

Abstract
Background
A patient with factor XI (fXI) deficiency was reported with an Arg184Gly substitution in the fXI A3 domain. The A3 domain contains an exosite required for binding of factor IX (fIX) to activated fXI (fXIa).
Objective
To test the effects of the Arg184Gly substitution on fIX activation, and to characterize the fIX binding site on fXIa.
Methods
Recombinant fXIa and fIX variants were used to identify residues involved in fIX activation by fXIa. Analysis of the fXI structure was used to identify potential fIX binding sites.
Results
Km for fIX activation by fXIa-Gly184 is ∼3-fold higher than for fXIa, suggesting Arg184 is part of the exosite. Arg184 and adjacent residues Ile183 and Asp185 contribute to charged and hydrophobic areas that are not present in the fXI homolog prekallikrein (PK). Replacing residues 183 to 185 with alanine abolished exosite activity, similar to replacement of the entire A3 domain with A3 from PK (fXIa/PKA3). Reintroducing fXI residues 183-185 into fXIa/PKA3 partially restored the exosite, while replacing residues 183-185 and 260-264 completely restored exosite function. FIX in which the Ω-loop (residues 4 to 11) is replaced with the factor VII Ω-loop was activated poorly by fXIa, suggesting the fIX Ω-loop binds to fXIa.
Conclusion
The results support a model in which the Ω-loop of fIX binds to an area on fXIa comprised of residues from the N- and C-termini of the A3 domain. These residues are buried in zymogen fXI, and must be exposed upon conversion to fXIa to permit fIX binding.
Keywords: Factor XI, factor XIa, factor IX, proteolysis, blood coagulation
Introduction
Factor XI (fXI) is the zymogen of factor XIa (fXIa), a plasma protease that converts factor IX (fIX) to factor IXaβ (fIXaβ) during blood coagulation [1,2]. FXI is a 160 kDa dimer of identical subunits, each containing four apple domains (A1 to A4) and a trypsin-like catalytic domain [3-7]. The apple domains form a disk-like structure on which the catalytic domain rests in a “cup-and-saucer” arrangement [7]. Congenital fXI deficiency is associated with a variable propensity for excessive trauma-induced bleeding [3,8,9]. The compact nature of fXI may render relatively susceptible to mutation-induced misfolding. FXI deficient patients typically have commensurate reductions in fXI activity and antigen (cross-reactive material negative [CRM–] deficiency) [3,10], and most mutations causing fXI deficiency compromise structure sufficiently to interfere with protein secretion in expression studies [3,4]. Of the ∼150 missense mutations identified in the fXI genes of fXI deficient patients, only twelve are associated with circulating dysfunctional protein (CRM+). Eight of these involve the catalytic domain (www.factorxi.org) [3,4]. Thus, little structure-function information on the apple domain disk has been gleaned from studying naturally occurring fXI variants.
FIX is initially cleaved by fXIa at the Arg145-Ala146 peptide bond to form factor IXα (fIXα), followed by cleavage of the Arg180-Val181 bond to form fIXaβ [2,11]. Substrate specificities for coagulation protease-mediated reactions are governed by binding interactions involving exosites on the protease that are distinct from the active site [12-14]. An exosite involved in fIX and fIXα binding is located within the fXIa A3 domain [11]. Replacement of the A3 domain results in a significant reduction in catalytic efficiency for cleavage of both fIX and fIXα [11]. Guella et al. described a 24 year old woman with 10% of the normal plasma fXI activity but with 50% of the normal antigen level [15]. This CRM+ patient has a missense mutation resulting in a Gly substitution for Arg184 at the N-terminus of the fXI A3 domain. Here we present a kinetic analysis of the functional consequences of the Gly184 substitution to the interaction with fIX. Guided by the results, we performed additional mutational analysis of the fXIa and fIX proteins. The results suggest a model for the fIX binding interaction with the fXIa A3 domain.
Experimental Procedures
Recombinant proteins
HEK293 fibroblasts (ATCC-CRL1573) were transfected with 40 μg pJVCMV containing human fXI [11,16,17] or fIX [18,19] cDNAs and 2 μg pRSVneo as described [11,16,17]. Protein was raised in serum-free medium (Cellgro Complete, Mediatech). For fIX, 10 μg/ml vitamin K1 was added to media. FXI was purified by antibody affinity chromatography [11,16,17]. In addition to wild type fXI (fXIWT), several variants were prepared. In fXI-Gly184, Arg184 is replaced with Gly, while in fXI-Ala183-185 Ile183, Arg184, and Asp185 are replaced with alanine. In fXI/PKA3 the A3 domain (residues 182 to 265) is replaced with the prekallikrein (PK) A3 domain [11,17]. Two modified forms of fXI/PKA3 were prepared. In fXI/PKA3-A, fXI sequence is restored at amino acids 183 to 185. In fXI/PKA3-B fXI sequence is restored at amino acids 183 to 185 and 260 to 264 [16]. FIX was purified by antibody affinity chromatography [18,19]. In addition to wild type fIX (fIXWT), four chimeric proteins were prepared in which Gla-domain residues are replaced with corresponding residues from human factor VII (fVII): (C1 – residues 4, 5, 9, 10 and 11; C2 – 13, 19 and 22; C3 – 30, 32, 33, and 34; and C4 – 41, 43, 44, and 46). Protein concentrations were determined by colorimetric assay (Bio-Rad,), and confirmed by densitometry on SDS-PAGE by comparison to standard plasma fXI or fIX preparations. FXI was converted to fXIa by incubation with fXIIa (20:1 substrate:enzyme) at 37°C for 24 hrs in 50 mM Tris-HCl pH 7.4, 100 mM NaCl (TBS). Complete activation was confirmed by SDS-PAGE.
Plasma clotting assay
FXI activities were determined in a partial thromboplastin time (PTT) assay. FXI deficient plasma (30 μl) was mixed with 30 μl fXI (0.3 to 30 nM) in TBS with 0.1% BSA (TBSA) and 30 μl PTT-A reagent (Diagnostica Stago). After incubation at 37°C for 5 min, 30 μl 25 mM CaCl2 was added, and time to clot formation was determined on an ST4 fibrometer (Diagnostica Stago). In a separate experiment, 30 μl of fXIa diluted in TBSA (0.3 to 30 nM) was mixed with 30 μl fXI-deficient plasma and 30 μl rabbit brain cephalin (Sigma). After incubation at 37 °C for 30 sec, 30 μl 25 mM CaCl2 was added, and time to clot formation determined. Results were compared to standard curves prepared with fXIWT or fXIaWT starting at 30 nM. Activities are percent of wild-type protein activity.
Hydrolysis of S-2366 by fXIa
FXIa (6 nM) was incubated with 50-2000 μM L-pyroglutamyl-L-prolyl-L-arginine p-nitroaniline (S-2366, Diapharma) in TBS at RT. In separate experiments, FXIa (6 nM) was incubated with 500 μM S-2366 and varying concentrations of fIXaβ (fIXaβ does not cleave S-2366) in TBS with 5 mM CaCl2 at RT. Generation of free p-nitroaniline (pNA) was followed by monitoring absorbance at 405 nm. Rates of pNA generation (nM/s) were determined using an extinction coefficient of 9920 M-1.cm-1 (405 nm). Km, kcat and Ki for S-2366 cleavage were determined by non-linear least squares fitting performed with MicroMath Scientific Software [11].
FIX activation by fXIa
The method for activation of fIX and fIX/fVII chimeras was previously reported [11]. FIX (50-3000 nM) in Assay Buffer (50 mM HEPES, pH 7.4, 125 mM NaCl, 5 mM CaCl2, 1 mg/ml PEG 8000) was incubated at RT with fXIa (1-500 nM active sites) in PEG 20,000-coated tubes. At various times samples were size fractionated on non-reducing 17% polyacrylamide-SDS gels, and stained with GelCode Blue (Pierce). Gels were imaged on an Odyssey Infrared Imaging System (LI-COR Biosciences). Conversion of fIX to fIXα and fIXaβ was assessed by densitometry. Full progress curves were constructed for disappearance of fIX, and generation of fIXα and fIXaβ.
Kinetic analysis
Steady-state kinetic parameters Kd, Km, kcat and Ki for cleavage of fIX and fIXα were obtained by numerical integration fitting of full progress curves of fIX depletion, and fIXα and fIXaβ formation, at varying substrate concentrations; and by analysis of the initial rate dependence of substrate depletion as a function of substrate concentration. Rates of cleavage of the fIX Arg145-Ala146 and Arg180-Val181 bonds were analyzed with KinTek Explorer Version 2.5 software [11,20] using the following reaction mechanism:
IXα* denotes fIXα bound to fXIa in a favorable orientation for cleavage after Arg180. For Kintek analysis, at least six data sets for fIX in the 25 to 3000 nM substrate concentration range, were analyzed simultaneously. For published data for fXIaWT and fXIa/PKA3, Ki1 and Ki2 were constrained to values determined using surface plasmon resonance (SPR) [11]. No constraints were imposed for analysis of other fXIa species. Results of numerical analyses were compared to those obtained by Michaelis-Menten analyses for cleavage after Arg145. Initial velocities (v0) of cleavage after fIX Arg145 were determined by measuring initial slopes of progress curves for disappearance of fIX, normalized to 1 nM fXIa active sites. Values for v0 were analyzed with the Michaelis-Menten equation, and Km and kcat were obtained from direct non-linear least squares analysis using Scientist Software.
Surface plasmon resonance (SPR)
Binding studies were performed on a Biacore T100 flow biosensor (Biacore, Uppsala, Sweden) as described [11]. Briefly, plasma fXIa with the active site irreversibly blocked by FPR-CMK was bound to CM5 sensorchips by standard amine coupling. Recombinant fIX (1-2000 nM) was passed over the chips at 10 μl/min using a 6 min association time and 10 min dissociation time. Data were corrected for nonspecific binding by subtracting signals obtained with analytes infused through a flow cell without coupled protein. Sensor-chips were regenerated by washing with 30 mM EDTA. Binding was analyzed with BIAevaluation software (Biacore) using a 1:1 binding model. Kd was calculated from the quotient of the derived dissociation (kd) and association (ka) rate constant.
Results
FXIa-Gly184
On non-reducing SDS-PAGE, fXIWT and fXI-Gly184 migrate as 160 kDa dimers (Fig.1, left panel). Conversion of a fXI subunit to fXIa requires cleavage of the Arg369-Ile370 bond between the heavy chain (apple domains) and the catalytic domain (Fig.1, right panel) [4-6]. The tripeptide S-2366 was cleaved similarly by fXIaWT (Fig.2A, Km 185 ± 40 μM, kcat 41 ± 2 sec-1) and fXIa-Gly184 (Fig.2B, Km 200 ± 40 μM, kcat 45 ± 2 sec-1), indicating the Gly184 substitution did not alter the catalytic active site. Previously, we showed that fIXaβ inhibits fXIa cleavage of S-2366 through a mixed mechanism involving interactions with the fXIa active site and areas remote from the active site [21]. Ki for inhibition of fXIaWT cleavage of S-2366 by fIXaβ (90 ± 20 nM, Fig.2C) was ∼2-fold lower than for fXIa-Gly184 (190 ± 30 nM, Fig.2D), indicating binding of fIXaβ to fXIa-Gly184 is modestly weaker than to fXIaWT. These values are in reasonable agreement with those determined by Kintek analysis for the entire reaction (Table 1, discussed below). In a PTT assay, fXI-Gly184 had ∼25% of fXIWT. In a second assay, fXIaWT or fXIa-Gly184 was added to plasma, bypassing the need for fXI activation. FXIa-Gly184 again displayed ∼25% of normal activity, indicating reduced plasma activity is due largely to a defect in fIX activation. These values are consistent with those reported by Guella et al. for the proband [15], who had plasma fXI activity that was ∼20% of what would be predicted based on the fXI antigen level.
Figure 1. FXI and fXIa.
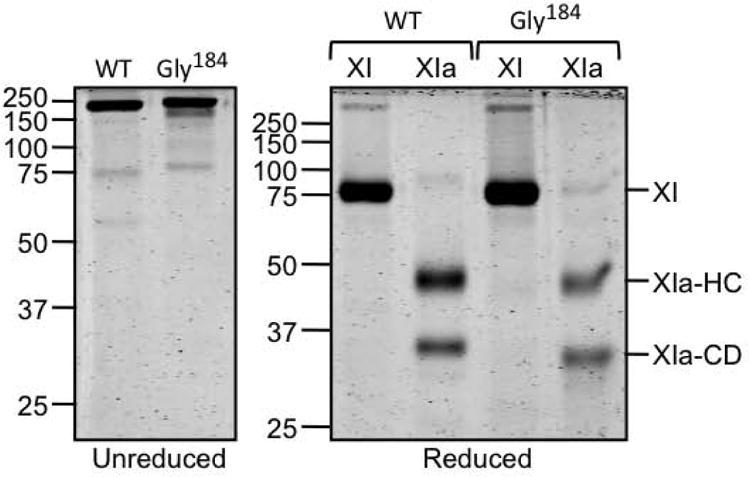
Left panel - Non-reducing SDS-PAGE stained with GelCode blue of recombinant fXIWT (WT) and fXI-Gly184. Right panel – Reducing SDS–PAGE of the zymogen (XI) and protease (XIa) forms of the proteins in the left hand panel. Positions of fXI zymogen (XI) and the heavy chain (XIa-HC) and catalytic domain (XIa-CD) are shown to the right of the panel. Positions of molecular mass standards for both gels are indicated on the left in kilodaltons.
Figure 2. S-2366 cleavage.
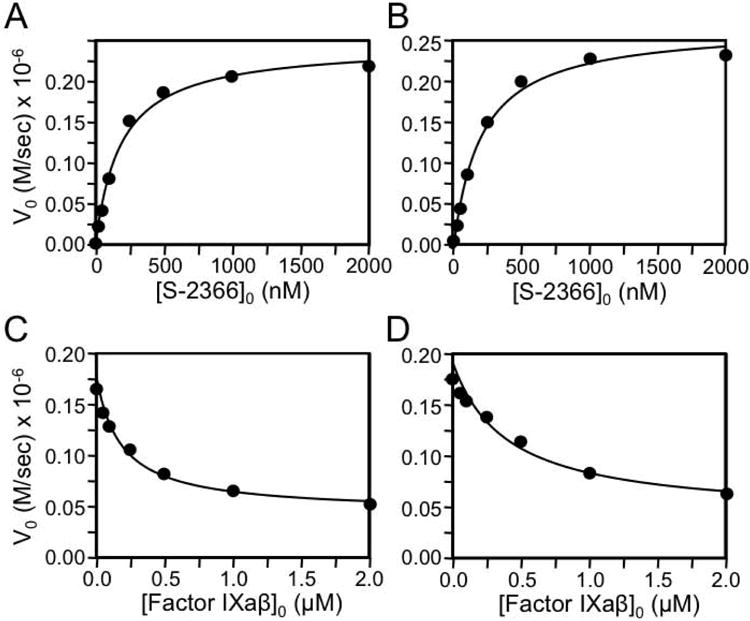
(A & B) Shown are the rates of cleavage of the tripeptide substrate S-2366 by 6 nM (A) fXIaWT or (B) fXI-Gly184. (C & D) Cleavage of S-2366 (500 nM) by 6 nM (C) fXIa fXIWT or (D) fXI-Gly184 in the presence of varying concentrations of fIXaβ. For all panels, data points represent averages of duplicate runs.
Table 1. Kinetic parameters for cleavage of fIX by fXIa.
Simulation of full-progress experimental traces was performed with KinTek software (KinTek Explorer Version 2.5) using the Equation shown in the Experimental Procedures section. Km and kcat for activation were calculated from individual rate constants for each step. Values are the mean ± standard deviation for each experiment. Ki was fixed for reactions with fXIa-WT and fXIa/PKA3 based on published SPR results.11
| Cleavage of fIX after Arg145 | ||||||
|---|---|---|---|---|---|---|
| Protease | Substrate | Kcat1 (min-1) | Kd1 (μM) | Km1 (μM) | Catalytic Efficiency 1 (μM-1 · min-1) | Ki1 (μM) |
| fXIaWTa | fIX | 12 ± 1 | 0.10 ± 0.01 | 0.20 ± 0.01 | 60 ± 6 | 0.14b |
| fXIa/PKA3a | fIX | 4.9 ± 0.1 | 4.9 ± 0.2 | 4.9 ± 0.2 | 1.0 ± 0.1 | 3b |
| fXIa-Gly184 | fIX | 13 ± 3 | 0.3 ± 0.1 | 0.7 ± 0.3 | 19 ± 10 | 0.4 ± 0.2 |
| fXIa-Ala183-185 | fIX | 9 ± 1 | 8 ± 1 | 10 ± 2 | 0.9 ± 0.2 | 6 ± 1 |
| fXIa/PKA3-A | fIX | 6.4 ± 0.2 | 1.3 ± 0.1 | 1.4 ± 0.1 | 4.6 ± 0.4 | 1.9 ± 0.1 |
| fXIa/PKA3-B | fIX | 19 ± 2 | 0.06 ± 0.01 | 0.09 ± 0.02 | 200 ± 30 | 0.5 ± 0.1 |
| Cleavage of fIX after Arg180 | ||||||
| Protease | Substrate | Kcat2 (min-1) | Kd2 (μM) | Km2 (μM) | Catalytic Efficiency 2 (μM-1 · min-1) | Ki2 (μM) |
| fXIaWTa | fIX | 35 ± 6 | 0.06 ± 0.01 | 0.08 ± 0.01 | 400 ± 100 | 0.06b |
| fXIa/PKA3a | fIX | 0.6 ± 0.1 | 7 ± 1 | 7 ± 1 | 0.09 ± 0.02 | 3b |
| fXIa-Gly184 | fIX | 40 ± 10 | 0.2 ± 0.1 | 0.2 ± 0.1 | 200 ± 50 | 0.5 ± 0.2 |
| fXIa-Ala183-185 | fIX | 0.4 ± 0.1 | 3 ± 1 | 3 ± 1 | 0.13 ± 0.01 | 0.29 ± 0.01 |
| fXIa/PKA3-A | fIX | 1.7 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 | 0.28 ± 0.02 |
| fXIa/PKA3-B | fIX | 39 ± 4 | 0.04 ± 0.01 | 0.07 ± 0.01 | 600 ± 100 | 0.30 ± 0.03 |
data for fXIaWT and fXIa/PKA3 are from reference 11.
fixed values for Ki are based on data from surface plasmon resonance studies [reference 11].
Cleavage of fIX by fXIa-Gly184
FIX conversion to fIXaβ involves sequential cleavage of the Arg145-Ala146 and Arg180-Val181 bonds [2,11]. We showed that catalytic efficiency for the Arg180-Val181 cleavage by fXIaWT is ∼7-fold greater than for the Arg145-Ala146 cleavage (Table 1), preventing fIXα accumulation (Fig.3A and 3B) [11]. Replacing the fXIa A3 domain with A3 from the homolog PK results in a protease (fXIa/PKA3) lacking exosites required for normal fIX and fIXα cleavage [11,17]. The PKA3 replacement markedly reduces catalytic efficiency for both cleavages (Table 1), but the second cleavage is compromised to a greater extent, leading to fIXα accumulation (Fig.3A). FXIa-Gly184 cleaves fIX more slowly than fXIaWT (Fig.3A, 3C-3E and Table 1), with Kms for cleavage after Arg145 and Arg180 that are 3- and 2-fold higher, respectively, than for fXIaWT (Table 1). As with fXIaWT, relatively little fIXα accumulates during fIX activation by fXIa-Gly184 (Fig.3A and 3C). Michaelis-Menten analyses of conversion of fIX to fIXα agree with the numerical analysis for cleavage after Arg145 (Table 2). The results support the hypothesis that Arg184 is a component of a fIX/fIXα binding site on the A3 domain. The magnitude in reductions in the catalytic efficiencies for the two bond cleavages with fXIa-Gly184 are consistent with the ∼75% overall reduction in activity observed in clotting assays, and with the original description of the discrepancy in fXI activity and antigen in the proband [15]. However, the defect in is modest compared to that in fXIa/PKA3, indicating the exosite is not completely disrupted.
Figure 3. FIX cleavage by fXIa.
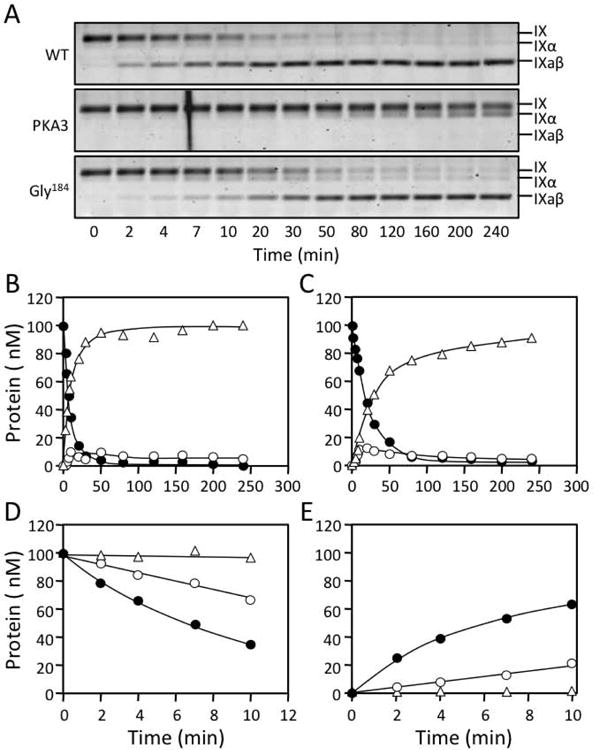
(A) Non-reducing 17% polyacrylamide-SDS gels of 100 nM fIX in Assay Buffer with Ca2+ incubated at RT with 3 nM active sites of fXIaWT (top), fXIa/PKA3 (middle) or, fXIa-Gly184 (bottom). Positions of standards for fIX, fIXα and fIXaβ are indicated at the right of each panel. (B & C) Progress curves of fIX disappearance (●), and fIXα (○) and fIXaβ (Δ) generation for (B) fXIaWT or (C) fXI-Gly184 from panel A. Lines represent least-squares fits to the data. (D) Initial rates of fIX disappearance for fXIaWT (●), fXIa-Gly184 (○) and fXIa/PKA3 (Δ) determined over the first 10 min of activation from panel A. (E) Initial rates of fIXaβ formation for fXIaWT (●), fXIa-Gly184 (○) and fXIa/PKA3 (Δ) determined over the first 10 min of activation from panel A.
Table 2. Kinetic parameters for cleavage of fIX by fXIa determined from initial velocities.
The initial velocities (v0) of cleavage of fIX after Arg145 was plotted against initial substrate concentration, and analyzed using the Michaelis-Menten equation. Km and kcat were obtained from direct non-linear least squares analysis using Scientist Software. Due to the low affinity between the enzyme and substrate in reactions using fXIa variants, we were not able to reach saturation reactions with fXIa-Ala183-185 or fXIa/PKA3. Thus, values for Km are approximate and the value for kcat for fXIa-Ala183-185 was not determined (ND).
| Protease | Substrate | Bond cleaved | Kcat1 (min-1) | Km1 (μM) | Catalytic Efficiency 1 (μM-1 · min-1) |
|---|---|---|---|---|---|
| fXIaWTa | fIX | Arg145 | 10 ± 1 | 0.27 ± 0.07 | 40 ± 10 |
| fXIa/PKA3a | fIX | Arg145 | 6 ± 1 | 3.9 ± 0.9 | 1.6 ± 0.4 |
| fXIa-Gly184 | fIX | Arg145 | 11 ± 1 | 0.8 ± 0.2 | 12 ± 4 |
| fXIa-Ala183-185 | fIX | Arg145 | ND | > 8 | 0.70 ± 0.10 |
| fXIa/PKA3-A | fIX | Arg145 | 5 ± 1 | 1.3 ± 0.4 | 4 ± 1 |
| fXIa/PKA3-B | fIX | Arg145 | 12 ± 1 | 0.08 ± 0.01 | 140 ± 20 |
data for fXIaWT and fXIa/PKA3 are from reference 11.
Structure of the fXI A3 domain
To gain insight into the potential effects of the Arg184Gly mutation we examined the structural context of Arg184 utilizing the fXI crystal structure [7]. The topology of the fXI A3 domain (Fig.4A) shows the Arg184 side chain projecting upward on one face of the β-sheet (yellow) adjacent to the Cys182-Cys265 disulfide bond connecting the N- and C- termini of A3. The structural analysis software Metapocket [22], which utilizes eight predictors to identify potential surface binding sites, identified three hydrophobic pockets on the surface of fXI A3 (Fig.4B). The alkyl groups of the Arg184 and Arg210 side chains contribute to pocket 1 with additional contributions from the side chains of Ile186, Leu262, and Phe221, and Phe206 from the α-helix (Fig.4A and 4B). Modeling of the PK structure was carried out previously utilizing the fXI crystal structure as a template (Fig.4C) [23]. The model indicates that pocket 1 is not present in PK (Fig.4C and 4D). In fXI, residue 209 on the α-helix is Gly (Fig.4A), while in PK it is Arg (Fig.4C). The PK Arg209 side chain is predicted to interfere with formation of a hydrophobic pocket in this location (Fig.4C and 4D).
Figure 4. The FXI A3 Domain and fIX Gla-domain.
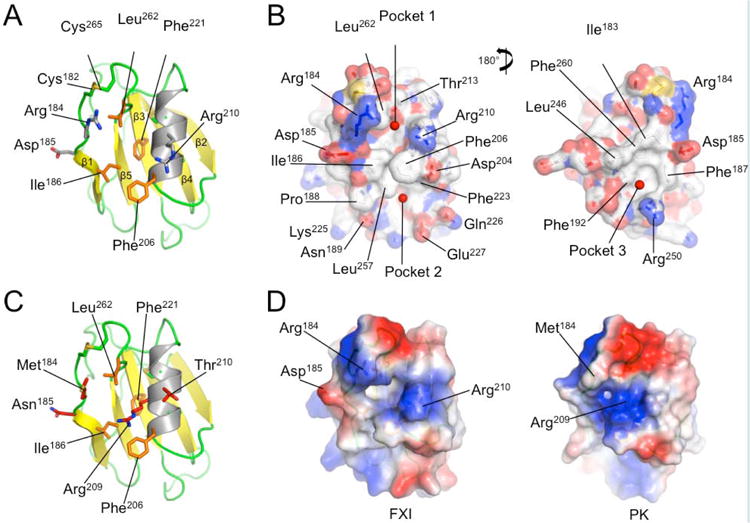
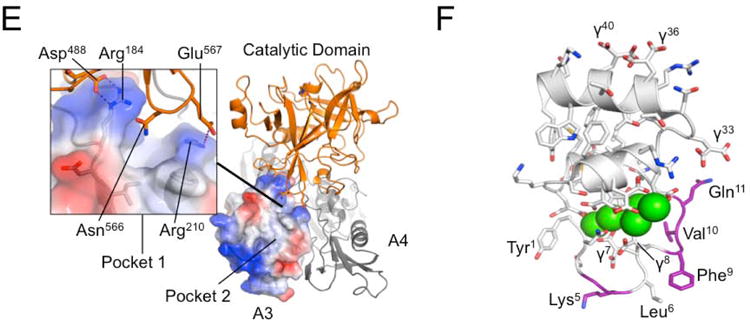
(A) Topological diagram showing the A3 domain of human fXI with the β-sheet indicated in yellow and α-helix in grey. Side chains of residues in the locality of Arg184 are shown as sticks. (B) Surface representations of the fXI A3 domain are shown with partial transparency to highlight side chains of specific residues that are colored by atom type. Two rotations are shown related by 180 degrees. The software program Metapocket identified three pockets on the surface of A3 (shown as red balls) (C) Topological diagram of the human PK A3 domain model with side chains of non-conserved residues in the area of the pocket in fXI shown as sticks. Note that the side chain of Arg209 in PK occupies the site of the hydrophobic pocket in fXI. (D) Charged surface representations of fXI (left) and PK (right) A3 domains. Blue indicates positive charge and red negative charge. Note the absence of pocket 1 in the PK A3 domain model due to the Arg209 side chain. (E) Structure of the zymogen fXI monomer showing the A3 domain as a charged surface representation, and the A4 and catalytic domains as ribbon drawings. Note that Arg184 and the adjacent hydrophobic pocket 1 are covered by the catalytic domain. The inset shows specific interactions between side chains of the protease domain and A3. (F) Topological diagram of the human fIX Gla-domain with residues in the Ω-loop (4 to 11) that differ from the corresponding region of the human fVII Gla-domain highlighted in magenta. Positions of certain γ-carboxyglutamic acid residues are indicated by the symbol “γ”. Calcium ions in the vicinity of the Ω-loop are represented by green spheres. The image is derived from a structure for a complex between the human fIX Gla-domain and the antibody 10C12.29 Figures prepared with Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC. http://www.pymol.org/citing).
Charged surface representations (Fig.4B and 4D) illustrate that Arg184 and Arg210 contribute to positively charged patches on either side of pocket 1. Arg184 and Asp185 run along one side of pocket 1, with the other side formed primarily by the α-helix. Leu262 is in proximity to residues 184 and 185 by virtue of the Cys182-Cys265 bond, and contributes to the wall of pocket 1 (Fig.4A). The Gly184 substitution would likely lead to loss of a positive charge that could contribute to electrostatic interactions with fIX, to the loss of hydrophobic interactions with the alkyl group of the Arg184 side chain, and would introduce main chain flexibility that could affect local loop conformation.
In fXI, Arg184 forms salt bridges with Asp488 and Asn566 from the catalytic domain [7]. As a result, Arg184 and hydrophobic pocket 1 are buried beneath the catalytic domain (Fig.4E), and are not accessible for fIX binding. The observation that fIX and fIXα bind to fXIa, but not to fXI [11,18], is consistent with the hypothesis that Arg184 and pocket 1 are part of a fIX binding site that must be exposed during fXI conversion to fXIa through repositioning of the catalytic domain relative to the A3 domain.
Hydrophobic pocket 2 is located on the opposite side of Phe206 from pocket 1 (Fig.4B left panel), with Asn189, Phe223, Ser225, Gln226, and Glu227 contributing to its formation. It would seem that this pocket is less likely to be a component of a fIX-binding site specific to fXIa, as a similar pocket (with some substitutions) is also likely to be present on the PK A3 domain. Furthermore, pocket 2 is exposed to the aqueous phase in the zymogen structure. Hydrophobic pocket 3, which is on the opposite side of A3 from pocket 1 (Fig.4B right panel), is formed by Asp185, Phe187, Phe192, Leu246, Arg250 and Phe260. While not predicted to be present in PK, this pocket is also unobstructed in zymogen fXI.
Cleavage of fIX by fXIa-Ala183-185
The kinetic data for fXIa-Gly184 are consistent with residue Arg184 contributing to a binding site for fIX and fIXα on the fXIa A3 domain. In fXIa-Ala183-185, Arg184 and the adjacent residues Ile183, and Asp185 are replaced with alanine. FXIa-Ala183-185 has <1% of the activity of fXIaWT in a PTT assay. Cleavage patterns and kinetic parameters for fIX activation by fXIa-Ala183-185 are shown in Fig.5A and in Tables 1 and 2. The protease exhibits a defect in fIX activation that is comparable to the defect observed with fXIa/PKA3 (Tables 1) [11]. This shows that replacement of Ile183, Arg184 and Asp185 completely disrupts the specific interaction between the A3 domain and fIX and fIXα, and suggests that these residues in combination are a major component of the fIX-binding site.
Figure 5. Progress curves of fIX activation by fXIa183-185 and fXIa/PKA3 gain-of-function variants.
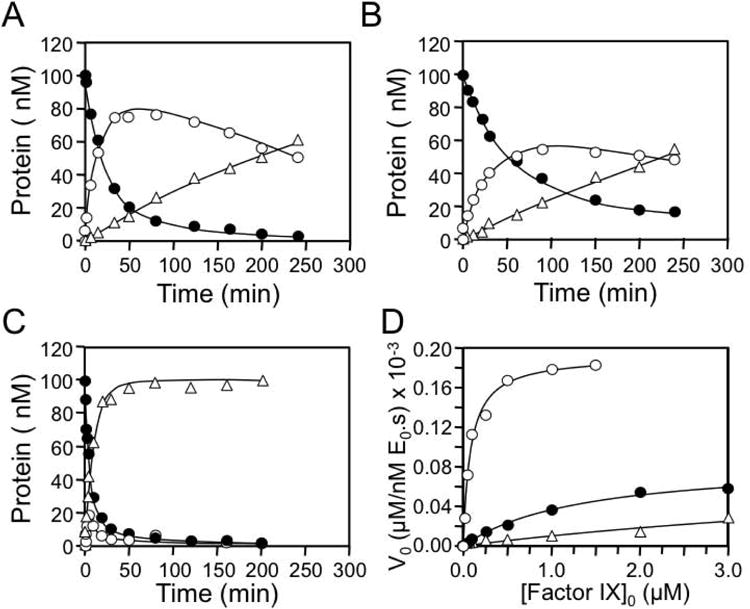
Shown are progress curves of fIX disappearance (●), and fIXα (○) and fIXaβ (Δ) generation for reactions containing 100 nM fIX and (A) 30 nM fXIa183-185, (B) 3 nM fXIa/PKA3-A, and (C) 3 nM fXIa/PKA3-B. Note the different fXIa concentrations used in the three reactions. FXIa concentrations represent the concentration of active subunits. (D) Initial velocities of cleavage of fIX after Arg145 (conversion of fIX to fIXα) by fXIa183-185 (Δ), fXIa/PKA3-A (●), and fXIa/PKA3-B (○) as a function of fIX concentration.
Cleavage of fIX by gain-of-function variants of fXIa/PKA3
FXI/PKA3 was used as a scaffold to reintroduce fXI sequence to restore activity toward fIX. The wild type PK sequence His183, Met184 and Asn185 in fXIa/PKA3 was replaced with Ile183, Arg184 and Asp185 from the fXIa sequence to create fXIa/PKA3-A. FXIa/PKA3-A has ∼5% of fXIaWT coagulant activity, with catalytic efficiencies for cleavage after Arg145 and Arg180 that are 4- and 10-fold greater, respectively, than for fXIa/PKA3 (Table 1). However, these values remain considerably lower than those for fXIaWT, and accumulation of fIXα confirms that the exosite and the activation mechanism are not completely restored (Fig.5B).
In the fXI A3 structure the Cys182-Cys265 bond brings amino acids at the N- (183-185) and C- (residues 260-264) termini into proximity with each other (Fig.4A) [6,7]. Leu262 and Phe260 contribute to formation of hydrophobic pockets 1 and 3, respectively. To determine if the C-terminus of the A3 domain contributes to formation of the fIX-binding site, we replaced the PK sequence Tyr260-Ser261-Leu262-Leu263-Thr264 in fXIa/PKA3-A with Phe260-Ser261-Leu262-Gln263-Ser264 from fXI, creating fXIa/PKA3-B. Superimposing these changes on the N-terminal changes at residues 183-185 restored normal fIX activation (Fig.5C and 5D) to fXIa/PKA3. Indeed, fXIa/PKA3-B is a modestly more efficient activator of fIX than fXIaWT (Tables 1 and 2), with catalytic efficiencies for the first and second cleavages that are 3.3- and 1.5-fold greater, respectively, than for fXIaWT. As with fXIaWT, cleavage of fIX by fXIa/PKA3-B was associated with little fIXα accumulation. Thus, replacing six amino acids in the PKA3 domain (residues 183,184,185, 260, 263 and 264) with the corresponding fXI residues is sufficient to restore exosite activity and the mechanism for fIX activation in fXIa/PKA3.
Cleavage of fIX/VII-Gla chimeras by fXIa
The fIX-Gla domain is required for fIX binding to fXIa [18]. Replacing the fIX Gla-domain with the fVII Gla-domain results in a molecule (fIX-VIIGla) that retains activity in phospholipid dependent reactions, but binds poorly to fXIa [18]. This indicates that fIX-specific elements in the Gla-domain are required for binding to fXIa. The primary sequences of the human fIX and fVII Gla-domains are shown in Fig.6A. Based on the comparison, fIX/VII chimeras (C1 to C4) were prepared in which three to five residues of fIX sequence are replaced with fVII sequence (Fig.6A and 6B). FXIa activated fIX-VIIGla significantly more slowly than fIXWT (Fig.6C), with substantial accumulation of fIXα (Fig.6D). FIXWT and chimeras C2, C3, and C4 were converted to fIXaβ at roughly comparable rates (Fig.6C), with little fIXα accumulation (Fig.6D). Chimera C1 demonstrated a pronounced defect in activation by fXIa (Fig.6C), with significant fIXα accumulation (Fig.6D). In surface plasmon resonance studies, C1 bound poorly to immobilized fXIa (Kd >2000 nM), compared to fIXWT (Kd 350 nM), C2 (Kd 400 nM), or C4 (Kd 350 nM). There was insufficient C3 to conduct an SPR study. The substitutions in C1 are in the phospholipid binding Ω-loop of the Gla-domain (Fig.4F) [1,2], indicating residues within this loop are required for fIX binding to fXIa.
Figure 6. Factor IX with fVII sequence in the Gla-domain.
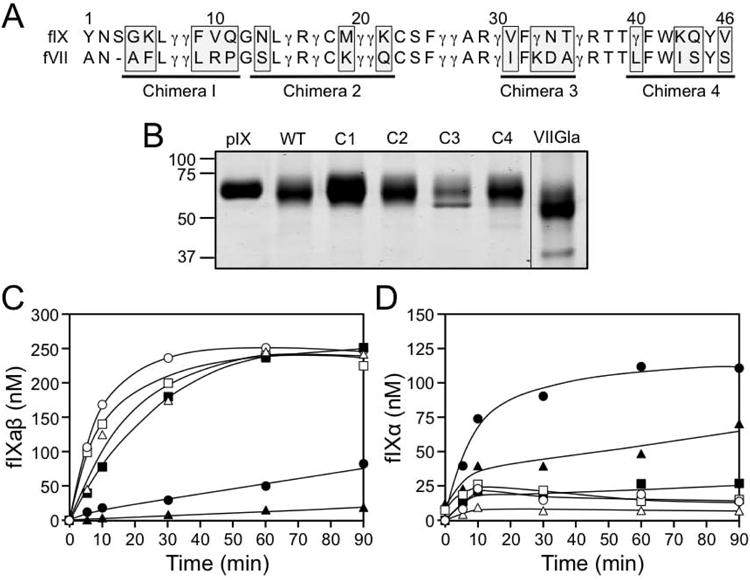
(A) Primary sequences of the human factor IX (fIX) and factor VII (fVII) Gla-domains. The numbering system shown is for fIX. The symbol γ indicates the position of γ-carboxyglutamic acid residues. Underlined sequences were changed from fIX sequence to fVII sequence to generate chimeras C1, C2, C3 and C4. The amino acids changed in each chimera are highlighted by the gray boxes. (B) Stained SDS-polyacrylamide gel of purified plasma fIX (pIX), recombinant wild type fIX (WT), fIX/fVII chimeras (C1, C2, C3, and C4) and fIX in which the Gla-domain from residues 1 through 46 are changed to fVII sequence (fIX/VII-Gla - abbreviated VIIGla). The weaker band running underneath fIX C3 is contaminating bovine serum albumin from conditioned media. (C and D) FXIa (2 nM) was incubated with 250 nM fIXWT (○), C1 (●), C2 (□), C3 (■), C4 (Δ) and fIX/VII-Gla (▲) (250 nM) in TBS with calcium, as described under methods. Shown are concentrations of (C) fIXaβ and (D) fIXα at various times as determined by densitometry of SDS-polyacrylamide gels.
Discussion
The fXI substitution Arg184Gly is located within the A3 domain [6,7], an area implicated in binding to heparin [24], platelet glycoprotein 1B [25], and fIX [11]. Recently, we presented evidence that A3 contains an exosite that binds fIX and the activation intermediate fIXα that is central to the mechanism of fIX activation by fXIa [11]. Consistent with the hypothesis that Arg184 is a component of this exosite, our kinetic analysis shows that fIX and fIXα have moderately reduced affinity for fXI-Gly184 compared to fXIaWT, consistent with the ∼4-fold lower fXI activity relative to antigen observed in the proband's plasma [15]. Arg184 is partially buried under the catalytic domain in fXI, and is not accessible for fIX binding (Fig.4E), consistent with the observation that fIX does not bind fXI [11,18], and the hypothesis that conformational changes must occur upon conversion to fXIa that exposes the fIX binding site. In fXI, the A3 domain forms contacts with the catalytic domain through salt bridges between Arg184 and Asp488 and Asn566, and between Arg210 and Glu567 (Fig.4E) [7]. These bonds are probably disrupted upon fXI activation, as structures for isolated fXIa catalytic domains show Asp488, Asn566 and Glu567 forming interactions within the active catalytic domain that are not present in fXI [26,27]. Asp488, Asn566 and Glu567 may function as a type of latch that keeps the fIX-binding exosite on A3 concealed, and that is released after cleavage of the fXI Arg369-Ile370 bond generates fXIa.
The functional defect caused by the Gly184 substitution is modest compared to the defect reported for fXIa/PKA3 [11,17], showing that residues in addition to Arg184 contribute to fIX binding. On A3, Arg184 and adjacent residues form a ridge between two hydrophobic pockets that are not predicted to be present in the homolog PK. Replacing residues 183 to 185 in fXIa eliminates exosite activity, while introducing fXI residues 183 to 185 into fXIa/PKA3 (fXIa/PKA3-A) partially restores activity, indicating these residues form part of the fIX binding exosite. The substitutions in fXIa-Ala183-185 could also disrupt the adjacent hydrophobic pockets, one or both of which could be involved in fIX binding. Interestingly, replacing PK residues Tyr260, Leu263, and Thr264 in the C-terminus of fXIa/PKA3-A with Phe260, Gln263, and Ser264 from fXI results in a protease, fXIa/PKA3-B, with activity toward fIX comparable to fXIaWT. Residues 260 to 265 run underneath the chain containing 183-185 (Fig.4A), with Leu262 and Phe260 (Fig.4B) contributing to hydrophobic pockets 1 and 3, respectively. Introducing the C-terminal sequence from fXI A3 in fXIa/PKA3-B could restore fIX binding by supplying missing components of the binding site, or by properly orienting structures such as residues 183 to 185 that are required for binding.
The topography of the A3 domain in the vicinity of Arg184 suggests that fIX binding may involve charged and hydrophobic interactions. In this regard, it is interesting to note that the phospholipid-binding Gla-domain of fIX is required for fIX binding to fXIa [18]. Replacing the fIX Gla-domain with the Gla-domain from fVII [18] or protein C [28] reduces the capacity of fXIa to bind to fIX, and to activate fIX, indicating structures specific to the fIX Gla-domain mediate binding to fXIa. Replacing residues 4 to 11 in fIX (the Ω-loop) with those from fVII, reduces fXIa-mediated fIX activation, and is associated with fIXα accumulation, similar to what is observed during fIX activation by fXIa-Ala183-185. The Gla-domain facilitates fIX binding to phosphatidylserine (PS)-rich surfaces in a Ca2+-dependent manner through a combination of charged and hydrophobic interactions involving the Ω-loop [2]. This interaction enhances fIX activation by the factor VIIa/tissue factor complex, and factor X activation by fIXaβ, by reducing Km for the reactions [1,2]. Ca2+-dependent binding of fIX to the fXIa A3 domain also determines Km for fIX activation [11], suggesting the interaction serves a similar purpose to fIX binding to phospholipids during activation by factor VIIa. The image of the fIX-Gla domain in Fig.4F is based on a structure reported by Huang et al. for the Gla-domain in complex with the conformation-specific anti-fIX IgG 10C12 [29]. The antibody contains a hydrophobic pocket that accommodates Leu6, Phe9 and Val10 from the Gla-domain. Taken as a whole, the data are consistent with a model in which residues in the Ω-loop of fIX and fIXα mediate binding to the fXIa A3 domain, through interactions involving residues at the N-terminus of A3, including Arg184, and an adjacent hydrophobic pocket.
The environments in which fXIa activates fIX during coagulation in vivo are not established. Unlike many other protease-substrate interactions involved in hemostasis, fXIa activation of fIX is not enhanced by phosphatidylserine-rich phospholipids, such as those found on the surface of activated platelets [3]. However, fXI and fXIa do bind to the platelet receptors glycoprotein 1b [25] and ApoER2' [30], suggesting fXIa-mediated fIX activation may occur on the platelet surface. The resulting fIXaβ would then be in a position to participate in factor X activation. Platelets also release polymers of inorganic phosphate upon activation that enhance fXI activation [31]. We observed that other polyanions that enhance fXI activation in vitro, such as dextran sulfate or heparin, have deleterious effects on fIX activation by fXIa (unpublished observations). However, polyphosphate does not inhibit fIX activation by fXIa, suggesting that fIX activation by polyphosphate-bound fXIa may occur at sites of platelet activation.
The gene for fXI is the product of a duplication event involving the PK gene [32]. The fXI subunit and PK (a monomeric protein) share identical domain structure, and are 58% identical in amino acid sequence in humans [5-7,23,33]. Despite the similarities, the active form of PK (α-kallikrein) is a poor fIX activator. The amino acid sequences at the N- and C-termini of fXIa A3 implicated in fIX binding are highly conserved across mammalian species [5,32], and are distinctly different from the corresponding sequence in PK, consistent with this area of the domain contributing to a fXIa specific function. The data presented here support the conclusion that residues 183-185 and 260-264 on fXIa are critical to formation of a binding site for fIX and its activation intermediate fIXα that are required for normal conversion of fIX to fIXaβ, and that the defect in the naturally occurring variant fXI-Gly184 is related to an alteration in this binding site.
Acknowledgments
The authors wish to acknowledge support from awards HL81326 and HL58837 (D.G.), HL80018 (I.M.V.), and HL36365 (S.P.B) from the National Heart, Lung and Blood Institute; and RG/12/9/29775JE (J.E.) from the British Heart Foundation.
Addendum
Authorship: Y.G. performed the experiments investigating fIX activation, and wrote the manuscript I.M.V. contributed to design of experiments for fIX activation, and performed kinetic analyses of data. M-f.S. prepared recombinant proteins and tested their activity against chromogenic substrates and in clotting assays. S.P.B contributed to experimental design, structural interpretation, and writing of the manuscript. J.E. performed structural analysis of the effects of the Arg184Gly substitution on A3 domain structure. D.G. was responsible for oversight of the project and preparation of the final manuscript.
Footnotes
Conflict of interest disclosure: D.G. is a consultant to several pharmaceutical companies, Merck, Novartis, Isis, and Aronora, and receives consultant's fees.
References
- 1.Brummel-Ziedens K, Mann KG. Hematology: basic principles and practice. 6th. Churchill Livingstone-Elsevier; Philadelphia: 2012. Molecular basis of blood coagulation; pp. 1821–41. [Google Scholar]
- 2.Vadivel K, Schmidt AE, Marder VJ, Krishnaswamy S, Bajaj SP. Hemostasis and Thrombosis: basic principles and clinical practice. 6th. Lippincott, Williams & Wilkins; Philadelphia: 2012. Structure and function of vitamin K-dependent coagulation and anticoagulation proteins; pp. 208–32. [Google Scholar]
- 3.Gailani D, Renné T, Emsley J. Factor XI and the Plasma contact system. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; NY, NY: 2010. [Google Scholar]
- 4.Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–77. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujikawa K, Chung D, Hendrickson L, Davie E. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–24. doi: 10.1021/bi00357a018. [DOI] [PubMed] [Google Scholar]
- 6.McMullen B, Fujikawa K, Davie E. Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry. 1991;30:2056–60. doi: 10.1021/bi00222a008. [DOI] [PubMed] [Google Scholar]
- 7.Papagrigoriou E, McEwan PA, Walsh PN, Emsley J. Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat Struct Mol Biol. 2006;13:557–8. doi: 10.1038/nsmb1095. [DOI] [PubMed] [Google Scholar]
- 8.Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009 Jul;7(Suppl 1):84–7. doi: 10.1111/j.1538-7836.2009.03395.x. [DOI] [PubMed] [Google Scholar]
- 9.Bolton-Maggs PH. Factor XI deficiency – resolving the enigma? Hematology Am Soc Hematol Educ Program. 2009:97–105. doi: 10.1182/asheducation-2009.1.97. [DOI] [PubMed] [Google Scholar]
- 10.Saito H, Ratnoff OD, Bouma BN, Seligsohn U. Failure to detect variant (CRM+) plasma thromboplastin antecedent (factor XI) molecules in hereditary plasma thromboplastin antecedent deficiency: a study of 125 patients of several ethnic backgrounds. J Lab Clin Med. 1985;106:718–22. [PubMed] [Google Scholar]
- 11.Geng Y, Verhamme IM, Messer A, Sun MF, Smith SB, Bajaj SP, Gailani D. A Sequential Mechanism for Exosite-Mediated Factor IX Activation by Factor XIa. J Biol Chem. 2012;287:38200–9. doi: 10.1074/jbc.M112.376343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnaswamy S. Exosite-driven substrate specificity and function in coagulation. J Thromb Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 13.Page MJ, Macgillivray RT, Di Cera E. Determinants of specificity in coagulation proteases. J Thromb Haemost. 2005;3:2401–8. doi: 10.1111/j.1538-7836.2005.01456.x. [DOI] [PubMed] [Google Scholar]
- 14.Bock PE, Panizzi P, Verhamme IM. Exosites in the substrate specificity of blood coagulation reactions. J Thromb Haemost. 2007;5:81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guella I, Soldà G, Spena S, Asselta R, Ghiotto R, Tenchini ML, Gastaman G, Duga S. Molecular characterization of two novel mutations causing factor XI deficiency: a splicing defect and a missense mutation responsible for a CRM+ defect. Thromb Haemost. 2008;99:523–30. doi: 10.1160/TH07-12-0723. [DOI] [PubMed] [Google Scholar]
- 16.Sun MF, Zhao M, Gailani D. Identification of amino acids in the factor XI apple 3 domain required for activation of factor IX. J Biol Chem. 1999;274:36373–8. doi: 10.1074/jbc.274.51.36373. [DOI] [PubMed] [Google Scholar]
- 17.Sun Y, Gailani D. Identification of a factor IX binding site on the third apple domain of activated factor XI. J Biol Chem. 1996;271:29023–8. doi: 10.1074/jbc.271.46.29023. [DOI] [PubMed] [Google Scholar]
- 18.Aktimur A, Gabriel MA, Gailani D, Toomey JR. The factor IX gamma-carboxyglutamic acid (Gla) domain is involved in interactions between factor IX and factor XIa. J Biol Chem. 2003;278:7981–7. doi: 10.1074/jbc.M212748200. [DOI] [PubMed] [Google Scholar]
- 19.Smith SB, Verhamme IM, Sun MF, Bock PE, Gailani D. Characterization of Novel Forms of Coagulation Factor XIa: independence of factor XIa subunits in factor IX activation. J Biol Chem. 2008;283:6696–6705. doi: 10.1074/jbc.M707234200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson KA, Simpson ZB, Blom T. Global Kinetic Explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal Biochem. 2009;387:20–9. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Ogawa T, Verhamme IM, Sun MF, Bock PE, Gailani D. Exosite-mediated substrate recognition of factor IX by factor XIa. The factor XIa heavy chain is required for initial recognition of factor IX. J Biol Chem. 2005;280:23523–30. doi: 10.1074/jbc.M500894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Li Y, Lin B, Schroeder M, Huang B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics. 2011;27:2083–8. doi: 10.1093/bioinformatics/btr331. [DOI] [PubMed] [Google Scholar]
- 23.Hooley E, McEwan PA, Emsley J. Molecular modeling of the prekallikrein structure provides insights into high-molecular-weight kininogen binding and zymogen activation. J Thromb Haemost. 2007;5:2461–6. doi: 10.1111/j.1538-7836.2007.02792.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhao M, Abdel-Razek T, Sun MF, Gailani D. Characterization of a heparin binding site on the heavy chain of factor XI. J Biol Chem. 1998;273:31153–9. doi: 10.1074/jbc.273.47.31153. [DOI] [PubMed] [Google Scholar]
- 25.Baglia FA, Gailani D, López JA, Walsh PN. Identification of a binding site for glycoprotein Ib-alpha in the Apple 3 domain of factor XI. J Biol Chem. 2004;279:45470–6. doi: 10.1074/jbc.M406727200. [DOI] [PubMed] [Google Scholar]
- 26.Navaneetham D, Jin L, Pandey P, Strickler JE, Babine RE, Abdel-Meguid SS, Walsh PN. Structural and mutational analyses of the molecular interactions between the catalytic domain of factor XIa and the Kunitz protease inhibitor domain of protease nexin 2. J Biol Chem. 2005;280:36165–75. doi: 10.1074/jbc.M504990200. [DOI] [PubMed] [Google Scholar]
- 27.Jin L, Pandey P, Babine RE, Weaver DT, Abdel-Meguid SS, Strickler JE. Mutation of surface residues to promote crystallization of activated factor XI as a complex with benzamidine: an essential step for the iterative structure-based design of factor XI inhibitors. Acta Crystallogr D Biol Crystallogr. 2005;61:1418–25. doi: 10.1107/S0907444905024340. [DOI] [PubMed] [Google Scholar]
- 28.Ndonwi M, Broze GJ, Agah S, Schmidt AE, Bajaj SP. Substitution of the Gla domain in factor X with that of protein C impairs its interaction with factor VIIa/tissue factor: lack of comparable effect by similar substitution in factor IX. J Biol Chem. 2007;282:15632–44. doi: 10.1074/jbc.M701908200. [DOI] [PubMed] [Google Scholar]
- 29.Huang M, Furie BC, Furie C. Crystal structure for the calcium-stabilized human factor IX Gla domain bound to a conformation-specific anti-factor IX antibody. J Biol Chem. 2004;279:14338–46. doi: 10.1074/jbc.M314011200. [DOI] [PubMed] [Google Scholar]
- 30.White-Adams TC, Berny MA, Tucker EI, Gertz JM, Gailani D, Urbanus RT, de Groot PG, Gruber A, McCarty OJ. Identification of coagulation factor XI as a ligand for platelet apolipoprotein E receptor 2 (ApoER2) Arterioscler Thromb Vasc Biol. 2009;29:1602–7. doi: 10.1161/ATVBAHA.109.187393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118:6963–70. doi: 10.1182/blood-2011-07-368811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ponczek MB, Gailani D, Doolittle RF. Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost. 2008;6:1876–83. doi: 10.1111/j.1538-7836.2008.03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human plasma prekallikrein: the presence of four novel apple domains in the amino-terminal portion of the molecule. Biochemistry. 1991;30:2050–6. doi: 10.1021/bi00222a007. [DOI] [PubMed] [Google Scholar]