

Abstract
Endoplasmic reticulum stress is closely involved in the early stage of diabetic retinopathy. In the present study, a streptozotocin-induced diabetic animal model was given an intraperitoneal injection of tauroursodeoxycholic acid. Results from immunofluorescent co-localization experiments showed that both caspase-12 protein and c-Jun N-terminal kinase 1 phosphorylation levels significantly in-creased, which was associated with retinal ganglion cell death in diabetic retinas. The C/ERB mologous protein pathway directly contributed to glial reactivity, and was subsequently responsible for neuronal loss and vascular abnormalities in diabetic retinopathy. Our experimental findings dicate that endoplasmic reticulum stress plays an important role in diabetes-induced retinal neu-ronal loss and vascular abnormalities, and that inhibiting the activation of the endoplasmic reticulum stress pathway provides effective protection against diabetic retinopathy.
Keywords: neural regeneration, peripheral nerve injury, endoplasmic reticulum stress, diabetic retinopathy, injury of retinal ganglion cells, Müller cells, astrocytes, c-Jun N-terminal kinase, caspase-12 protein, C/ERB homologous protein, retinal microcirculation, glial fibrillary acidic protein, grant-supported paper, neuroregeneration
Research Highlights
-
(1)
Endoplasmic reticulum stress plays an important role in the hyperglycemia-induced death of ganglion cells and impairment of retinal microvessels.
-
(2)
Tauroursodeoxycholic acid treatment effectively inhibited the activation of the endoplasmic reti-culum stress pathway, and provided effective protection against diabetic retinopathy.
-
(3)
Both caspase-12 protein and phosphorylated c-Jun N-terminal kinase 1 levels significantly in-creased, and they were associated with retinal ganglion cells in diabetic retinas.
-
(4)
The C/ERB homologous protein was shown to co-localize with glial fibrillary acidic tein-positive glial cells in the retina of diabetic rats, and subsequently provided evidence for the tivation of glial cells in diabetic retinopathy.
INTRODUCTION
Diabetic retinopathy is one of the most common complications associated with diabetes mellitus and the leading cause of blindness in adults[1]. Mechanisms underlying diabetic retinopathy onset and progression are not fully elucidated. Both neuronal and vascular abnormalities are associated with the pathogenesis of early diabetic retinopathy[2]. The death of neurons is irreversible and directly affects visual function. The most severely affected neurons in diabetic retinopathy are retinal ganglion cells[3]. Therefore, understanding the etiology of neuronal loss and vascular abnormalities will lead to better treatments for diabetic retinopathy. The endoplasmic reticulum (ER) has received much attention for its role in signal transduction relevant to cell survival and death. A number of pathophysiological insults lead to ER stress[4]. Persistent and intense ER stress can trigger apoptosis by induction of the caspase-12-dependent pathway[5], c-Jun NH2-terminal kinase pathway[6] and C/ERB homologous protein pathway[7]. ER stress is reported to be involved in the pathogenesis of various neuronal diseases in the brain and retina[6,8,9,10]. Growing evidence also indicates that ER stress is closely involved in the early stage of diabetic retinopathy[11]. Over-stimulation of neurons by glutamate, which is elevated in the vitreous of diabetic patients[12], may activate ER stress in retinal ganglion cells and subsequently lead to death of retinal ganglion cells in the mouse[13]. An in vitro study showed that a low or sudden reduction of glucose levels activated ER stress and resulted in apoptosis of cultured retinal pericytes[14]. Afterwards, ER stress regulated the over-expression of vascular endothelium growth factor, which was responsible for vascular leakage and neovascularization[15].
Tauroursodeoxycholic acid is a hydrophilic bile acid that is normally produced endogenously in humans at very low levels. Tauroursodeoxycholic acid is formed in the conjugation pathway of ursodeoxycholic acid, which is commonly used as a bile acid replacement therapy for treatment of certain cholestatic syndromes[16]. Tauroursodeoxycholic acid also functions as a chemical chaperone and alleviates ER stress both in vivo and in vitro[17,18]. Although major progress has been achieved, there is still no clear evidence directly associating ER stress with the pathogenesis of diabetic retinopathy. We propose that ER stress plays an important role in diabetic retinopathy, and this study demonstrated that (1) the expression of ER stress marker proteins was significantly elevated in streptozotocin (STZ)-induced diabetic retinas; and (2) tauroursodeoxycholic acid treatment effectively inhibited diabetes-induced ER stress marker protein expression, and appeared to protect diabetic retinas from neuronal cell death and vascular abnormality.
RESULTS
Quantitative analysis of experimental animals
Fifty-four Sprague-Dawley rats aged 8 weeks, weighing 180 g, were randomly divided into three groups: control group (normal feeding), diabetic group (diabetes mellitus models were established with intraperitoneal injection of STZ), and tauroursodeoxycholic acid treatment group (diabetes mellitus was established and tauroursodeoxycholic acid 100 mg/kg was administered per day). Each group contained 18 rats. Rats were examined at 2, 4, 6 and 8 weeks after injection for measurements of body weight and blood glucose levels. All experimental rats were involved in the final analysis without any loss.
Changes of body weight and blood glucose levels in diabetic rats
From 2 to 8 weeks after onset of diabetes, the age-matched control rats had a 34% gain in body weight, whereas diabetic rats had a 12% gain in body weight, and tauroursodeoxycholic acid-treated diabetic rats had a 14% gain in body weight. Diabetic rats treated with or without tauroursodeoxycholic acid weighed significantly less than control rats (P < 0.001). Blood glucose levels differed significantly between control and diabetic rats with or without tauroursodeoxycholic acid treatment at all ages studied (P < 0.001). However, the difference between diabetic rats with or without tauroursodeoxycholic acid treatment was not significant in terms of blood glucose levels and body weight (Table 1).
Table 1.
Average weights and blood glucose levels of control and diabetic rats
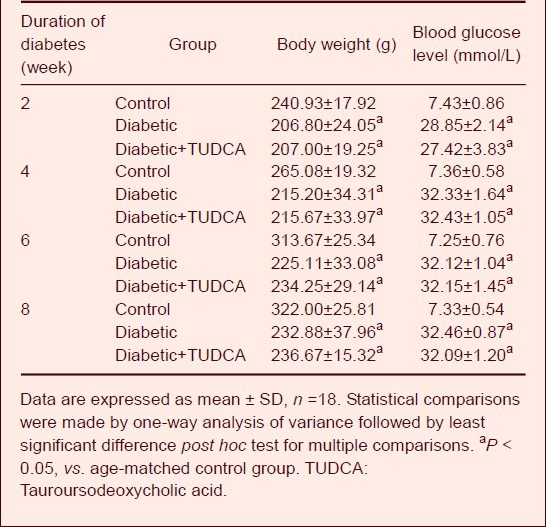
Tauroursodeoxycholic acid treatment significantly prevented retinal ganglion cell death in diabetic rats
Retinas of control rats (Figure 1A) and tauroursodeoxycholic acid-treated rats (Figure 1C) had a ganglion cell layer, where cells were densely packed with little space intervening between cells. In contrast, diabetic retinas demonstrated a loss of cells in the ganglion cell layer (Figure 1B). The numbers of cells in the ganglion cell layer were counted using a double blind method (Figure 1D). The cell count in the ganglion cell layer in diabetic rats was significantly reduced when compared with that in control rats (P < 0.001). After treatment with tauroursodeoxycholic acid, the cell count increased by 55% relative to that in un-treated retinas (P < 0.001), and there was no significant difference when compared with age-matched control retinas.
Figure 1.
Morphology of cells in the ganglion cell layer (GCL) of age-matched control and diabetic rats (hematoxylin-eosin staining; A–C: light microscopy; scale bar: 50 μm).
(A) In control retinas, the cells in the GCL were densely packed. (B) In diabetic retinas (at 8 weeks after onset of diabetes), cellular dropout was evident in the GCL (bold arrows), (C) After tauroursodeoxycholic acid treatment (intraperitoneal injection, 100 mg/kg per day, from 5 days after STZ injection) for 8 weeks, morphologic alterations induced by diabetes were ameliorated.
(D) Number of cells in the GCL of diabetic and age-matched control retinas. aP < 0.001, vs. diabetic group. Data are expressed as mean ± SD in six rats for each group (one-way analysis of variance and least significant difference test). STZ: Streptozotocin; TUDCA: tauroursodeoxycholic acid.
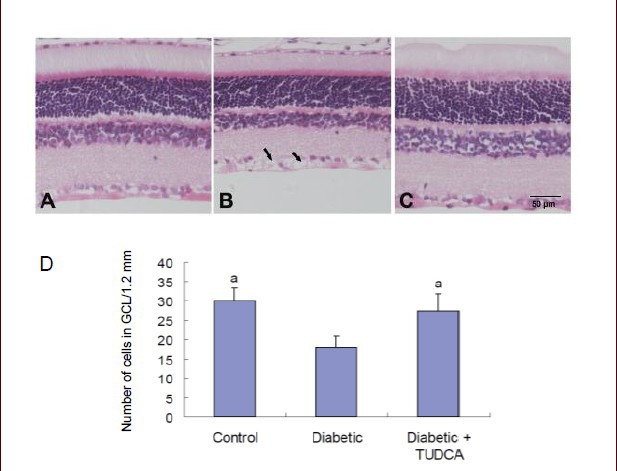
Tauroursodeoxycholic acid decreased leukocyte adhesion in the retina of diabetic rats
In the age-matched control retina, very little ConA binding was shown. At 8 weeks after onset of diabetes, some ConA labeling appeared in the branches of large venules, postcapillary venules, and arterioles in the ganglion cell layer (Figure 2A). Systemic treatment with tauroursodeoxycholic acid markedly reduced the number of adherent leukocytes in the retinas of diabetic rats (Figure 2B). The labeled static leukocytes often clustered in localized areas, rather than being evenly distributed throughout the whole retina. (Figure 2C) shows the total number of adherent leukocytes per retina in the experimental model. The number of adherent leukocytes significantly increased in diabetic retinas at 8 weeks (P < 0.001). Systemic treatment with tauroursodeoxycholic acid markedly reduced the number of adherent leukocytes in the retinas of diabetic rats (P < 0.001).
Figure 2.
Leukocyte adhesion in the retina of diabetic rats with or without tauroursodeoxycholic acid (TUDCA) treatment (scale bars: 10 μm).
(A) Static leukocytes were shown in retinal vasculature with diabetes. The labeled static leukocytes often clustered in local area, other than evenly distributed throughout the whole retina. (B) After 8-week TUDCA treatment (intraperitoneal injection, 100 mg/kg per day, from 5 days after streptozotocin injection), the number of adherent leukocytes in the retina of diabetic rats reduced. (C) Quantification of leukostasis. The number of static leukocytes was counted. Diabetic rats demonstrated a marked increase in the number of static leukocytes in retinal vasculature. Systemic administration of TUDCA significantly decreased the number of static leukocytes in diabetic retinas. Data are represented as the number of static leukocytes in retinal vasculature each retina. aP < 0.001, vs. diabetic group. Data were expressed as mean ± SD in six rats for each group (one-way analysis of variance and least-significant difference test).
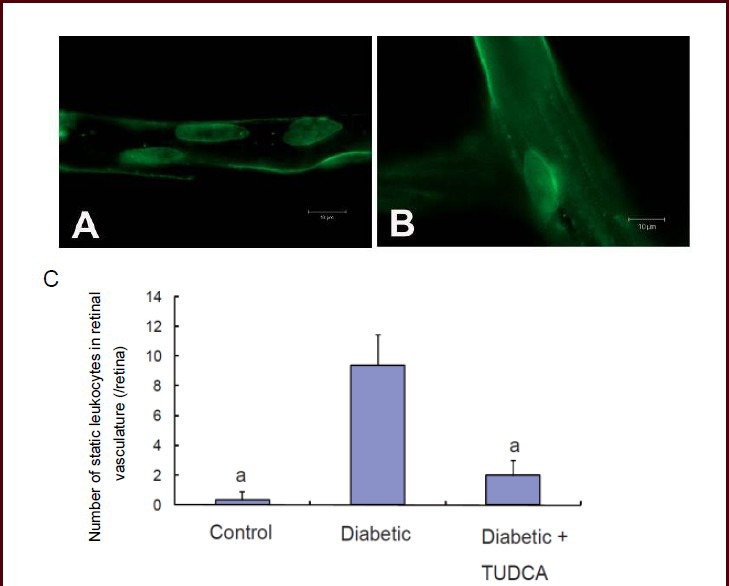
Tauroursodeoxycholic acid alleviated vascular abnormalities in the retina of diabetic rats
Vascular changes, including breakdown of the blood-retinal barrier, thickening of the capillary basement membrane, and reduction in the number of pericytes and an increment in the number of acellular capillaries, have been widely documented in diabetic retinopathy. In the present study, retinal vascular abnormalities were measured by infusion of FITC-Dextran. Diabetic rats (Figure 3A) showed significant lesions in the retinal capillary network as well as large vessels when compared with age-matched controls (data not shown) and tauroursodeoxycholic acid-treated rats (Figure 3B). The diabetic retinas demonstrated focal dilation of retinal capillaries, vessel tortuosity, and sac-like bulges in vessel walls.
Figure 3.
Change in retinal microvessels in diabetic rats with or without tauroursodeoxycholic acid treatment (fluorescein isothiocyanate-dextran angiograph, light microscopy; scale bars: 100 μm).
(A) At 8 weeks after onset of diabetes, the retina from diabetic rats exhibited focal dilation of retinal capillaries, with vessels tortuosity and sac-like bulges in the vessel wall.
(B) After 8-week tauroursodeoxycholic acid treatment (intraperitoneal injection, 100 mg/kg per day), no apparent vessel abnormalities were observed.
Tauroursodeoxycholic acid reduced caspase-12 expression in the retina of diabetic rats
At 8 weeks after diabetic onset, caspase-12 expression was determined by immunofluorescence, western blot analysis and real-time PCR analysis. Immunofluorescence results showed that no positive caspase-12 staining was shown in the age-matched control retina (Figure 4A). At 8 weeks, a marked increase of positive caspase-12 staining was observed throughout the whole ganglion cell layer, and the inner nuclear layer also presented scattered staining (Figure 4B). After tauroursodeoxycholic acid treatment, caspase-12-positive labeling was much weaker than that in un-treated retinas (Figure 4C). Representative western blot analysis of caspase-12 expression in the different groups is shown in Figure 4D. Results showed that the age-matched control retinas constitutively expressed moderate quantities of procaspase-12 protein, and its expression remained stable in diabetic retinas treated with or without tauroursodeoxycholic acid. However, control retinas expressed very low levels of cleaved caspase-12. In diabetic rats, caspase-12 expression was significantly up-regulated (P < 0.001); however, after tauroursodeoxycholic acid treatment, expression was significantly down-regulated (P < 0.001; Figure 4D–E).
Figure 4.
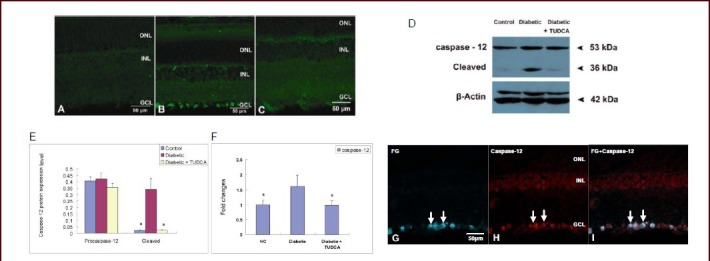
Changes in caspase-12 expression in the retina of control rats and diabetic rats with or without tauroursodeoxycholic acid (TUDCA) treatment (scale bars: 50 μm).
(A–B) Immunofluorescent labeling of caspase-12 in control and diabetic rats revealed a marked increase of positive caspase-12 staining, which was observed throughout the whole ganglion cell layer (Leica DM4000). (C) After 8-week TUDCA treatment (intraperitoneal injection, 100 mg/kg per day), immunofluorescent caspase-12-positive labeling significantly decreased (Leica DM4000).
Western blot analysis (D–E) and real-time PCR analysis (F) demonstrated that protein and mRNA expression of cleaved caspase-12 expression was significantly up-regulated in diabetic retinas, and TUDCA treatment markedly reduced diabetes-induced cleaved caspase-12 expression. Data represent caspase-12 protein or mRNA expression levels (absorbance caspase-12/β-actin). aP < 0.001, vs. diabetic group. Data are expressed as mean ± SD of six rats for each group (one-way analysis of variance and least-significant difference test).
(G–I) Double immunofluorescent labeling of caspase-12 and fluogold (FG)-retro-labeled ganglion cells in the diabetic retina. FG in blue, and caspase-12 in red. Overlay of labeling for both FG and caspase-12 in orange. (G) Retro-labeled FG-positive ganglion cells. (H) Caspase-12-positive staining was demonstrated in the ganglion cell layer. (I) Co-localization of FG and caspase-12 showed that caspase-12 was mainly expressed by ganglion cells. ONL: Outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer.
Reverse transcription-PCR analysis indicated that the age-matched control retinas expressed low levels of caspase-12 mRNA. At 8 weeks after onset of diabetes, caspase-12 expression was significantly up-regulated (P < 0.001). After tauroursodeoxycholic acid treatment, its expression was significantly down-regulated when compared with the diabetic group (Figure 4F). The co-localization study showed that most caspase-12- positive staining was expressed on fluogold (FG)-retro- labeled ganglion cells (Figure 4G–I).
Tauroursodeoxycholic acid reduced phosphorylated c-Jun N-terminal kinase 1 expression in diabetic rats
The phosphorylation of c-Jun N-terminal kinase 1 was determined by immunofluorescence and western blot analysis. The expression of c-Jun N-terminal kinase 1 mRNA was determined by real-time PCR analysis. In the age-matched control retina, only a few positive phosphorylated c-Jun N-terminal kinase 1 cells were observed (Figure 5A). At 8 weeks after the onset of diabetes, a marked increase in phosphorylated c-Jun N-terminal kinase 1-positive staining was observed throughout the whole ganglion cell layer (Figure 5B). After treatment with tauroursodeoxycholic acid, the phosphorylated c-Jun N-terminal kinase 1-positive labeling was much weaker than that in un-treated retinas (Figure 5C).
Figure 5.
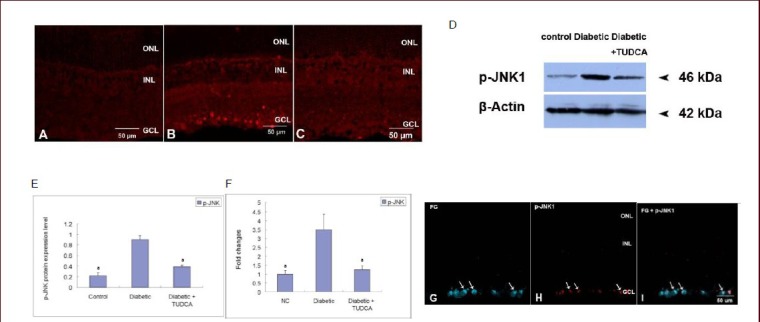
Phosphorylation of c-Jun N-terminal kinase 1 (p-JNK1) in the retina of control rats and diabetic rats with or without tauroursodeoxycholic acid (TUDCA) treatment (scale bars: 50 μm).
(A–B) Immunofluorescent labeling of the phosphorylated c-Jun N-terminal kinase 1 in control rats and diabetic rats at 8 weeks after onset of diabetes revealed a marked increase in phosphorylated c-Jun N-terminal kinase 1 levels throughout the whole ganglion cell layer. (C) After 8-week TUDCA treatment (intraperitoneal injection, 100 mg/kg per day), immunofluorescent labeling of phosphorylated c-Jun N-terminal kinase 1 significantly decreased.
Western blot analysis (D–E) and real-time PCR analysis (F) demonstrated that protein or mRNA expression of phosphorylated c-Jun N-terminal kinase 1 was significantly up-regulated in diabetic retinas, and TUDCA treatment markedly reduced high glucose-induced expression. Data are presented as the phosphorylated c-Jun N-terminal kinase 1 protein or mRNA expression level (absorbance of phosphorylated c-Jun N-terminal kinase 1/β-actin). aP < 0.001, vs. diabetic group. Data are expressed as mean ± SD of six rats for each group (one-way analysis of variance and least significant difference test).
(G–I) Double immunofluorescent labeling of phosphorylated c-Jun N-terminal kinase 1 and fluogold (FG)-retro-labeled ganglion cells in the diabetic retina. FG in blue, and phosphorylated c-Jun N-terminal kinase 1 in red. Overlay of labeling for both FG and phosphorylation of c-Jun N-terminal kinase 1 in orange. (G) Retro-labeled FG-positive ganglion cells (arrows). (H) Phosphorylated c-Jun N-terminal kinase 1 staining in the ganglion cell layer (arrows). (I) Co-localization of FG and phosphorylation of c-Jun N-terminal kinase 1 showed that phosphorylated c-Jun N-terminal kinase 1 was mainly expressed by ganglion cells (arrows). ONL: Outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer.
A representative western blot analysis of the expression of phosphorylated c-Jun N-terminal kinase 1 in the different groups is shown in Figure 5D. Results showed that the age-matched control retinas expressed low levels of phosphorylated c-Jun N-terminal kinase 1. At 8 weeks after onset of diabetes, its expression was markedly up-regulated (P < 0.001). After treatment with tauroursodeoxycholic acid, its expression was significantly down-regulated when compared with the diabetic group (P < 0.001; (Figure 5D–F). The co-localization study showed that the vast majority of phosphorylated c-Jun N-terminal kinase 1-positive staining was expressed on FG-retro-labeled ganglion cells (Figure 5G–I).
Tauroursodeoxycholic acid reduced C/ERB homologous protein expression in diabetic rats
The expression of C/ERB homologous protein was determined by immunofluorescence, western blot analysis and real-time PCR analysis. In the age-matched control retina, only a few positive C/ERB homologous protein cells were observed (Figure 6A). At 8 weeks after the onset of diabetes, a marked increase in positive C/ERB homologous protein staining was observed throughout the whole nerve fiber layer and ganglion cell layer (Figure 6B). After tauroursodeoxycholic acid treatment, C/ERB homologous protein-positive labeling was much weaker than that in un-treated retinas (Figure 6C). Representative western blot analysis of C/ERB homologous protein expression in the different groups was shown in Figure 6D. Results showed that the age-matched control retinas constitutively expressed moderate quantities of C/ERB homologous protein. At 8 weeks after the onset of diabetes, its expression was markedly up-regulated (P < 0.001). After tauroursodeoxycholic acid treatment, its expression was significantly down-regulated when compared with the diabetic group (P < 0.001; (Figure 6D–F). To determine whether C/ERB homologous protein-positive staining was expressed on ganglion cells or glial cells, we labeled C/ERB homologous protein on FG-retro-labeled diabetic retinas and double labeled C/ERB homologous protein with glial fibrillary acidic protein on diabetic retinas. The co-localization study showed that most C/ERB homologous protein-positive staining was not expressed on ganglion cells (data not shown), but was expressed on glial fibrillary acidic protein-positive glial cells (Figure 6G-I).
Figure 6.
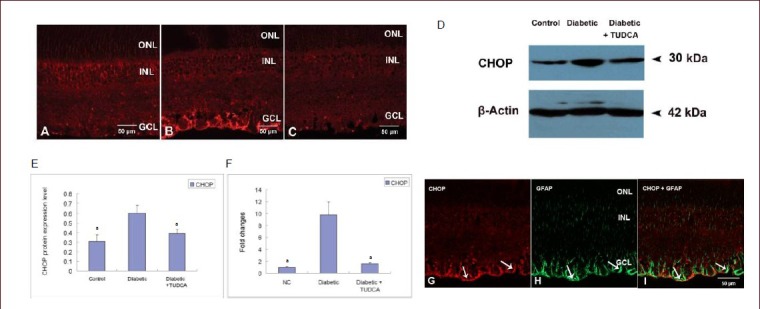
Changes in C/ERB homologous protein (CHOP) expression in the retina of control rats and diabetic rats with or without tauroursodeoxycholic acid (TUDCA) treatment (scale bars: 50 μm).
(A) Immunofluorescent labeling of C/ERB homologous protein in control rats. (B) Immunofluorescent labeling of C/ERB homologous protein in diabetic rats at 8 weeks after onset of diabetes revealed a marked increase in C/ERB homologous protein staining throughout the whole nerve fiber layer and ganglion cell layer. (C) After 8-week TUDCA treatment (intraperitoneal injection, 100 mg/kg per day), immunofluorescence of C/ERB homologous protein significantly decreased.
Western blot analysis (D-E) and real-time PCR analysis (F) demonstrated that protein or mRNA expression of C/ERB homologous protein was significantly up-regulated in diabetic retinas, and TUDCA treatment markedly reduced diabetes-induced C/ERB homologous protein expression. Data are presented as the C/ERB homologous protein or mRNA expression level (absorbance of C/ERB homologous protein/β-actin). aP < 0.001, vs. diabetic group. Data are expressed as mean ± SD of six rats for each group (one-way analysis of variance and least significant difference test).
(G–I) Double immunofluorescent labeling of C/ERB homologous protein and GFAP in the diabetic retina. C/ERB homologous protein in red, and GFAP in green. Overlay of labeling for both C/ERB homologous protein and GFAP in orange. (G) C/ERB homologous protein-positive staining was demonstrated in the nerve fiber layer and ganglion cell layer (arrows). (H) GFAP-positive staining of both astrocytes and Müller cells (arrows). (I) Co-localization of C/ERB homologous protein and GFAP showed that C/ERB homologous protein was mainly expressed by astrocytes and Müller cells (arrows). GFAP: Glial fibrillary acidic protein; ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer.
Glial fibrillary acidic protein expression in the retina of diabetic rats
The expression of glial fibrillary acidic protein in age- matched control and diabetic retinas was determined by immunofluorescence studies both on frozen sections and whole-mount retinas. In the age-matched control retina, glial fibrillary acidic protein staining was confined in the inner most retinal layer (Figure 7A), where starlike astrocytes were located (Figure 7D). Müller cells were not labeled. Astrocytes had processes that wrapped around retinal blood vessels, forming a glia limitans (Figure 7D). In diabetic retinas, both astrocytes and Müller cell end feet were stained (Figure 7B). The starlike astrocyte cell body, as well as cell processes, which wrapped around retinal blood vessels, were irregularly distributed (Figure 7E). After treatment with tauroursodeoxycholic acid, the positive glial fibrillary acidic protein labeling was much weaker, mainly restricted to the inner most astrocytes (Figure 7C), which were no different when compared with the control retina (Figure 7F). Glial fibrillary acidic protein-positive Müller cells were observed throughout the whole retinal layer, and the tortuous Müller cell end feet surrounded the vascular layer (Figure 7G–I).
Figure 7.
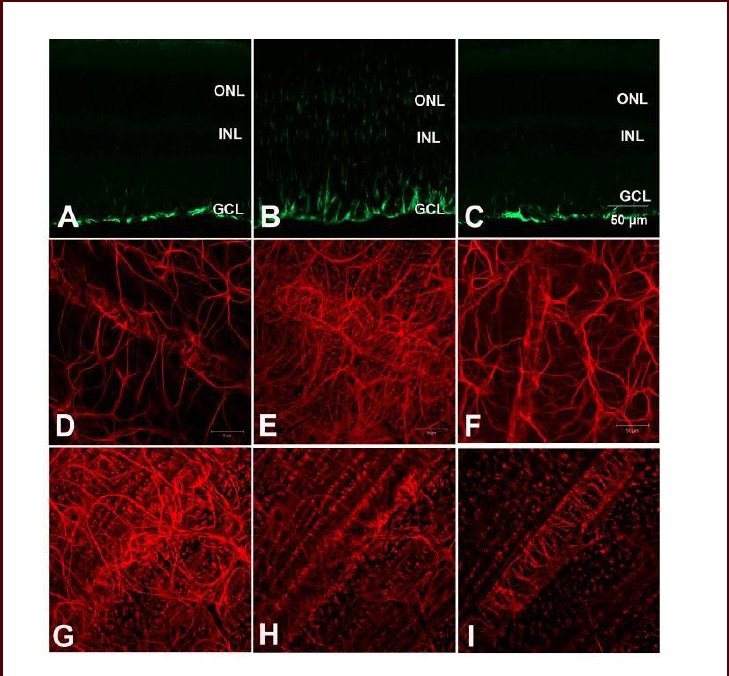
Immunofluorescent labeling of glial fibrillary acidic protein in control rats and diabetic rats with or without tauroursodeoxycholic acid (TUDCA) treatment on retinal sections (A–C) and whole-mounts (D–I) (scale bar: 50 μm).
(A, B) Retinas from control and diabetic rats (at 8 weeks after onset of diabetes) exhibited strong glial fibrillary acidic protein-positive staining in both astrocytes and Müller cells. (C) After 8-week TUDCA treatment, glial fibrillary acidic protein-positive labeling was significantly decreased. (D) In the control whole-mount retina, starlike astrocytes extended processes wrapped around retinal blood vessels. (E) In diabetic retinas, both astrocytes and Müller cell end feet were stained. The astrocyte cell bodies, as well as the processes, were irregularly distributed. (F) After 8-week TUDCA treatment, positive glial fibrillary acidic protein labeling showed no difference when compared with the controls. (G–I) Representative glial fibrillary acidic protein-positive Müller cell staining from the nerve fiber layer to the inner nuclear layer. Glial fibrillary acidic protein-positive Müller cells were observed throughout the whole retinal layer, and the tortuous Müller cell end feet surrounded the vascular layer. ONL: Outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer.
DISCUSSION
Our studies demonstrated that: (1) the expression of ER stress marker proteins including caspase-12, C/ERB homologous protein and phosphorylated c-Jun N-terminal kinase 1 was significantly elevated in STZ- induced diabetic retinas. The expression of caspase-12 and phosphorylated c-Jun N-terminal kinase 1 protein correlated with retinal ganglion cell death, whereas expression of C/ERB homologous protein correlated directly with glial reactivity; and (2) tauroursodeoxycholic acid treatment effectively inhibited diabetes-induced ER stress marker protein expression, and appeared to have protected diabetic retinas from neuronal cell death and vascular abnormalities.
Ultrastructural observation demonstrated that the cytoplasm of retinal ganglion cells contained abundant ER, suggesting active synthesis of secretory proteins and a susceptibility to ER stress[19]. Glutamate, which is known to be elevated in the vitreous of diabetic patients[14], caused calcium influx from the ER to the cytosol and resulted in ER calcium depletion and ER stress[20]. However, to date, there has been no investigation concerning the involvement of caspase-12 in the pathogenesis of diabetic retinopathy. ER stress-induced apoptosis also involves activation of the c-Jun N-terminal kinase pathway. In response to ER stress, activated IRE-1 recruits tumor necrosis factor receptor-associated factor 2 and apoptosis signal-regulating kinase 1 to form the IRE-1/tumor necrosis factor receptor-associated factor 2/apoptosis signal-regulating kinase 1 complex, which then activates c-Jun N-terminal kinase and triggers cell death[21]. From previous studies, phosphorylated c-Jun N-terminal kinase is pro-apoptotic in NMDA-induced retinal ganglion cell death[22]. Our present study demonstrated that both caspase-12 protein and phosphorylated c-Jun N-terminal kinase 1 were significantly increased in diabetes stressed retinal ganglion cells. Thus, not only is the caspase-12 pathway associated with retinal ganglion cell death in diabetic retinas, but also the c-Jun N-terminal kinase pathway. Consequently, it seems reasonable that ER stress-mediated cell death pathways are related to retinal ganglion cell loss in diabetic retinopathy.
Glial processes make contact with both blood vessels and neurons, and are one of the components in the blood-retinal barrier[23]. Our current studies demonstrated that astrocytes extend processes in the control retina and form a glia limitans on the retinal vasculature[24]. However, astrocytes shrank and were irregularly distributed in diabetic retinas. The role of astrocytes in retinal function is complicated[25,26,27,28]. In the present study, diabetes- induced Müller cells underwent gliosis, indicated by up-regulation of intermediate filament glial fibrillary acidic protein.
Thus, glial changes are involved and subsequently may be related to both neuronal and vascular abnormalities in diabetic retinopathy.
C/ERB homologous protein contributed greatly to the onset and progression of apoptotic retinal ganglion cell death in glaucoma and C/ERB homologous protein expression was significantly increased in the diabetic retina. In the present study, our results demonstrated that C/ERB homologous protein was greatly up-regulated in diabetic retinas, and it was co-localized with glial fibrillary acidic protein-positive glial cells. Thus, it is reasonable to assume that C/ERB homologous protein plays an important role in glial reactivity in diabetic retinopathy. Considering the glial function in the diabetic retina, our studies indicated that C/ERB homologous protein expression directly contributes to glial reactivity and may also be responsible for neuronal and vascular abnormalities in diabetic retinopathy.
Tauroursodeoxycholic acid has been shown to be protective in several models of neuronal disease. It also was found to preserve rod and cone function and overall photoreceptor number during retinal degeneration rd10 mice, and to prevent lens epithelial cell death and cataract formation in galactosemic rats. In the present study, tauroursodeoxycholic acid treatment effectively reduced diabetes-induced ER stress marker protein expression, and subsequently preserved retinal ganglion cells and retinal vasculature from diabetes-induced damage.
In conclusion, our studies demonstrate for the first time that both the caspase-12 and JNK pathways were directly associated with retinal ganglion cell death in diabetic retinas. C/ERB homologous protein expression directly contributed to glial reactivity and might be secondarily responsible for neuronal and vascular abnormalities in diabetic retinopathy. The ER stress modulator tauroursodeoxycholic acid effectively inhibited ER stress marker protein expression and provided protection against diabetic retinopathy, further suggesting that targeting ER function may result in therapeutic gain.
MATERIALS AND METHODS
Design
A randomized, controlled animal experiment.
Time and setting
This research was conducted in the Animal Center of Peking University Health Science Center and Peking University Third Hospital Eye Institute during October 2009 to December 2011.
Materials
Sprague-Dawley female rats, 8 weeks old, weighing 180 g, were provided by the Animal Center of Peking University Health Science Center, China (License No. SYXK (Jing) 2011-0039). All experiments were performed in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China[29].
Methods
Establishment of diabetic rat model using STZ injection
Diabetic models in rats were induced as previously described[30]. In brief, rats with blood glucose levels > 16.7 mmol/L for 5 days after receiving STZ (60 mg/kg; Sigma-Aldrich, St. Louis, MO, USA; #S0130, dissolved in 10 mmol/L sodium citrate buffer, pH 4.2) were considered to be successful diabetic models. Body weight and blood glucose levels (One Touch® brand blood glucose monitors, LifeScan) were checked every 2 weeks. For the in vivo tauroursodeoxycholic acid (TCI, Tokyo, Japan) treatment, rats were treated by intraperitoneal administration (100 mg/kg per day) beginning 5 days after STZ injection.
Retinal leukostasis quantified with concanavalin A
The retinal leukostasis study was performed as described previously[31]. In brief, rats were deeply anesthetized using a ketamine-xylazine mixture. The chest cavity was opened and a 14-gauge perfusion cannula was introduced into the left ventricle. Drainage was achieved using a 16-gauge needle placed in the right atrium. The animals were perfused with 250 mL PBS per kg body weight to remove erythrocytes and nonadherent leukocytes. After PBS perfusion, fixation with 1% (w/v) paraformaldehyde and 0.5% (w/v) glutaraldehyde was achieved using 200 mL/kg body weight of perfusate for 3 minutes. PBS perfusion was performed at a physiological pressure because the pumping heart provided motive force. All subsequent perfusions were given post mortem at 100 mmHg pressure (1 mmHg = 0.133 kPa). Nonspecific binding was blocked with 1% (w/v) albumin in PBS (total volume 100 mL/kg body weight) followed by perfusion with FITC-coupled concanavalin A lectin (20 μg/mL in PBS, pH 7.4, 5 mg/kg body weight; Vector Laboratories, Burlingame, CA, USA). Concanavalin A was used to label adherent leukocytes and vascular endothelial cells. Residual unbound lectin was removed with a 1% (w/v) albumin in PBS perfusion for 1 minute followed by a PBS perfusion for 4 minutes. The retina were carefully removed and flat mounts prepared using a fluorescence anti-fading medium (Sigma-Aldrich).
The retinae were then imaged under a confocal laser scanning microscope at 488 nm (510 META; Carl Zeiss, Oberkochen, Germany). The total number of leukocytes in the retinal arterioles, venules, and capillaries was then determined.
Vessel leakage labeled by FITC-dextran perfusion
For leakage studies, rats were deeply anesthetized and were perfused through the left ventricle using a FITC-Dextran stock solution, 10 mg/mL (Sigma-Aldrich) at 50 mL/kg body weight concentration. Animals were killed 1 minute after perfusion. The eyes were enucleated and fixed in 4% (w/v) paraformaldehyde overnight. The corneas and lens were then removed, and peripheral retinas were dissected and flat-mounted on microscope slides for examination under a confocal laser scanning microscope (Carl Zeiss).
Retinal ganglion cells retrograde labeled by FG
Retinal ganglion cells were identified by retrograde labeling with FG (Molecular Probes, Eugene, OR, USA). Briefly, rats were anesthetized by intraperitoneal administration of 5 mg/kg xylazine and 50 mg/kg ketamine (Sigma-Aldrich). The skin over the cranium was incised, and the skull was exposed. A total of four holes 1 mm in diameter were drilled in the skull. These positions correspond to the superior colliculi and lateral geniculate bodies as determined from a stereotactic rat brain atlas. Two holes were located at 6.8 mm posterior to bregma, 1.6 mm lateral to the midline, with a depth of 4.0 mm on both sides of the midline raphe. The other two holes were located at 4.6 mm posterior to bregma, 3.8 mm lateral to the midline, with a depth of 5.6 mm on both sides. Five microliters of FG at a concentration of 10 mg/mL was injected into each hole. The skull opening was then sealed. The overlying skin was sutured and antibiotic ointment was applied externally.
Immunocytochemical identification of caspase-12, phosphorylated c-Jun N-terminal kinase 1, C/ERB homologous protein and glial fibrillary acidic protein in the retina
Animals were euthanized with an overdose of pentobarbital, and their eyes were immediately enucleated and fixed in 4% (w/v) paraformaldehyde in PBS for 1 hour. The anterior segments were removed and posterior segments were further fixed in the same fixative for an additional 5 hours. Tissue samples were transferred to 20% (w/v) sucrose buffer overnight at 4°C for cryoprotection, and then embedded in OCT compound. Frozen sections were cut into 8 μm thickness through the optic nerve head and ora serrata with a cryostat, and the sections were kept in a −80°C freezer until ready for use. The number of retinal ganglion cells was counted 600 μm away from the optic nerve head. Flat-mounted retinas were prepared by bisecting the eyes at the ciliary body into anterior and posterior sections. The lens and vitreous were removed, and the retina was subsequently separated from the underling retinal pigment epithelium and choroid. Flat-mounted retinas were kept at 4°C until ready for use. For immunofluorescence, frozen sections were incubated with rabbit polyclonal antibody to caspase-12 (1:200, AB3612, Chemicon, Temecula, CA, USA), mouse monoclonal antibody to C/ERB homologous protein (1:50, SC-7351, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-active antibody to c-Jun N-terminal kinase 1 (1:100, V7931, Promega, Madison, WI, USA), or rabbit polyclonal antibody to glial fibrillary acidic protein (1:100, Z0334, DAKO Corp, Carpinteria, CA, USA). The secondary antibodies (Cy™3-conjugated goat anti-rabbit antibody and Cy™2- conjugated goat anti-mouse antibody) were bought from Jackson ImmunoResearch Laboratory (West Grove, PA, USA).
Results were examined under a confocal laser scanning microscope (Carl Zeiss). Control sections were treated in the same way with omission of primary antibodies.
Real-time fluorescent quantitative PCR for caspase-12, phosphorylated c-Jun N-terminal kinase 1 and C/ERB homologous protein expression
Retinal RNA was isolated with TRIzol (Gibco/Invitrogen Corporation, Carlsbad, CA, USA). Real-time PCR was performed in 96-well plates using standard protocols with SYBR® Green as a fluorescent detection dye in a Bio-Rad iCycler (Bio-Rad, Hercules, CA, USA). All PCR reactions were at a final volume of 30 μL comprising SYBR Green PCR mix, 600 μmol/L forward and reverse primers, and 1 ng of cDNA. Fluorescence was measured at 72°C.
The quantity of mRNA was calculated by normalizing the threshold cycle of the target gene to the threshold cycle of the β-actin housekeeping gene in the same sample. The relative quantification of the target gene was determined using 2−ΔΔCT.
Western blot assays for caspase-12, phosphorylated c-Jun N-terminal kinase 1 and C/ERB homologous protein expression
Retinal protein was extracted with 0.5 mL ice-cold lysis buffer (50 mmol/L Tris-Cl, pH 8.0, 0.02% (w/v) sodium azide, 1 μg/mL aprotinin, 1% (v/v) NP-40, and 100 μg/mL phenylmethylsulfonyl fluoride). A total of 40 μg protein was loaded onto each lane. Antibodies specific for caspase-12 (rabbit polyclonal antibody, 1:1 000, Chemicon), C/ERB homologous protein (mouse monoclonal antibody, 1:500, Santa Cruz Biotechnology) and phosphorylated c-Jun N-terminal kinase 1 (rabbit polyclonal antibody, 1:500, Promega) were used for immunodetection. Blots were visualized using an enhanced chemiluminescent technique (SC-2048; Santa Cruz Biotechnology). As a control for equal loading of proteins, a β-actin antibody (mouse monoclonal antibody, 1:2 000, A2228; Sigma- Aldrich) was used. For quantitative evaluation of western blot studies, the films were scanned and optical densities were quantified with Quantity One 1-D analysis software (Bio-Rad). The western blot experiments were repeated four times from separate samples for each time point.
Statistical analysis
Data were expressed as mean ± SD. Statistical comparisons were made by one-way analysis of variance followed by least significant difference test for multiple comparisons with SPSS 18.0 software (SPSS, Chicago, IL, USA). A value of P < 0.05 was considered statistically significant.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, No. 81170877, and the National Basic Research Program of China, No. 2007CB512203.
Conflicts of interest: None declared.
Ethical approval: The research was approved by the Institutional Research Board at Peking University Health Science Center, China (Approval ID LA2011-037).
REFERENCES
- [1].Gardner TW, King GL. Diabetic retinopathy. Diabetes Care. 1998;21(1):143–156. doi: 10.2337/diacare.21.1.143. [DOI] [PubMed] [Google Scholar]
- [2].Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008;586(Pt18):4401–4408. doi: 10.1113/jphysiol.2008.156695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kohzaki K, Vingrys AJ, Bui BV. Early inner retinal dysfunction in streptozotocin-induced diabetic rats. Invest Ophthalmol Vis Sci. 2008;49(8):3595–3604. doi: 10.1167/iovs.08-1679. [DOI] [PubMed] [Google Scholar]
- [4].Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13(3):363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- [5].Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- [6].Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ma Y, Brewer JW, Diehl JA, et al. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318(5):1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- [8].Marhfour I, Lopez XM, Lefkaditis D, et al. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia. 2012;55(9):2417–2420. doi: 10.1007/s00125-012-2604-3. [DOI] [PubMed] [Google Scholar]
- [9].Balasubramanyam M, Singh LP, Rangasamy S. Molecular intricacies and the role of ER stress in diabetes. Exp Diabetes Res 2012. 2012 doi: 10.1155/2012/958169. 958169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang LP, Wu LM, Guo XJ, et al. Activation of endoplasmic reticulum stress in degenerating photoreceptors of the rd1 mouse. Invest Ophthalmol Vis Sci. 2007;48(11):5191–5198. doi: 10.1167/iovs.07-0512. [DOI] [PubMed] [Google Scholar]
- [11].Hu WK, Liu R, Pei H, et al. Endoplasmic reticulum stress-related factors protect against diabetic retinopathy. Exp Diabetes Res 2012. 2012 doi: 10.1155/2012/507986. 507986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ambati J, Chalam KV, Chawla DK, et al. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997;115(9):1161–1166. doi: 10.1001/archopht.1997.01100160331011. [DOI] [PubMed] [Google Scholar]
- [13].Shimazawa M, Miwa A, Ito Y, et al. Involvement of endoplasmic reticulum stress in optic nerve degeneration following N-methyl-D-aspartate-induced retinal damage in mice. J Neurosci Res. 2012;90(10):1960–1969. doi: 10.1002/jnr.23078. [DOI] [PubMed] [Google Scholar]
- [14].Nakayama M, Aihara M, Chen YN, et al. Neuroprotective effects of flavonoids on hypoxia-, glutamate-, and oxidative stress-induced retinal ganglion cell death. Mol Vis. 2011;17:1784–1793. [PMC free article] [PubMed] [Google Scholar]
- [15].Roybal CN, Yang S, Sun CW, et al. Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J Biol Chem. 2004;279(15):14844–14852. doi: 10.1074/jbc.M312948200. [DOI] [PubMed] [Google Scholar]
- [16].Heuman DM, Mills AS, McCall J, et al. Conjugates of ursodeoxycholate protect against cholestasis and hepatocellular necrosis caused by more hydrophobic bile salts. In vivo studies in the rat. Gastroenterology. 1991;100(1):203–211. doi: 10.1016/0016-5085(91)90602-h. [DOI] [PubMed] [Google Scholar]
- [17].Chen Y, Liu CP, Xu KF, et al. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am J Nephrol. 2008;28(6):1014–1022. doi: 10.1159/000148209. [DOI] [PubMed] [Google Scholar]
- [18].Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- [20].Munemasa Y, Ohtani-Kaneko R, Kitaoka Y, et al. Contribution of mitogen-activated protein kinases to NMDA-induced neurotoxicity in the rat retina. Brain Res. 2005;1044(2):227–240. doi: 10.1016/j.brainres.2005.03.014. [DOI] [PubMed] [Google Scholar]
- [21].Fernandes KA, Harder JM, Fornarola LB, et al. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012;46(2):393–401. doi: 10.1016/j.nbd.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kamalden TA, Ji D, Osborne NN. Rotenone-induced death of RGC-5 cells is caspase independent, involves the JNK and p38 pathways and is attenuated by specific green tea flavonoids. Neurochem Res. 2012;37(5):1091–1101. doi: 10.1007/s11064-012-0713-5. [DOI] [PubMed] [Google Scholar]
- [23].Choi YK, Kim KW. Blood-neural barrier: its diversity and coordinated cell-to-cell communication. BMB Rep. 2008;41(5):345–352. doi: 10.5483/bmbrep.2008.41.5.345. [DOI] [PubMed] [Google Scholar]
- [24].Fletcher EL, Phipps JA, Ward MM, et al. Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr Pharm Des. 2007;13(26):2699–2712. doi: 10.2174/138161207781662920. [DOI] [PubMed] [Google Scholar]
- [25].Kimelberg HK. Functions of mature mammalian astrocytes: a current view. Neuroscientist. 2010;16(1):79–106. doi: 10.1177/1073858409342593. [DOI] [PubMed] [Google Scholar]
- [26].Caprara C, Grimm C. From oxygen to erythropoietin: relevance of hypoxia for retinal development, health and disease. Prog Retin Eye Res. 2012;31(1):89–119. doi: 10.1016/j.preteyeres.2011.11.003. [DOI] [PubMed] [Google Scholar]
- [27].Jadhav AP, Roesch K, Cepko CL. Development and neurogenic potential of Müller glial cells in the vertebrate retina. Prog Retin Eye Res. 2009;28(4):249–262. doi: 10.1016/j.preteyeres.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kaur C, Foulds WS, Ling EA. Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res. 2008;27(6):622–647. doi: 10.1016/j.preteyeres.2008.09.003. [DOI] [PubMed] [Google Scholar]
- [29].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006 Sep 30; [Google Scholar]
- [30].Yang LP, Sun HL, Wu LM, et al. Baicalein reduces inflammatory process in a rodent model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50(5):2319–2327. doi: 10.1167/iovs.08-2642. [DOI] [PubMed] [Google Scholar]
- [31].Coxon A, Bready J, Min H, et al. Context-dependent role of angiopoietin-1 inhibition in the suppression of angiogenesis and tumor growth: implications for AMG 386, an angiopoietin-1/2-neutralizing peptibody. Mol Cancer Ther. 2010;9(10):2641–2651. doi: 10.1158/1535-7163.MCT-10-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]