

Abstract
Background
Inflammation during inflammatory bowel disease (IBD) may alter nutrient availability to adherent mucosal bacteria and impact their metabolic function. Microbial metabolites may regulate intestinal CD4+ T cell homeostasis. We investigated the relationship between inflammation and microbial function by inferred metagenomics of the mucosal microbiota from colonic pinch biopsies of IBD patients.
Methods
Paired pinch biopsy samples of known inflammation states were analyzed from UC (23), CD (21) and controls (24) by 16S ribosomal sequencing, histopathology and flow cytometry. PICRUSt was used to generate metagenomic data, and derive relative Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway abundance information. Leukocytes were isolated from paired biopsy samples and analyzed by multi-color flow cytometry. Active inflammation was defined by neutrophil infiltration into the epithelium
Results
Carriage of metabolic pathways in the mucosal microbiota was relatively stable among IBD patients despite large variations in individual bacterial community structures. However, microbial function was significantly altered in inflamed tissue of UC patients, with a reduction in carbohydrate and nucleotide metabolism in favor of increased lipid and amino acid metabolism. These differences were not observed in samples from CD patients. In CD, microbial lipid, carbohydrate, and amino acid metabolism was tightly correlated with frequency of CD4+Foxp3+ Tregs, whereas in UC these pathways were correlated with frequency of CD4+IL-22+ (TH22) cells.
Conclusions
Metabolic pathways of the mucosal microbiota in CD do not vary as much as UC with inflammation state, indicating a more systemic perturbation of host-bacteria interactions in CD compared to more localized dysfunction in UC.
Introduction
Inflammatory bowel diseases (IBD) are characterized by chronic inflammation of the human gastrointestinal tract, driven by abnormal interactions between the immune system and the intestinal microbiota 1-3. For both ulcerative colitis (UC) and Crohn's disease (CD), the two main subtypes of IBD, recent studies using 16S ribosomal sequencing have shown disease associations with dysbiosis or abnormal compositions of the gut microbiota 4-8. Dysbiosis includes increases in bacterial numbers, decreased bacterial biodiversity, increased abundance of the phyla Proteobacteria and Bacteroidetes, and a decreased abundance of Firmicutes 9-11. This dysbiosis is observed in colonic pinch biopsy samples as well as fecal samples 6,12. Characterization of the mucosal microbiota from pinch biopsies reflects the communities adhering to the intestinal epithelium, which is quite distinct from the luminal bacteria. Analyses of pinch biopsies enable investigation of the microbiota at different locations in the colon, which can also be directly compared with pathology samples in order to correlate disease activity with adherent bacterial communities in IBD. There are large differences in the microbiota between stool and mucosal biopsies 6,13,14 and differences in colonic pH may result in differences at various locations of the gut 6,15.
While sequencing 16S ribosomal RNA reveals alterations to taxonomic groups and species composition of IBD patients, it does not inform us of the metabolic activity and function of the microbial communities. Microbial function in communities can be assessed using shotgun metagenomics 13,16, but this approach is dependent on the isolation of sufficient quantities of bacterial DNA, which is not possible from colonic pinch biopsy samples. Recently, an approach was developed to investigate microbiota functional profiles by inferring the metagenome of the closest available whole genome sequences using 16S gene sequence profiles 6,17. While this approach has some limitations 18, it provides a way to detect changes to microbial function when the quantity of bacterial DNA is very limited (e.g. in pinch biopsies). Studies on microbial function in healthy individuals have shown that although bacterial communities vary considerably among individuals, overall microbial functions of these communities are largely conserved 13. However, in IBD, functional differences were more pronounced than taxonomic differences 6. While a few studies have now analyzed the changes in microbial function in IBD based on metagenomic sequencing 6,19, alterations in microbial function during different inflammation states of the sampled tissues has not been examined.
In this study, we utilized inferred metagenomics by PICRUSt 17 to investigate functional differences in the microbiota of patients with UC and CD with biopsies taken from regions of known inflammation states. By pairing biopsies used for analyses of microbial communities with pathology results and flow cytometry profiles of CD4+ effector cytokine production, we compared the effect of inflammation state on the mucosal microbiota metabolic function for UC and CD patients and relate this information to the homeostasis of CD4+ T cell populations in the same sampled tissue.
Materials and Methods
Participants
Pinch biopsies were taken from IBD patients and healthy subjects undergoing surveillance colonoscopies at the Mount Sinai Medical Center and the Manhattan campus of the VA New York Harbor Healthcare System. The CD and UC patient cohorts were of similar age (CD: 38±11yrs, UC: 41±11yrs), race (CD: 84.5% Caucasian, UC: 85.7%), sex (CD: 84.6% male, UC: 64%) and disease duration (CD: 15.8±11.1yrs, UC: 15.6±9.2yrs) (Supplemental Table 2). The healthy subjects were significantly older (61±7yrs) as they were undergoing surveillance colonoscopy for colorectal cancer. Institutional review board approval was obtained before involving patients in the study.
16S rRNA analyses, QIIME, and PICRUSt
DNA was isolated from pinch biopsy material. The V4 region of the 16S rRNA gene was PCR amplified for sequencing with region specific barcoded primers 20 and sequenced on a MiSeq sequencer 21 along with other barcoded samples. Data files were demultiplexed and converted from the fastq format to the .fna format using QIIME and reads shorter than 140bp were discarded 22. Sequences were assigned to operational taxonomic units (OTUs) with a threshold of 97% pair-wise identity and classified taxonomically according to the Ribosomal Database Project (RDP) for use in taxonomic analysis. OTUs were subsequently picked by UCLUST against a closed reference table, the latest version of the Greengenes OTU database 23,24. PICRUSt was then used on the Greengene picked OTUs to generate metagenomic data, and derive relative Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway abundance 17.
KEGG data was analyzed with QIIME, and LEfSe 25. Cluster was used to generate cladograms and heatmaps of functional data. Beta diversity was calculated using the Bray-Curtis method and visualized using Principal Coordinate Analysis (PCoA). LEfSe analysis was performed with an LDA score threshold of >3.0, a .05 alpha value for the factorial Kruskal-Wallis test and a one-against-all multi-class analysis strategy. LEfSe uses the Kruskal-Wallis (KW) sum-rank test to detect features with significantly different abundances between assigned classes, and performs LDA to estimate the effect size of each feature 25. Statistical analysis was performed using Prism 6.0 (GraphPad Software, La Jolla, CA). The unpaired t-test was used to assess statistical significance for all the samples.
Isolation of cells
Pinch biopsies from the colonic mucosa of healthy individuals and IBD patients were digested at 37°C for 1 hour in 100 units/ml collagenase Type VIII (Sigma) and 0.150 mg/ml DNase (Sigma) in complete RPMI 1640 (Invitrogen) containing 10% FCS, 2 mM L-glutamine, 100 units/ml penicillin, 0.1 mg/ml streptomycin, and 0.05 mM 2-Mercaptoethanol. Cells were filtered through a 50 micron filter, washed with 5 ml PBS, and pelleted. Cells were then resuspended in 40% Percoll (GE Healthcare) in complete RPMI, under-layed with 80% Percoll, and centrifuged at 2,200 rpm for 20 min at room temperature. Lamina propria mononuclear cells were collected at the interface and used for subsequent flow cytometry analyses.
FACS staining
Cells were stimulated with 50 ng/ml phorbol 12-myristate 13-acetate (PMA) and 500 ng/ml ionomycin for 4 hours at 37°C in the presence of brefeldin A (GolgiPlug, BD). Following this in vitro stimulation, cells were stained with anti-CD3, anti-CD4, anti-CD8, and fixed in 4% paraformaldehyde in PBS. Cells were permeabilized in Perm/Wash buffer (BD) and stained with anti-IL-22 and anti-Foxp3. Cells were acquired on an LSRII (BD) and analyzed with FlowJo (Tree Star, Inc.) software. Positive gates for cytokine and nuclear antigen expression are drawn based on control experiments performing Fluorescence Minus One (FMO) controls with PBMC samples stained with the same intracellular cytokine panels.
Results
Functional variation and taxonomic abundances in mucosal biopsies
To investigate the mucosal microbiota communities in pinch biopsies from UC (N=21) and CD (N=23) patients relative to healthy subjects (N=24), we performed deep sequencing analysis on the variable region 4 (V4) region of bacterial 16S rRNA. A total of 868,044 quality-filtered sequences were obtained from the samples with a mean of 12,226 ± 4493 (SD) sequences per sample. At the phylum level, the relative abundance of the three most prevalent bacterial phyla of Bacteroidetes, Firmicutes, and Proteobacteria was slightly different between UC, CD, and healthy controls (Figure 1A). Samples from CD patients had a significantly higher proportion of Bacteroidetes compared to healthy controls (Figure 1B). Although UC and CD biopsies showed slightly decreased abundance of Firmicutes and slightly increased abundance of Proteobacteria, these differences were not statistically significant (Figure 1A and Figure S1).
Figure 1. Taxonomic and functional diversity of the mucosal microbiota in IBD and healthy patients.

(A) Pie charts representing the mean relative abundances of the major (>1%) phyla present in the mucosal microbiota from healthy, UC and CD patients. (B) Scatterplot showing significantly higher relative abundance of the Bacteroidetes phyla in biopsies from CD patients (p < 0.05). (C) Stacked bar charts showing the individual variability of the relative abundances of the major bacterial Phyla among individual patients. (D) Relative abundances of metabolic pathways encoded in the mucosal microbiota are evenly distributed and similar across both individuals and disease states.
The relative abundance of different clades among individual samples within the same group varied considerably for IBD patients as well as healthy individuals (Figure 1C). However, when we assessed the microbial metabolic and functional KEGG pathways of these communities by inferred metagenomics using PICRUSt 6,17, the pathways were more evenly distributed and consistent between individuals (Figure 1D). This observation is consistent with results from the Human Microbiome Project demonstrating stability of metabolic pathways despite variability of microbial taxa 13.
Metabolic alterations to the mucosal microbiota based on inflammation status in UC and CD
The pinch biopsies from patients with IBD were taken from various locations in the colon and small intestine, and for each location a separate biopsy was sent to pathology for characterization of inflammation state. Active chronic colitis was defined by neutrophil infiltration into the epithelium, in the setting of epithelial cell damage 26. To determine the relationship between the relative abundance of KEGG metabolic pathways present in the mucosal biopsies in relation to the inflammation state of the tissues, we utilized both unsupervised and supervised approaches.
By performing unsupervised hierarchical clustering analysis on the relative abundance values of KEGG pathways represented from the mucosal microbiota of UC patients, we found that there is a clear distinction between the clustering of inflamed samples and non-inflamed samples (Figure 2A and S2). The separate clustering of inflamed and non-inflamed samples was also observed by principal component analysis (Figure 2B). To identify which groups of microbial functional pathways were driving the differences based on inflammation status, we determined the KEGG pathways that were enriched or under represented in inflamed tissue using the LEfSe approach 25 (Figure 2C).
Figure 2. Functional divergence between the mucosal microbiota of inflamed and non-inflamed samples from UC patients.

(A) Unsupervised hierarchical clustering analysis of abundance values for KEGG pathways shows distinctive clustering of inflamed (red) and non-inflamed (green) samples. Each column is a separate sample and row is a particular pathway. Blue = less than the mean, Yellow = greater than mean. (B) Principal Component Analysis of KEGG pathways confirms the segregation of inflamed and non-inflamed samples along PC1 and PC2. (C) Supervised comparison identifies differential abundance of specific KEGG pathways using LEfSe (LDA > 3.0). (D) Genes in carbohydrate and nucleotide metabolism pathways are more abundant in non-inflamed normal tissue from UC patients, with a shift towards more abundance in lipid and amino acid metabolism genes for the mucosal microbiota of inflamed tissues. ***P<0.0001.
In UC, 5 of the 8 functional pathways identified (LDA score > 3.0) that were more abundant for bacteria inhabiting areas of inflamed tissue were related to metabolism; particularly amino acid, terpenoid, lipid, and xenobiotic metabolism (Figure 2C and 2D). The other pathways were related to cell motility and signaling. Many pathways that were more highly represented in non-inflamed tissue were also involved in metabolism; namely carbohydrate, nucleotide, and energy metabolism (Figure 2C and 2D). Hence, the mucosal microbiota of actively inflamed tissue from UC patient may utilize different metabolic pathways then those in non-inflamed tissues.
The same analyses were performed on samples from CD patients (Figure 3). In contrast to UC, hierarchical clustering analysis of samples from CD patients did not segregate well by inflammation state (Figure 3A). This was consistent with the principal component analysis (Figure 3B), which showed that normal and inflamed samples were mostly intermingled. Finally, when we conducted a supervised comparison of inflamed and non-inflamed samples by LEfSe analysis, a reduced significance threshold (LDA > 2.0) was required to identify an increased abundance for energy metabolism in inflamed tissue (Figure 3C). There was also an increased abundance for genes in the nervous system pathway, but this is difficult to interpret. There were no KEGG pathways that were underrepresented in inflamed CD samples. Amino acid, lipid and carbohydrate metabolism was unaltered (Figure 3D). Hence, metabolic pathways of the mucosal microbiota in CD do not vary as much as UC with inflammation state.
Figure 3. Limited differences between the mucosal microbiota of inflamed and non-inflamed samples from CD patients.
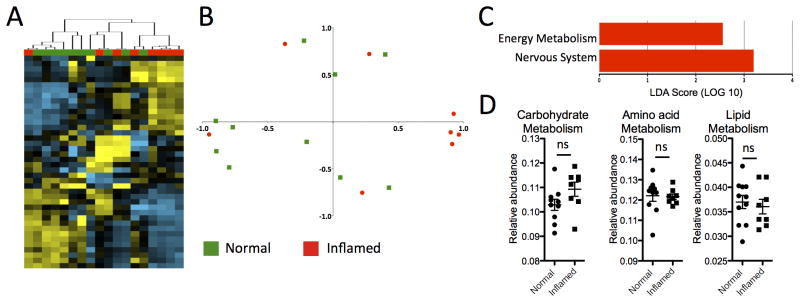
(A) Hierarchical clustering analysis of samples from CD patients shows limited segregation of inflamed (red) and non-inflamed (green) samples. (B) Principal Component Analysis shows limited segregation of inflamed and non-inflamed samples. (C) Supervised comparison identifies only two differentially abundant KEGG pathways using LEfSe (LDA > 2.0). (D) Genes involved in amino acid, lipid and nucleotide metabolism pathways are not significantly different between inflamed and normal tissue of CD patients, but there is a significant increase in the energy metabolism pathway. *P<0.05.
Differences in bacteria functional pathways in inflamed tissue compared to healthy controls
We next compared the functional pathways for the mucosal microbiota of inflamed samples from both UC and CD patients with healthy subjects (Figure 4). Non-inflamed samples were not included because they should be more similar to healthy tissue. Unsupervised hierarchical clustering analysis showed that inflamed UC samples and CD samples clustered distinctly in two major branches (Figure 4A and S3). Some healthy samples clustered with UC samples and others with CD samples. The distinct separation of UC and CD inflamed samples was confirmed by PCoA plots where they clustered distinctly (Figure 4B), intermingled with samples from healthy subjects. This indicates that microbial function of inflamed tissue is more distinct between UC and CD patients, than in relation to healthy subjects.
Figure 4. KEGG metabolic pathways are significantly different in the mucosal microbiota of inflamed tissue from UC and CD patients.

(A) UC and CD patient samples with active inflammation cluster distinctly into separate arms. Some samples from healthy patients cluster with UC samples, whereas other healthy samples cluster with CD samples. (B) Principal Component Analysis confirms the segregation of CD and UC inflamed samples, although there is no clear separation from healthy subjects. (C) LEfSe analysis (LDA > 3.0) shows that amino acid and lipid metabolism was more abundant in the mucosal microbiota of inflamed UC samples, whereas carbohydrate, energy and nucleotide metabolism pathways was more abundant in inflamed CD samples.
To determine the functional KEGG pathways that are driving these differences, we again performed supervised comparisons by LEfSE (LDA >3.0). Different metabolic pathways were enriched in the mucosal microbiota of UC and CD patients (Figure 4C). Whereas the KEGG pathways of amino acid and lipid metabolism were enriched in UC samples, CD samples were enriched in carbohydrate, energy and nucleotide metabolism (Figure 4C).
Relationship between microbiota metabolic profiles and homeostasis of CD4+ T cell populations
Recently, microbial metabolites were demonstrated to regulate the homeostasis of intestinal regulatory T cells (Tregs) 27. We measured the relative frequency of CD4+Foxp3+ Tregs among lamina propria mononuclear cells (LPMCs) isolated from pinch biopsies paired with the samples that were sequenced (Figure 5A and 5B) and compared them with KEGG microbial metabolic pathways (Figure 5C). We previously found by flow cytometry that actively inflamed samples from UC patients were depleted of T helper type 22 (Th22) cells and enriched for mono-IL-17 producing Th17 cells 28. Hence, we also compared the frequency of CD4+IL-22+ cells (Figure 5A and 5D) with the relative abundance values of the KEGG metabolic pathways (Figure 5E).
Figure 5. Correlation of microbial function with the homeostasis of CD4+ effector cells.

(A) Representative gating strategy for the FACS analysis of lamina propria mononuclear cells (LPMCs), showing live CD3+ CD4+ lymphocytes with intracellular staining for interleukin (IL)- IL-22 production and Foxp3 expression. (B) Frequency of CD4+Foxp3+Tregs from LPMCs of CD patients in Inflamed (red) and Non-Inflamed (blue) biopsy sites. (C) Linear regression of KEGG pathways (lipid, carbohydrate and amino acid metabolism) with the percentage of CD4+Foxp3+ present in the same biopsy location taken from CD patients. (D) Frequency of CD4+IL-22+ cells of UC patients in Inflamed (red) and Non-Inflamed (blue) biopsy sites. (E) Linear regression of KEGG pathways with the percentage of CD4+IL-22+ cells present in the same biopsy location taken from UC patients.
For CD patients, the frequency of Foxp3+ Tregs was not significantly different between inflamed and non-inflamed biopsy samples (Figure 5B). However, there was a tight correlation between Treg frequency and microbial lipid metabolism, and to a lesser extent carbohydrate and amino acid metabolism (Figure 5C). These relationships between Treg frequency and microbial metabolism pathways were not significant for UC patients (data not shown). In contrast, UC patients had significantly fewer CD4+IL-22+ cells in inflamed biopsy samples (Figure 5D) and the frequency of these CD4+IL-22+ cells was significantly correlated with microbial metabolism pathways (Figure 5E). The relationship between CD4+IL-22+ cells microbial metabolism pathways was not observed for CD samples (data not shown). These results suggest that host-microbial interactions for UC and CD patients may be quite different and that the tissue microenvironment, as reflected by the types of effector CD4+ cells present, may influence the functional phenotype of the mucosal microbiota. Alternatively, metabolites produced by different microbial metabolic pathways may influence the homeostasis of CD4+Foxp3+ and CD4+IL-22+ cells differently for UC and CD patients.
Discussion
In this study, we apply the approach of inferred metagenomics using PICRUSt on colon biopsy samples of known inflammation states from patients with UC, CD and controls, in order to determine if there are metabolic alterations to the mucosal microbiota during inflammatory bowel diseases. We then determined that alterations to microbial metabolism are directly correlated with the homeostasis of CD4+Foxp3+ and CD4+IL-22+ cells. Furthermore, the relationship between inflammation status and CD4+ cells with metabolic alterations to the mucosal microbiota is completely different for patients with UC and CD.
Changes to the composition of the microbiome and its interaction with the immune system plays a crucial role in the pathogenesis of CD and UC, but this relationship is still poorly understood 1-3,29,30. Alterations to the microbiome in IBD include decreased biodiversity, increased numbers of bacteria, and changes in the relative abundance of Firmicutes and Proteobacteria. 9-11. In this study on the mucosal microbiota, we found that only the relative abundance of Bacteroidetes was significantly different from healthy controls and only in CD. Contrary to published data, we found no phylum-level significant differences in Firmicutes or Proteobacteria. However, there are many inconsistencies between studies. For example, no significant changes at the phylum level for Bacteroidetes, Firmicutes, or Proteobacteria was found in pediatric IBD patients at the time of diagnosis, compared to healthy patients 31, which raises the possibility that microbial alterations could be a function of previous treatment or disease progression, rather than preceding disease. Since the microbiome profiles are influenced by age, medication history, smoking status, and BMI 6,32, we are still some distance away from unraveling these complex relationships in the context of inflammatory bowel diseases.
The tissue microenvironment can influence the nutrient availability for the microbiota, which is critical in defining the composition of the community. The metabolic activities of the microbiota are reflected in the genes encoded into their genomes, which has previously required deep metagenomic sequencing in order to be analyzed. Deep metagenomic sequencing is not possible on biopsy specimens because the relative content of bacterial DNA to host DNA is extremely small. Recently, a method called PICRUSt that can infer the metagenome through using 16S sequence data to extract information from the closest available whole-genome sequences has been developed 17. Using PICRUSt, we investigated functional perturbations to the mucosal microbiota communities of IBD patients. Consistent with findings in other body tissues 13, we found that overall function was largely conserved across samples, even with widespread differences in bacteria composition. However, important differences exist between bacteria found adhered to inflamed tissue compared to non-inflamed tissue.
We find that alterations in microbial function were more significant between inflamed and non-inflamed regions of UC patients, in comparison to CD patients. This is somewhat contrary to a previous study that found CD to have larger functional perturbations with inflammation then UC 6. The fact that normal and inflamed tissue is more different in patients with UC than CD is consistent with the hypothesis that UC is a more regionalized disease whereas CD is a more systemic disease. This observation is also consistent with our previous results comparing the profiles of immune effector CD4+ T cells in normal and inflamed tissues between patients with UC and CD 28. CD4+ T cell frequencies in normal non-inflamed tissue for UC patients were also more similar to healthy controls than non-inflamed tissue from patients with CD 28. Since we also observed here that there are greater differences between the mucosal microbiota of inflamed CD samples with inflamed UC and healthy samples, then between inflamed and non-inflamed CD samples, there may be a more systemic perturbation of host-bacteria interactions in CD that is independent of the inflammation status of the mucosa.
In inflamed regions of UC patients, the metabolic activities of adherent bacteria was altered to have a reduce abundance of genes utilized for carbohydrate and nucleotide metabolism, and an increased abundance of genes for lipid and amino acid metabolism. Intestinal bacteria compete for the limited supply of nutrients within the intestine, which should be reflected in genes that are encoded into their genomes and their relative abundance 33. Bacteria that are highly dependent on nutrients from the host environment may have a reduced genome lacking in genes for specific metabolic pathways. For example, segmented filamentous bacteria (SFB) that can promote differentiation of TH17 cells, lack genes involved in nucleotide and amino acid biosynthesis 34. One hypothesis is that inflamed tissue provides less supply of carbohydrates and bacteria that are able to undergo amino acid and lipid metabolism dominate. Hence, in actively inflamed regions of the gut of a UC patient, perhaps nucleotides and carbohydrates might become more readily available, favoring the expansion of carbohydrate auxotrophic bacteria that maintain biosynthetic pathways for amino acids and lipids. Recently, butyrate-producing bacterial taxa were found to be reduced in ulcerative colitis patients, together with a reduction in short-chain fatty acids (SCFA) 35.
Interestingly, we find that this functional shift from carbohydrate and nucleotide metabolism, towards increased amino acid and lipid metabolism in active inflammation of UC patients was also associated with frequency of CD4+IL-22+ cells. Alterations in microbial metabolic pathways could result in production of different metabolites, some of which (e.g. SCFA) have been shown to regulate intestinal Foxp3+ Treg populations in mice 27. Although there are few differences for microbial metabolic pathways between inflamed and non-inflamed regions of CD patients, there was a very strong association between the frequency of Foxp3+ Treg and microbial metabolism pathways. These observations suggest a link between bacterial metabolism and the intestinal homeostasis of Foxp3+ Tregs in CD patients.
The mucus layer in the intestinal tract is an important barrier to luminal bacteria, as well as a major substrate and adhesion component for bacteria living in this microenvironment. Alterations to the mucus layer may lead to breaches of the epithelial barrier by specific bacteria, resulting in immune responses that may result in chronic inflammation. Changes in the quantity and quality of this mucus layer will influence the constituency of attached microbial populations. Mucin production can be dysfunctional in IBD, and there is a particular paucity of mucin in UC 36. Since mucin is a significant source of carbohydrates for the commensal bacteria 33, mucosal microbiota in inflamed regions of the gut may shift toward bacteria that can metabolize lipids and amino acids for energy. Further support of this principle is that bacteria like Firmicutes 33 are unable to metabolize amino acids and are seen with decreased prevalence in IBD.
This study provides new insight into the differences between UC and CD, particularly with regards to microbiota composition and function. We have now found by both flow cytometry analysis of effector T cells and by 16S sequencing of the mucosal microbiota that alterations to the intestinal mucosa in CD are largely colon-wide, and do not vary as much as UC with inflammation state. Together this is indicative of a more systemic perturbation of mucosal immunity in CD patients, compared to a more localized dysfunction in UC patients. Therapeutic strategies designed to improve host-bacteria interactions during IBD may have to be selective for UC and CD patients, since there is clearly a different interplay between mucosal immune responses and the gut microbiota of these two classes of IBD patients.
Supplementary Material
Acknowledgments
We would like to thank the Knight and Huttenhower laboratories for making QIIME and PICRUSt easily available to the scientific community. We thank members of the Earth Microbiome Project at Argonne National Laboratories for assistance in microbial sequencing.
Grant support: PL is supported by NIH grants 5R01AI093811, 3R21AI094166, the Broad Medical Research Program of The Broad Foundation, Kevin and Masha Keating Family Foundation; LM is supported by NIH grant: P01 DK072201; IC is supported in part by NIH grant: UL1 TR000038. JP is a recipient of an AGA-Eli and Edythe Broad Student Research Fellowship.
References
- 1.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011 Jun 16;474(7351):307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nature reviews Gastroenterology & hepatology. 2012 Oct;9(10):599–608. doi: 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 3.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011 May;140(6):1729–1737. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank DN, Amand AL, St, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007 Aug 21;104(34):13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manichanh C, Rigottier-Gois L, Bonnaud E, et al. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut. 2006 Feb;55(2):205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome biology. 2012;13(9):R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fite A, Macfarlane S, Furrie E, et al. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. Journal of clinical microbiology. 2013 Mar;51(3):849–856. doi: 10.1128/JCM.02574-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annual review of immunology. 2012;30:759–795. doi: 10.1146/annurev-immunol-020711-074937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sokol H, Lay C, Seksik P, Tannock GW. Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflammatory bowel diseases. 2008 Jun;14(6):858–867. doi: 10.1002/ibd.20392. [DOI] [PubMed] [Google Scholar]
- 10.Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004 May;53(5):685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swidsinski A, Ladhoff A, Pernthaler A, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002 Jan;122(1):44–54. doi: 10.1053/gast.2002.30294. [DOI] [PubMed] [Google Scholar]
- 12.Lepage P, Hasler R, Spehlmann ME, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. 2011 Jul;141(1):227–236. doi: 10.1053/j.gastro.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012 Jun 14;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stearns JC, Lynch MD, Senadheera DB, et al. Bacterial biogeography of the human digestive tract. Scientific reports. 2011;1:170. doi: 10.1038/srep00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012 Jun 8;336(6086):1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012 Jun 14;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 2013 Sep;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer F, Trimble WL, Chang EB, Handley KM. Functional predictions from inference and observation in sequence-based inflammatory bowel disease research. Genome biology. 2012;13(9):169. doi: 10.1186/gb-2012-13-9-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jan 10;109(2):594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011 Mar 15;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012 Mar 8; doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010 May;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010 Oct 1;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 24.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 2006 Jul;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geboes K, De Hertogh G. Indeterminate colitis. Inflamm Bowel Dis. 2003 Sep;9(5):324–331. doi: 10.1097/00054725-200309000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013 Aug 2;341(6145):569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leung JM, Davenport M, Wolff MJ, et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. 2013 May 22; doi: 10.1038/mi.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elson CO, Cong Y. Host-microbiota interactions in inflammatory bowel disease. Gut microbes. 2012 Jul-Aug;3(4):332–344. doi: 10.4161/gmic.20228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown SJ, Mayer L. The immune response in inflammatory bowel disease. The American journal of gastroenterology. 2007 Sep;102(9):2058–2069. doi: 10.1111/j.1572-0241.2007.01343.x. [DOI] [PubMed] [Google Scholar]
- 31.Hansen R, Russell RK, Reiff C, et al. Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn's but not in ulcerative colitis. The American journal of gastroenterology. 2012 Dec;107(12):1913–1922. doi: 10.1038/ajg.2012.335. [DOI] [PubMed] [Google Scholar]
- 32.Mariat D, Firmesse O, Levenez F, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC microbiology. 2009;9:123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamada N, Chen GY, Inohara N, Nunez G. Control of pathogens and pathobionts by the gut microbiota. Nature immunology. 2013 Jun 18;14(7):685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sczesnak A, Segata N, Qin X, et al. The genome of th17 cell-inducing segmented filamentous bacteria reveals extensive auxotrophy and adaptations to the intestinal environment. Cell Host Microbe. 2011 Sep 15;10(3):260–272. doi: 10.1016/j.chom.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2013 Sep 10; doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 36.Boltin D, Perets TT, Vilkin A, Niv Y. Mucin function in inflammatory bowel disease: an update. Journal of clinical gastroenterology. 2013 Feb;47(2):106–111. doi: 10.1097/MCG.0b013e3182688e73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.