Δ/σ)max < 0.001 |
| S = 1.10 | Δρmax = 0.24 e Å−3 |
| 1833 reflections | Δρmin = −0.20 e Å−3 |
| 120 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 1 restraint | Extinction coefficient: 0.0115 (14) |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), 712 Friedel pairs |
| Secondary atom site location: difference Fourier map | Absolute structure parameter: 0.02 (3) |
(I) Potassium trifluorido(4-methoxyphenyl)borate. Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
(I) Potassium trifluorido(4-methoxyphenyl)borate. Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| K1 | 0.93636 (3) | 0.577078 (16) | 0.49116 (4) | 0.01659 (9) | |
| C1 | 0.48663 (17) | 0.69908 (7) | 0.3989 (2) | 0.0159 (3) | |
| C2 | 0.36313 (18) | 0.72309 (8) | 0.2628 (2) | 0.0195 (3) | |
| H2 | 0.2718 | 0.6875 | 0.2199 | 0.023* | |
| C3 | 0.36757 (18) | 0.79722 (8) | 0.1867 (2) | 0.0223 (3) | |
| H3 | 0.2813 | 0.8114 | 0.0938 | 0.027* | |
| C4 | 0.5004 (2) | 0.84983 (8) | 0.2493 (2) | 0.0208 (3) | |
| C5 | 0.62439 (19) | 0.82828 (8) | 0.3880 (2) | 0.0233 (3) | |
| H5 | 0.7139 | 0.8643 | 0.4324 | 0.028* | |
| C6 | 0.61673 (19) | 0.75407 (8) | 0.4608 (2) | 0.0206 (3) | |
| H6 | 0.7018 | 0.7402 | 0.5549 | 0.025* | |
| C7 | 0.3970 (3) | 0.94712 (11) | 0.0422 (3) | 0.0393 (5) | |
| H7A | 0.4278 | 1.0000 | 0.0044 | 0.059* | |
| H7B | 0.2674 | 0.9454 | 0.0865 | 0.059* | |
| H7C | 0.4105 | 0.9121 | −0.0622 | 0.059* | |
| B1 | 0.48502 (18) | 0.61227 (8) | 0.4743 (2) | 0.0153 (3) | |
| F1 | 0.57562 (10) | 0.55930 (5) | 0.35081 (13) | 0.01754 (18) | |
| F2 | 0.30347 (10) | 0.58074 (4) | 0.50165 (17) | 0.02096 (19) | |
| F3 | 0.58402 (11) | 0.60460 (5) | 0.64078 (13) | 0.02023 (19) | |
| O1 | 0.52053 (18) | 0.92364 (6) | 0.1842 (2) | 0.0297 (3) |
(I) Potassium trifluorido(4-methoxyphenyl)borate. Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| K1 | 0.01225 (13) | 0.02391 (15) | 0.01361 (16) | 0.00081 (8) | 0.00031 (12) | 0.00042 (12) |
| C1 | 0.0132 (5) | 0.0193 (6) | 0.0152 (6) | 0.0009 (5) | 0.0007 (5) | −0.0009 (5) |
| C2 | 0.0199 (6) | 0.0204 (6) | 0.0183 (7) | −0.0015 (5) | −0.0044 (5) | −0.0014 (6) |
| C3 | 0.0233 (6) | 0.0229 (6) | 0.0206 (7) | 0.0011 (5) | −0.0043 (5) | 0.0017 (6) |
| C4 | 0.0234 (6) | 0.0167 (6) | 0.0222 (7) | 0.0007 (5) | 0.0007 (6) | 0.0011 (6) |
| C5 | 0.0223 (6) | 0.0200 (6) | 0.0277 (8) | −0.0036 (5) | −0.0054 (5) | −0.0028 (6) |
| C6 | 0.0181 (5) | 0.0211 (6) | 0.0226 (8) | 0.0002 (5) | −0.0046 (5) | −0.0010 (6) |
| C7 | 0.0421 (9) | 0.0273 (7) | 0.0484 (13) | 0.0011 (7) | −0.0085 (8) | 0.0167 (9) |
| B1 | 0.0121 (5) | 0.0185 (6) | 0.0153 (7) | 0.0000 (4) | 0.0002 (6) | 0.0008 (6) |
| F1 | 0.0182 (4) | 0.0196 (4) | 0.0148 (5) | 0.0031 (3) | 0.0025 (3) | −0.0007 (4) |
| F2 | 0.0129 (3) | 0.0209 (3) | 0.0291 (5) | −0.0015 (2) | 0.0028 (4) | −0.0004 (3) |
| F3 | 0.0204 (3) | 0.0271 (4) | 0.0132 (5) | −0.0004 (3) | −0.0022 (3) | 0.0025 (4) |
| O1 | 0.0352 (5) | 0.0176 (5) | 0.0362 (8) | −0.0009 (4) | −0.0037 (6) | 0.0052 (5) |
(I) Potassium trifluorido(4-methoxyphenyl)borate. Geometric parameters (Å, º)
| K1—F3i | 2.6156 (10) | C4—C5 | 1.398 (2) |
| K1—F2ii | 2.6211 (7) | C5—C6 | 1.390 (2) |
| K1—F1iii | 2.6550 (10) | C5—H5 | 0.9500 |
| K1—F1iv | 2.7568 (10) | C6—H6 | 0.9500 |
| K1—F3 | 2.7836 (8) | C7—O1 | 1.423 (2) |
| K1—F1 | 2.7887 (8) | C7—H7A | 0.9800 |
| K1—F2iv | 2.8885 (8) | C7—H7B | 0.9800 |
| K1—F3iv | 3.4886 (9) | C7—H7C | 0.9800 |
| C1—C2 | 1.3940 (19) | B1—F3 | 1.4162 (19) |
| C1—C6 | 1.4036 (18) | B1—F2 | 1.4196 (14) |
| C1—B1 | 1.5987 (18) | B1—F1 | 1.4403 (17) |
| C2—C3 | 1.398 (2) | B1—K1v | 3.2930 (14) |
| C2—K1i | 3.5183 (15) | F1—K1i | 2.6550 (10) |
| C2—H2 | 0.9500 | F1—K1v | 2.7568 (10) |
| C3—C4 | 1.3910 (19) | F2—K1vi | 2.6211 (7) |
| C3—H3 | 0.9500 | F2—K1v | 2.8885 (8) |
| C4—O1 | 1.3696 (17) | F3—K1iii | 2.6157 (10) |
| F3i—K1—F2ii | 94.59 (3) | O1—C4—C5 | 115.78 (13) |
| F3i—K1—F1iii | 173.67 (3) | C3—C4—C5 | 119.79 (13) |
| F2ii—K1—F1iii | 90.34 (3) | C6—C5—C4 | 119.99 (13) |
| F3i—K1—F1iv | 79.00 (3) | C6—C5—H5 | 120.0 |
| F2ii—K1—F1iv | 70.82 (2) | C4—C5—H5 | 120.0 |
| F1iii—K1—F1iv | 106.45 (2) | C5—C6—C1 | 121.78 (14) |
| F3i—K1—F3 | 107.78 (2) | C5—C6—H6 | 119.1 |
| F2ii—K1—F3 | 152.47 (3) | C1—C6—H6 | 119.1 |
| F1iii—K1—F3 | 66.40 (3) | O1—C7—H7A | 109.5 |
| F1iv—K1—F3 | 128.21 (2) | O1—C7—H7B | 109.5 |
| F3i—K1—F1 | 66.83 (3) | H7A—C7—H7B | 109.5 |
| F2ii—K1—F1 | 159.35 (3) | O1—C7—H7C | 109.5 |
| F1iii—K1—F1 | 108.89 (2) | H7A—C7—H7C | 109.5 |
| F1iv—K1—F1 | 95.79 (2) | H7B—C7—H7C | 109.5 |
| F3—K1—F1 | 47.98 (3) | F3—B1—F2 | 107.30 (13) |
| F3i—K1—F2iv | 100.35 (3) | F3—B1—F1 | 104.95 (10) |
| F2ii—K1—F2iv | 110.489 (19) | F2—B1—F1 | 104.73 (10) |
| F1iii—K1—F2iv | 81.59 (3) | F3—B1—C1 | 112.45 (11) |
| F1iv—K1—F2iv | 47.24 (3) | F2—B1—C1 | 114.56 (11) |
| F3—K1—F2iv | 81.63 (2) | F1—B1—C1 | 112.10 (12) |
| F1—K1—F2iv | 66.60 (3) | B1—F1—K1i | 122.39 (8) |
| C2—C1—C6 | 116.59 (12) | B1—F1—K1v | 98.47 (7) |
| C2—C1—B1 | 121.46 (12) | K1i—F1—K1v | 117.24 (3) |
| C6—C1—B1 | 121.90 (12) | B1—F1—K1 | 96.46 (7) |
| C1—C2—C3 | 122.95 (12) | K1i—F1—K1 | 112.53 (3) |
| C1—C2—K1i | 86.24 (8) | K1v—F1—K1 | 106.81 (3) |
| C3—C2—K1i | 114.99 (10) | B1—F2—K1vi | 156.48 (8) |
| C1—C2—H2 | 118.5 | B1—F2—K1v | 93.39 (6) |
| C3—C2—H2 | 118.5 | K1vi—F2—K1v | 107.73 (2) |
| K1i—C2—H2 | 68.1 | B1—F3—K1iii | 146.64 (8) |
| C4—C3—C2 | 118.88 (13) | B1—F3—K1 | 97.29 (7) |
| C4—C3—H3 | 120.6 | K1iii—F3—K1 | 113.95 (3) |
| C2—C3—H3 | 120.6 | C4—O1—C7 | 117.09 (13) |
| O1—C4—C3 | 124.43 (14) | ||
| C6—C1—C2—C3 | 1.2 (2) | C2—C1—C6—C5 | −1.1 (2) |
| B1—C1—C2—C3 | −176.35 (14) | B1—C1—C6—C5 | 176.46 (14) |
| C6—C1—C2—K1i | 118.72 (12) | C2—C1—B1—F3 | −164.66 (12) |
| B1—C1—C2—K1i | −58.84 (12) | C6—C1—B1—F3 | 17.90 (18) |
| C1—C2—C3—C4 | −0.2 (2) | C2—C1—B1—F2 | −41.81 (19) |
| K1i—C2—C3—C4 | −102.69 (14) | C6—C1—B1—F2 | 140.76 (15) |
| C2—C3—C4—O1 | 179.10 (14) | C2—C1—B1—F1 | 77.36 (16) |
| C2—C3—C4—C5 | −0.9 (2) | C6—C1—B1—F1 | −100.07 (15) |
| O1—C4—C5—C6 | −178.99 (14) | C3—C4—O1—C7 | −0.4 (2) |
| C3—C4—C5—C6 | 1.0 (2) | C5—C4—O1—C7 | 179.62 (15) |
| C4—C5—C6—C1 | 0.0 (2) |
Symmetry codes: (i) −x+3/2, y, z−1/2; (ii) x+1, y, z; (iii) −x+3/2, y, z+1/2; (iv) x+1/2, −y+1, z; (v) x−1/2, −y+1, z; (vi) x−1, y, z.
(I) Potassium trifluorido(4-methoxyphenyl)borate. Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the C1–C6 benzene ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···F2vii | 0.95 | 2.50 | 3.3359 (17) | 147 |
| C7—H7a···O1viii | 0.98 | 2.72 | 3.496 (1) | 137 |
| C3—H3···Cg1vii | 0.95 | 2.85 | 3.7171 (15) | 152 |
Symmetry codes: (vii) −x+1/2, y, z−1/2; (viii) −x+1, −y+2, z−1/2.
(II) Potassium trifluorido(4-fluorophenyl)borate. Crystal data
| K+·C6H4BF4− | F(000) = 800 |
| Mr = 202.00 | Dx = 1.751 Mg m−3 |
| Orthorhombic, Pbca | Mo Kα radiation, λ = 0.71075 Å |
| Hall symbol: -P 2ac 2ab | Cell parameters from 8210 reflections |
| a = 7.1317 (5) Å | θ = 3.2–27.5° |
| b = 7.3757 (5) Å | µ = 0.70 mm−1 |
| c = 29.129 (2) Å | T = 100 K |
| V = 1532.22 (18) Å3 | Block, colourless |
| Z = 8 | 0.07 × 0.05 × 0.01 mm |
(II) Potassium trifluorido(4-fluorophenyl)borate. Data collection
| Rigaku CCD diffractometer | 1726 independent reflections |
| Radiation source: fine-focus sealed tube | 1435 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.037 |
| ω scans | θmax = 27.5°, θmin = 3.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2012) | h = −9→8 |
| Tmin = 0.953, Tmax = 0.993 | k = −9→7 |
| 9537 measured reflections | l = −32→37 |
(II) Potassium trifluorido(4-fluorophenyl)borate. Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.027 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.065 | H-atom parameters constrained |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0274P)2 + 0.6658P] where P = (Fo2 + 2Fc2)/3 |
| 1726 reflections | (Δ/σ)max = 0.001 |
| 109 parameters | Δρmax = 0.27 e Å−3 |
| 0 restraints | Δρmin = −0.23 e Å−3 |
(II) Potassium trifluorido(4-fluorophenyl)borate. Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
(II) Potassium trifluorido(4-fluorophenyl)borate. Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| K1 | 0.06924 (4) | 0.02356 (5) | 0.205013 (12) | 0.01668 (10) | |
| C1 | 0.5118 (2) | 0.1023 (2) | 0.13156 (5) | 0.0161 (3) | |
| C2 | 0.3892 (2) | 0.0245 (2) | 0.09978 (6) | 0.0213 (3) | |
| H2 | 0.3130 | −0.0745 | 0.1091 | 0.026* | |
| C3 | 0.3750 (2) | 0.0869 (2) | 0.05506 (6) | 0.0245 (4) | |
| H3 | 0.2905 | 0.0324 | 0.0339 | 0.029* | |
| C4 | 0.4867 (2) | 0.2301 (2) | 0.04213 (5) | 0.0236 (4) | |
| C5 | 0.6112 (2) | 0.3121 (2) | 0.07145 (6) | 0.0250 (4) | |
| H5 | 0.6871 | 0.4105 | 0.0616 | 0.030* | |
| C6 | 0.6226 (2) | 0.2463 (2) | 0.11604 (6) | 0.0207 (3) | |
| H6 | 0.7085 | 0.3011 | 0.1367 | 0.025* | |
| B1 | 0.5200 (2) | 0.0373 (2) | 0.18355 (6) | 0.0147 (3) | |
| F1 | 0.43225 (11) | 0.16564 (12) | 0.21396 (3) | 0.0178 (2) | |
| F2 | 0.70288 (11) | 0.01321 (13) | 0.20154 (3) | 0.0210 (2) | |
| F3 | 0.42107 (11) | −0.12680 (12) | 0.19106 (3) | 0.0193 (2) | |
| F4 | 0.47218 (15) | 0.29343 (14) | −0.00157 (3) | 0.0327 (3) |
(II) Potassium trifluorido(4-fluorophenyl)borate. Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| K1 | 0.01228 (15) | 0.01386 (17) | 0.02390 (18) | 0.00022 (12) | 0.00079 (12) | 0.00020 (13) |
| C1 | 0.0143 (6) | 0.0139 (7) | 0.0200 (8) | 0.0019 (6) | 0.0007 (6) | −0.0006 (6) |
| C2 | 0.0214 (7) | 0.0201 (8) | 0.0225 (8) | −0.0029 (7) | −0.0016 (6) | 0.0002 (7) |
| C3 | 0.0268 (8) | 0.0256 (9) | 0.0211 (9) | −0.0019 (7) | −0.0056 (7) | −0.0016 (7) |
| C4 | 0.0306 (9) | 0.0243 (9) | 0.0160 (8) | 0.0046 (7) | 0.0013 (6) | 0.0020 (7) |
| C5 | 0.0296 (9) | 0.0221 (9) | 0.0234 (9) | −0.0056 (7) | 0.0052 (6) | 0.0014 (7) |
| C6 | 0.0222 (7) | 0.0189 (8) | 0.0211 (8) | −0.0050 (7) | 0.0007 (6) | −0.0014 (7) |
| B1 | 0.0116 (7) | 0.0130 (8) | 0.0194 (8) | 0.0014 (6) | 0.0006 (6) | −0.0008 (6) |
| F1 | 0.0184 (4) | 0.0152 (5) | 0.0198 (5) | 0.0027 (4) | 0.0027 (3) | −0.0012 (4) |
| F2 | 0.0125 (4) | 0.0295 (5) | 0.0209 (5) | 0.0032 (4) | −0.0024 (3) | −0.0012 (4) |
| F3 | 0.0205 (4) | 0.0125 (5) | 0.0247 (5) | −0.0017 (4) | 0.0003 (4) | 0.0026 (4) |
| F4 | 0.0474 (6) | 0.0332 (6) | 0.0174 (5) | −0.0013 (5) | −0.0007 (5) | 0.0061 (4) |
(II) Potassium trifluorido(4-fluorophenyl)borate. Geometric parameters (Å, º)
| K1—F3i | 2.6116 (10) | C4—F4 | 1.3597 (18) |
| K1—F2ii | 2.6159 (9) | C4—C5 | 1.373 (2) |
| K1—F1iii | 2.6527 (9) | C5—C6 | 1.389 (2) |
| K1—F1iv | 2.7612 (10) | C5—H5 | 0.9500 |
| K1—F3 | 2.7732 (9) | C6—H6 | 0.9500 |
| K1—F1 | 2.8050 (9) | B1—F2 | 1.4166 (17) |
| K1—F2iv | 2.8853 (10) | B1—F3 | 1.4182 (19) |
| K1—F3iv | 3.3927 (10) | B1—F1 | 1.4393 (18) |
| C1—C2 | 1.397 (2) | B1—K1v | 3.2665 (18) |
| C1—C6 | 1.399 (2) | F1—K1i | 2.6527 (9) |
| C1—B1 | 1.590 (2) | F1—K1v | 2.7612 (9) |
| C2—C3 | 1.385 (2) | F2—K1vi | 2.6159 (9) |
| C2—H2 | 0.9500 | F2—K1v | 2.8852 (10) |
| C3—C4 | 1.375 (2) | F3—K1iii | 2.6116 (10) |
| C3—H3 | 0.9500 | F3—K1v | 3.3928 (10) |
| F3i—K1—F2ii | 92.82 (3) | F4—C4—C3 | 118.44 (15) |
| F3i—K1—F1iii | 176.44 (3) | C5—C4—C3 | 122.87 (16) |
| F2ii—K1—F1iii | 88.32 (3) | C4—C5—C6 | 117.74 (16) |
| F3i—K1—F1iv | 76.56 (3) | C4—C5—H5 | 121.1 |
| F2ii—K1—F1iv | 71.99 (3) | C6—C5—H5 | 121.1 |
| F1iii—K1—F1iv | 107.00 (2) | C5—C6—C1 | 122.34 (15) |
| F3i—K1—F3 | 110.37 (2) | C5—C6—H6 | 118.8 |
| F2ii—K1—F3 | 152.42 (3) | C1—C6—H6 | 118.8 |
| F1iii—K1—F3 | 67.67 (3) | F2—B1—F3 | 107.08 (12) |
| F1iv—K1—F3 | 126.67 (3) | F2—B1—F1 | 104.80 (12) |
| F3i—K1—F1 | 67.74 (3) | F3—B1—F1 | 104.48 (11) |
| F2ii—K1—F1 | 159.45 (3) | F2—B1—C1 | 115.08 (12) |
| F1iii—K1—F1 | 111.49 (2) | F3—B1—C1 | 112.70 (13) |
| F1iv—K1—F1 | 96.05 (2) | F1—B1—C1 | 111.86 (12) |
| F3—K1—F1 | 47.78 (3) | F2—B1—K1v | 61.95 (7) |
| F3i—K1—F2iv | 99.45 (3) | F3—B1—K1v | 82.74 (8) |
| F2ii—K1—F2iv | 111.45 (2) | F1—B1—K1v | 57.03 (7) |
| F1iii—K1—F2iv | 83.24 (3) | C1—B1—K1v | 163.77 (11) |
| F1iv—K1—F2iv | 47.18 (2) | F2—B1—K1 | 145.97 (10) |
| F3—K1—F2iv | 80.14 (3) | F3—B1—K1 | 57.04 (6) |
| F1—K1—F2iv | 67.51 (3) | F1—B1—K1 | 58.42 (6) |
| F3i—K1—F3iv | 118.03 (3) | C1—B1—K1 | 98.93 (9) |
| F2ii—K1—F3iv | 73.38 (3) | K1v—B1—K1 | 85.12 (4) |
| F1iii—K1—F3iv | 65.53 (2) | B1—F1—K1i | 126.56 (8) |
| F1iv—K1—F3iv | 41.51 (2) | B1—F1—K1v | 97.04 (8) |
| F3—K1—F3iv | 106.37 (3) | K1i—F1—K1v | 117.58 (3) |
| F1—K1—F3iv | 109.07 (3) | B1—F1—K1 | 95.66 (8) |
| F2iv—K1—F3iv | 41.62 (2) | K1i—F1—K1 | 111.04 (3) |
| C2—C1—C6 | 116.82 (15) | K1v—F1—K1 | 105.32 (3) |
| C2—C1—B1 | 122.05 (14) | B1—F2—K1vi | 158.46 (9) |
| C6—C1—B1 | 121.08 (14) | B1—F2—K1v | 92.38 (8) |
| C3—C2—C1 | 122.18 (15) | K1vi—F2—K1v | 107.01 (3) |
| C3—C2—H2 | 118.9 | B1—F3—K1iii | 148.62 (8) |
| C1—C2—H2 | 118.9 | B1—F3—K1 | 97.55 (8) |
| C4—C3—C2 | 118.06 (15) | K1iii—F3—K1 | 113.33 (3) |
| C4—C3—H3 | 121.0 | B1—F3—K1v | 72.76 (8) |
| C2—C3—H3 | 121.0 | K1iii—F3—K1v | 100.12 (3) |
| F4—C4—C5 | 118.69 (15) | K1—F3—K1v | 91.16 (3) |
| C6—C1—C2—C3 | −0.7 (2) | C2—C1—C6—C5 | 0.8 (2) |
| B1—C1—C2—C3 | 176.65 (15) | B1—C1—C6—C5 | −176.56 (15) |
| C1—C2—C3—C4 | 0.1 (3) | C2—C1—B1—F2 | 135.24 (15) |
| C2—C3—C4—F4 | −179.32 (15) | C6—C1—B1—F2 | −47.6 (2) |
| C2—C3—C4—C5 | 0.4 (3) | C2—C1—B1—F3 | 12.0 (2) |
| F4—C4—C5—C6 | 179.43 (15) | C6—C1—B1—F3 | −170.75 (13) |
| C3—C4—C5—C6 | −0.3 (3) | C2—C1—B1—F1 | −105.32 (17) |
| C4—C5—C6—C1 | −0.3 (3) | C6—C1—B1—F1 | 71.88 (18) |
Symmetry codes: (i) −x+1/2, y+1/2, z; (ii) x−1, y, z; (iii) −x+1/2, y−1/2, z; (iv) x−1/2, y, −z+1/2; (v) x+1/2, y, −z+1/2; (vi) x+1, y, z.
(II) Potassium trifluorido(4-fluorophenyl)borate. Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C6—H6···F2vii | 0.95 | 2.53 | 3.4099 (19) | 154 |
Symmetry code: (vii) −x+3/2, y+1/2, z.
References
- Bruker (2012). SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Conole, G., Clough, A. & Whiting, A. (1995). Acta Cryst. C51, 1056–1059.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Fei, Z., Zhu, D.-R., Yang, X., Meng, L., Lu, Q., Ang, W. H., Scopelliti, R., Hartinger, C. G. & Dyson, P. J. (2010). Chem. Eur. J. 16, 6473–6481. [DOI] [PubMed]
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Quach, T. D., Batey, R. A. & Lough, A. J. (2001). Acta Cryst. E57, o688–o689.
- Rigaku (2010). CrystalClear. Rigaku Inc., Tokyo, Japan.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, II, global. DOI: 10.1107/S1600536814009684/wm0001sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814009684/wm0001Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S1600536814009684/wm0001IIsup3.hkl
Additional supporting information: crystallographic information; 3D view; checkCIF report