

Abstract
Drosophila melanogaster embryonic and larval tissues often contain a highly heterogeneous mixture of cell types, which can complicate the analysis of gene expression in these tissues. Thus, to analyze cell-specific gene expression profiles from Drosophila tissues, it may be necessary to isolate specific cell types with high purity and at sufficient yields for downstream applications such as transcriptional profiling and chromatin immunoprecipitation. However, the irregular cellular morphology in tissues such as the central nervous system, coupled with the rare population of specific cell types in these tissues, can pose challenges for traditional methods of cell isolation such as laser microdissection and fluorescence-activated cell sorting (FACS). Here, an alternative approach to characterizing cell-specific gene expression profiles using affinity-based isolation of tagged nuclei, rather than whole cells, is described. Nuclei in the specific cell type of interest are genetically labeled with a nuclear envelope-localized EGFP tag using the Gal4/UAS binary expression system. These EGFP-tagged nuclei can be isolated using antibodies against GFP that are coupled to magnetic beads. The approach described in this protocol enables consistent isolation of nuclei from specific cell types in the Drosophila larval central nervous system at high purity and at sufficient levels for expression analysis, even when these cell types comprise less than 2% of the total cell population in the tissue. This approach can be used to isolate nuclei from a wide variety of Drosophila embryonic and larval cell types using specific Gal4 drivers, and may be useful for isolating nuclei from cell types that are not suitable for FACS or laser microdissection.
Keywords: Biochemistry, Issue 85, Gene Expression, nuclei isolation, Drosophila, KASH, GFP, cell-type specific
Introduction
Drosophila tissues such as the central nervous system contain a complex mixture of cell types. Thus, to analyze cell-specific gene expression profiles from Drosophila tissues, it is first necessary to isolate a homogenous population of specific cells in sufficient quantities to enable downstream applications. Methods to isolate cells from intact tissues include laser microdissection, and fluorescence-activated cell sorting (FACS) of whole cells. While FACS has been used to isolate cells and nuclei from Drosophila embryos and from Caenorhabditis elegans for gene expression and chromatin profiling1-3, FACS and laser microdissection can be difficult to perform successfully in tissues that contain highly intermixed cell types or that contain cells with irregular morphology, such as neurons. To overcome this difficulty, nuclei rather than cells can be isolated from specific cell types and used for subsequent gene expression profiling. Importantly, microarray-based mRNA expression analysis using nuclear RNA samples shows generally comparable results with that performed using total RNA4,5. Moreover, gene expression analysis using nuclear RNA has been successfully used to study gene expression in multiple organisms including C. elegans, Arabidopsis thaliana, Drosophila, and humans4,52,3.
Several approaches have recently been described for the isolation of specific populations of labeled nuclei from Drosophila tissues that are suitable for gene expression analysis and/or chromatin immunoprecipitation. The batch isolation of tissue-specific chromatin for immunoprecipitation (BiTS-ChIP) method utilizes FACS to isolate fixed nuclei on the basis of cell-type specific expression of nuclear-localized GFP2. This approach has been successfully used to analyze the distribution of histone modifications and transcription factors using chromatin immunoprecipitation of isolated nuclei from the mesoderm of Drosophila embryos2. However, FACS-based approaches may be less suitable for isolating labeled nuclei that constitute only a small proportion of a mixed population due to the increased sort time needed to obtain suitable numbers for downstream applications. To overcome these limitations, several groups have utilized affinity-based isolation techniques to purify nuclei that are labeled with a specific epitope in a particular cell type. The isolation of nuclei tagged in specific cell types (INTACT) method developed for use in Arabidopsis thaliana6,7 has recently been adapted for use in Drosophila8. In this method, a nuclear envelope fusion protein that is a substrate for in vivo biotinylation is coexpressed with the Escherichia coli biotin ligase BirA in specific cell types. Biotin-labeled nuclei can be subsequently purified from mixed populations using streptavidin-based affinity isolation. Using this approach, nuclei were successfully labeled and isolated from the mesoderm of Drosophila embryos in which a nuclear envelope fusion protein was expressed under control of a mesoderm-specific enhancer8. The authors also generated nuclear envelope fusion proteins that can be expressed in any cell type under control of the Gal4 regulatory sequence, UAS9. This approach is capable of rapidly isolating subsets of labeled nuclei from mixed populations, but requires three separate transgenic constructs and may therefore be unsuitable for particular genetic applications. Recently, an approach has been described in which SUN (Sad1 and UNC-84) domain-containing proteins that localize to the inner membrane of the nuclear envelope were tagged with fluorescent proteins and expressed under control of the Gal4/UAS system10. Nuclei were isolated in the presence of nonionic detergent to remove the outer membrane of the nuclear envelope, and affinity-purified using magnetic beads coupled to anti-GFP antibodies. This approach was successfully used to isolate small populations of labeled nuclei from specific neuronal subtypes within the adult brain of Drosophila10.
Here, a protocol for isolation of labeled nuclei from a mixed population of cells from Drosophila larval tissue is described. This method was developed independently, but is similar to the approach described by Henry et al.10 First, nuclei were labeled with a fluorescent tag that is expressed only in specific cell types in Drosophila melanogaster to facilitate the subsequent isolation of tagged nuclei from mixed populations using an affinity-based approach. To label nuclei, the KASH (Klarsicht/Anc-1/Syne homology) domain was utilized. The KASH domain is a transmembrane domain that localizes to the outer membrane of the nuclear envelope, in part through interactions with SUN domain-containing proteins within the perinuclear space11. The C-terminal KASH domain of proteins such as Drosophila Msp-300 and Klarsicht anchors these proteins to the outer nuclear membrane, while their N-terminal domains interact with cytoskeleton proteins such as actin or microtubules in the cytoplasm12-14. Constructs were generated in which the KASH domain of Drosophila Msp-300 was fused to the C-terminus of EGFP, under control of the Gal4 regulatory sequence, UAS in the pUAST-attB vector15,16. Using phiC31 site-specific integration, transgenic flies were generated in which the UAS-EGFP::Msp-300KASH transgene was inserted in the attP2 loci on chromosome 3L17. The UAS-EGFP::Msp-300KASH flies can be crossed with flies that express the Gal4 driver in a particular cell type, resulting in the targeted expression of EGFP on the outer nuclear membrane in the cell type of interest. Labeled nuclei can then be purified from mixed cell populations using antibodies against GFP coupled to magnetic beads. In this approach, the use of nonionic detergent is not required because the EGFP tag is localized to the cytoplasmic side of the outer nuclear membrane, and is therefore accessible to antibodies.
The protocol described below can be used to isolate/enrich EGFP-labeled nuclei from specific cell types in Drosophila larval tissues, to quantify the purity and yield of isolated nuclei, and to extract nuclear RNA suitable for quantitative reverse-transcription polymerase chain reaction (qRT-PCR) gene expression analysis. The isolation and affinity-purification of nuclei (excluding tissue dissection) can be performed in less than one hour. Results are presented showing that glial nuclei can be successfully isolated from the larval optic lobe and eye imaginal disc, and used for subsequent gene expression analysis. It is anticipated that this approach will be useful for the isolation/enrichment of labeled nuclei from embryonic and larval tissues in which the target cells constitute less than 5% of the overall population. All Drosophila stocks and plasmids generated in this study are available from the authors upon request.
Protocol
1. Generation and Characterization of EGFP::KASH Drosophila
Cross Gal4 driver flies to UAS-EGFP::Msp-300KASHflies. Alternatively, recombinant flies that carry both the Gal4 driver and the UAS-EGFP::Msp-300KASH transgene can be generated using standard genetic techniques. This second approach is advantageous if large numbers of progeny are required.
- Characterize expression of the EGFP::KASH marker using standard microscopy techniques. Note: EGFP expression can be observed by standard microscopy techniques in dissected, fixed Drosophila tissues, or in whole larvae, and does not require use of an antibody.
- Determine the expression pattern of the EGFP::KASH marker in the entire organism under a fluorescence dissecting microscope (see Materials PDF). Note: Many Gal4 drivers, including the repo-GAL4 driver shown in Figure 1, exhibit nonspecific patterns of expression in other larval tissues such as the salivary gland (see Results).
- Analyze the EGFP::KASH expression pattern in the dissected tissue of interest using confocal microscopy.
- Fix the tissue of interest using formaldehyde at a final concentration of 4%, and stain DNA using DAPI at a final concentration of 0.1 μg/ml to visualize the localization of the EGFP::KASH tag relative to DNA.
- Acquire images using either a 40X / 1.30 oil immersion objective or a 20X / 0.75 oil immersion objective, depending on the size of the tissue of interest. The following excitation/emission conditions can be used for image acquisition: 408 nm/425-475 nm (DAPI); 488 nm /525-550 nm (GFP).
2. Isolate Nuclei from Drosophila Larval Tissues
- Prepare equipment for the larval tissue dissection and subsequent nuclei isolation. Note that whole dechorionated embryos could also be used as starting material if desired. It is recommended to use partially dissected larval tissues rather than entire larvae if the cell type of interest is present in a specific tissue such as the imaginal disc. This reduces nonspecific binding, and also eliminates problems that can be caused by expression of some Gal4 drivers in nontarget cells.
- Prepare two pairs of sharp forceps (see Materials PDF), one siliconized 9-well glass plate (see Materials PDF), and PBT (1x PBS, pH 7.4; 0.1% Tween-20).
- Prebind the GFP antibody to the magnetic beads. This step should be started before beginning the dissection. Add 10 μl of the magnetic beads (Invitrogen, see Materials PDF) and 2 μg of GFP antibody (Roche, see Materials PDF) to 400 μl of wash buffer (1x PBS, pH 7.4; 2.5 mM MgCl2) in a 1.5 ml tube. The amount of beads and antibody used can be optimized if necessary based on the amount of target in the sample and the binding affinity of the antibody to the beads.
- Incubate the beads and antibody with rotation for at least 30 min at room temperature.
- Place the 1.5 ml tube containing the beads and antibody in the magnetic rack (see Materials PDF) and allow the beads to bind to the magnetic for approximately 1-2 min. Remove the supernatant containing the unbound antibody and wash buffer using a 1 ml pipette. The beads will remain bound to the wall of the 1.5 ml tube on the side adjacent to the magnet.
- Rinse the beads twice with 1 ml of wash buffer each time as described in section 2.1.4. Remove the 1.5 ml tube from the magnetic rack and invert briefly to mix the beads and wash buffer during each wash step.
- Store the antibody-bound beads in wash buffer until ready to continue with step 2.7. The beads should not be allowed to dry out at any stage of the protocol.
Dissect larval tissues in PBT and transfer dissected tissues to one well of the 9-well dish. Typically, dissection of the optic lobe and eye imaginal disc from 100 third instar larvae provides sufficient material for downstream gene expression analysis following nuclei isolation.
Rinse the isolated tissue in nuclear extraction buffer (10 mM HEPES-KOH, pH 7.5; 2.5 mM MgCl2; 10 mM KCl) in a 1.5 ml tube. Transfer isolated larval tissues to a 1 ml Dounce Homogenizer (see Materials PDF) on ice that contains 1 ml of fresh nuclear extraction buffer. Incubate on ice for 5 min. It is not necessary to add protease inhibitors if RNA is to be extracted following nuclei isolation.
Disrupt the tissue by 20 strokes with the loose pestle and then incubate the sample on ice for 10 min to allow the cells to swell. Extract the nuclei from the cells using 15 strokes with the tight pestle. The number of strokes with the loose and tight pestles needs to be optimized for different tissues and cell types; excess homogenization can result in sheared nuclei, while insufficient homogenization will not extract all nuclei from the tissue.
Filter the homogenate, which contains the nuclei and cellular debris, through a 40 μm pore-size strainer (see Materials PDF) that has been prerinsed with nuclear homogenization buffer. Collect the filtered homogenate in a fresh tube.
- Collect pre-isolation samples for subsequent analysis to determine nuclei yield, nuclei integrity, and to determine transcript levels of target genes prior to nuclei isolation.
- Transfer 50 μl of the filtered homogenate to a fresh 1.5 ml tube using a pipette. This is the pre-isolation sample. Add 500 μl of Trizol reagent (see Materials PDF, CAUTION), invert to mix, and store at -20 °C for subsequent RNA isolation (step 3.1).
- Transfer 20 μl of the filtered homogenate to a fresh 1.5 ml tube.
- Add DAPI to a final concentration of 0.1 μg/ml, and incubate sample on ice for 10 min.
- Transfer DAPI-stained nuclei to a poly-lysine coated coverslip, and place on a microscope slide. Seal the coverslip using clear nail polish.
- Check the nuclei integrity, and presence of GFP-tagged nuclei of interest using fluorescent microscope. Note: It is not necessary to fix the nuclei, or to stain for GFP if these slides are analyzed reasonably quickly following preparation (within 24 hr).
- Transfer 10 μl of the filtered homogenate to a fresh 1.5 ml tube and add 90 μl of wash buffer (1x PBS, pH 7.4; 2.5 mM MgCl2). Add DAPI to a final concentration of 0.1 μg/ml, and incubate on ice for 10 min. Determine the nuclei yield in the pre-isolation sample using a hemocytometer to count the DAPI-positive nuclei using epifluorescence under a 10X air objective.
Place the 1.5 ml tube containing the antibody-bound beads (prepared in section 2.1.6) in a magnetic rack, and allow the magnetic beads to bind to the magnet for at least 2 min as described in section 2.1.4. Remove the wash buffer from the antibody-bound beads using a 1 ml pipette, and add the filtered homogenate from step 2.5 to the 1.5 ml tube using a 1 ml pipette.
Gently invert the tube several times to mix, and incubate at 4 °C for 30 min with end-over-end agitation. Avoid bubbles in the tube if possible since these can result in clumping of the nuclei.
Place the 1.5 ml tube containing the antibody-bound beads and homogenate in a magnetic rack, and allow the magnetic beads to bind to the magnet for at least 2 min as described in section 2.1.4. Remove the homogenate containing the unbound nuclei fraction using a 1 ml pipette.
Wash the sample 3x with 1 ml of wash buffer each time. Remove the 1.5 ml tube containing the beads, bound nuclei and wash buffer from the rack and invert to mix several times, then place the tube in the magnetic rack as described in step 2.9 and remove the supernatant using a 1 ml pipette.
- Add 150 μl of wash buffer to the beads, which should be bound to the target nuclei. This is the post-isolation sample. Prepare samples for microscopy analysis and subsequent RNA extraction.
- Transfer 20 μl of the post-isolation sample to a fresh 1.5 ml tube and stain with DAPI and analyze as described in section 2.6.2. Count the GFP+/DAPI+ and GFP-/DAPI+ nuclei captured by magnetic beads to determine the purity of the enriched GFP+ nuclei in the post-isolation sample.
- Add 1 ml of Trizol reagent to the remainder of the post-isolation sample, invert to mix, and store at -20 °C for subsequent RNA isolation (step 3.1). Note: Samples in Trizol can be stored for several weeks if necessary at -20 °C. It is not necessary to elute the nuclei from the magnetic beads prior to RNA extraction using Trizol reagent, because the beads do not interfere with the subsequent RNA isolation.
3. Determine Transcript Levels in Nuclei Isolated from Larval Tissues
- Isolate RNA from the pre-isolation and post-isolation samples prepared in sections 2.6.1 and 2.11.2 using the standard protocol described for Trizol-based RNA extraction (see Materials PDF) with the following modifications:
- Following precipitation of the RNA pellet, add 19.2 μl of RNase-free water and 0.8 μl of RNASecure reagent (see Materials PDF) to the pellet and incubate at 60 °C for 20 min to inactivate RNAses.
Determine the RNA concentration with a fluorescent-RNA binding dye such as the Qubit RNA assay kit (see Materials PDF) using the QubitR 2.0 Fluorometer following the manufacturer’s protocol.
Synthesize cDNA using an amplifying reverse transcriptase such as the EpiScript Reverse Transcriptase kit and oligodT primers following the manufacturer’s instructions. Note: Typically, 50 to 100 ng of RNA generates sufficient cDNA to perform multiple gene expression analyses. Equivalent amounts of pre-isolation and post-isolation RNA should be used to generate cDNA.
Add 180 μl of nuclease-free water to the 20 μl cDNA sample to dilute this 10-fold prior to real-time quantitative PCR (qPCR) analysis. This dilution is an important step, because undiluted cDNA samples can inhibit the qPCR.
- Prepare a qPCR master mix with polymerase and dNTPs, 4 pmoles each of forward and reverse primer, made up with sterile water to the final reaction volume of 20 μl.
- Design qPCR primers to target genes that are expected to be expressed in the cell type of interest, and at the developmental stage at which the tissue is collected. Design primers to span an intron if possible, so that genomic and cDNA PCR products can be distinguished. Note: If the gene expression profile of the target cell type is not known, primers against eGFP, which should be expressed specifically in the tagged nuclei population, can be used to optimize the protocol for a particular cell type and tissue (Table 1).
Determine the relative transcript levels of each gene of interest in the pre-isolation and post-isolation samples by comparison to a dilution series of cDNA prepared from the whole tissue. Transcript levels of target genes should be normalized to a reference gene such as Rpl32 (or preferably several reference genes).
Representative Results
The repo-GAL4 driver (Bloomington stock number 7415) is specifically expressed in glial cells in the Drosophila nervous system at multiple stages of development18. Flies were generated that stably express UAS-EGFP::Msp-300KASHunder control of the repo-GAL4 driver using standard genetic techniques. To characterize the expression pattern of the EGFP::KASH transgene in these flies, the pattern of GFP expression in the dissected optic lobe and eye imaginal disc from third instar larvae was examined using confocal microscopy. Dissected tissues were counterstained with DAPI to mark DNA and with α-chaoptin19 to label photoreceptor axons (mAb24B10,see Materials PDF). Figure 1A shows that the repo-GAL4 driven EGFP::KASH transgene is expressed in glia in the optic lobes and eye imaginal disc in the expected expression pattern. Under higher magnification, EGFP expression is observed in a circular pattern surrounding the DAPI-positive DNA, consistent with nuclear-envelope localization of the EGFP::KASH tag (Figure 1B). EGFP expression can be detected both in fixed and in unfixed tissues without the need for signal amplification using an antibody against GFP; antibodies against GFP were not used to visualize the EGFP::KASH expression in the images shown in Figures 1 and 2. To determine if the EGFP::KASH transgene is expressed nonspecifically in any other tissues in Drosophila larvae, GFP expression was examined in whole third instar larvae using a fluorescence dissecting microscope. Notably, nonspecific expression of the EGFP::KASH transgene was observed at high levels in the salivary glands of third instar larvae. This indicates that the repo-GAL4 driver has activity in some cell types other than glia in the larval stage of development. Thus, to specifically isolate glial nuclei, the optic lobe and eye imaginal disc from third instar larvae were isolated using dissection prior to homogenization and affinity-isolation.
To confirm that this protocol is suitable for the extraction of nuclei from Drosophila tissues, and to estimate the percentage of tagged nuclei present in a given tissue, the pre-isolation sample (section 2.6.2) from homogenates obtained from the optic lobe and eye imaginal disc of repo-GAL4, UAS-EGFP::Msp-300KASH third instar larvae was examined. Figure 2A shows a representative image of the pre-isolation sample obtained using confocal microscopy. In this image, the presence of nuclei is indicated by DAPI-positive events. A small number of sparsely scattered GFP-positive, DAPI-positive glial nuclei are also observed. We estimate that the GFP-positive nuclei account for less than 2% of the total nuclei population in this sample (Figure 2A). The total nuclei yield from optic lobe and eye imaginal discs that were dissected from 100 larvae was determined for the pre-isolation sample as described in section 2.6.3 (Table 2). A typical pre-isolation sample from 100 dissected optic lobe and eye imaginal discs contained between 0.8 and 1.2 x 107 nuclei, of which approximately 2% were GFP-positive. Figure 2B shows a representative image of DAPI-positive nuclei in a defined region of the hemocytometer. Note that the nuclei are intact, and maintain a consistent circular shape under the extraction conditions used in the protocol (Figures 2A and 2B).
To determine if this protocol can be used to isolate and/or highly enrich specific populations of labeled nuclei from tissues that contain mixed cell populations, α-GFP antibody-coated magnetic beads were used to isolate tagged nuclei from homogenates obtained from the optic lobe and eye imaginal disc of 100 repo-GAL4, UAS-EGFP::Msp-300KASH third instar larvae. In Figures 2C and 2D, it can be seen that the EGFP::KASH tagged glial nuclei bind to the α-GFP coated magnetic beads and are observed in the post-isolation sample (section 2.11.1) following magnetic separation. Although the magnetic beads exhibit some autofluorescence in the GFP channel, these can be readily differentiated from bead-bound nuclei by their smaller size (2.8 μm for the magnetic beads versus approximately 5 μm for the nuclei) and lack of DAPI staining. The GFP+/DAPI+ nuclei account for more than 95% of DAPI positive events in the post-isolation sample. Thus, the target tagged nuclei population are highly enriched in the post-isolation sample relative to the pre-isolation sample. The majority of the nuclei in the post-isolation sample show strong nuclear-envelope localized GFP fluorescence, and are usually surrounded by beads. However, it is possible that a small number of untagged nuclei could also be present in this sample. Thus, although the results in Figure 2C and in the gene expression analysis in Figure 3 (see below) indicate that the target tagged glial nuclei population is highly enriched, the possibility that some nuclei from some other nonspecific cell types are also present in the sample cannot be excluded. It is recommended that users optimize this protocol for their particular cell type and tissue of interest to maximize the enrichment of the target nuclei population, and to eliminate nonspecific nuclei binding. In particular, the ratio of starting material relative to antibody and beads is likely to be important for optimal enrichment of target nuclei. For this reason, the ranges of total nuclei numbers present in our typical pre-isolation samples are shown in Table 2. Based on the total nuclei numbers present in the pre-isolation sample, and the estimated proportion of glial cells in the mixed population from the optic lobe and eye imaginal disc, the maximum nuclei yield in the post-isolation sample was expected to be between 1.6 x 105 and 2.4 x 105 nuclei. However, using the hemocytometer to determine the nuclei yield as described in step 2.6.3, the actual nuclei yield obtained in the post-isolation sample was between 1.3 x 104 and 2.7 x 104 nuclei. These nuclei yields represent very rough estimates because the beads present in the post-isolation sample appear to affect accurate loading of the hemocytometer. Some GFP-positive nuclei are observed in the unbound homogenate, indicating that not all GFP-positive nuclei in the sample bind to the beads efficiently under the conditions used in this protocol. Increasing the time of binding or performing sequential isolation steps could potentially increase this yield, but this might come at the cost of both purity and RNA integrity. Using the conditions described in this protocol, sufficient quantities of RNA can be obtained in the post-isolation sample for downstream gene expression analysis by qRT-PCR (Table 2). Thus, this protocol is capable of rapidly and stringently isolating target nuclei from tissues that contain mixed populations of cells. Moreover, it is able to specifically isolate target nuclei that constitute only a small proportion, less than 5%, of the cells in a given population.
To further confirm that the post-isolation sample was highly enriched for our target glial nuclei population, the levels of transcripts for 4 different genes in the pre-isolation and post-isolation samples were compared by RT-qPCR. The transcript levels of elav, nrv2 and eGFP were determined in the two samples, and normalized to the reference gene, Rpl32, which is expressed at equivalent levels in glial cells and in the surrounding central nervous system (Figure 3). The fold difference in the post-isolation sample is shown relative to the pre-isolation sample, which is set to one. In Figure 3, the post-isolation sample shows significantly lower transcript levels of the neuronal-specific gene elav relative to the pre-isolation sample. This indicates that nuclei from neuronal cells are under-represented in the post-isolation sample. In contrast, higher transcript levels of both the glial-enriched nrv2 gene10 and eGFP are present in the post-isolation sample relative to the pre-isolation sample. This result indicates that the post-isolation sample is highly enriched for the target glial nuclei population. It is important to note that the transcript levels measured in this protocol represent nuclear transcripts, and therefore are likely to indicate levels of active transcription rather than steady-state mRNA. Reproducible expression of repo was not detected in either our pre-isolation or post-isolation samples, suggesting that the Gal4 protein expressed by the repo-GAL4 driver may persist after active transcription of the endogenous repo gene has ceased. This is consistent with results from other studies in which nuclei labeled using the twi promoter do not show enriched levels of twi transcripts, perhaps due to differences in the developmental timing of transcription versus protein stability8. Taken together, these results demonstrate that the described protocol can be successfully used to highly enrich target nuclei from tissues that contain mixed populations of cells, and that the isolated nuclei are suitable for gene expression analysis.
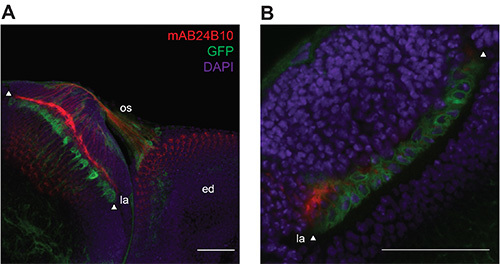 Figure 1. Expression pattern of repo-GAL4, UAS-EGFP::Msp-300KASHfly line. (A) In third instar larvae, repo-GAL4 drives expression of the UAS-EGFP::Msp-300KASHtransgene in glial cells in the optic lobe, optic stalk (os) and eye imaginal disc (ed). The nuclear envelope in glial cells is labeled with EGFP::KASH (green). R1 - R8 photoreceptor cell axons are labeled with α-chaoptin (mAB24B10, red) for comparison, and DNA is stained with DAPI (purple). GFP-labeled glial nuclei are visible along the lamina (la) within the optic lobe (marked by arrows). Scale bar, 50 μm. (B) Higher magnification image of the lamina ganglia of the optic lobe that contains EGFP::KASH labeled glial cell nuclei. Scale bar, 50 μm. Confocal images were acquired using either a 40X / 1.30 oil or a 20X / 0.75 oil immersion objective, and using excitation/emission wavelengths of 408 nm/425-475 nm (DAPI); 488 nm/525-550 nm (GFP); 561 nm/595-620 nm (Alexa5,6,8). Please click here to view a larger version of this figure.
Figure 1. Expression pattern of repo-GAL4, UAS-EGFP::Msp-300KASHfly line. (A) In third instar larvae, repo-GAL4 drives expression of the UAS-EGFP::Msp-300KASHtransgene in glial cells in the optic lobe, optic stalk (os) and eye imaginal disc (ed). The nuclear envelope in glial cells is labeled with EGFP::KASH (green). R1 - R8 photoreceptor cell axons are labeled with α-chaoptin (mAB24B10, red) for comparison, and DNA is stained with DAPI (purple). GFP-labeled glial nuclei are visible along the lamina (la) within the optic lobe (marked by arrows). Scale bar, 50 μm. (B) Higher magnification image of the lamina ganglia of the optic lobe that contains EGFP::KASH labeled glial cell nuclei. Scale bar, 50 μm. Confocal images were acquired using either a 40X / 1.30 oil or a 20X / 0.75 oil immersion objective, and using excitation/emission wavelengths of 408 nm/425-475 nm (DAPI); 488 nm/525-550 nm (GFP); 561 nm/595-620 nm (Alexa5,6,8). Please click here to view a larger version of this figure.
 Figure 2. Characterization of nuclei in “pre-isolation” and “post-isolation” samples. (A) Representative image from a repo-GAL4, UAS-EGFP::Msp-300KASHpre-isolation sample analyzed using a X 20 / 0.75 oil immersion objective and standard confocal microscope. DNA is stained with DAPI (blue), and EGFP::KASH tagged glial nuclei are shown in green (marked by arrows). Scale bar, 50 μm. (B) Representative image of DAPI-stained nuclei from a pre-isolation sample on a hemocytometer using a X 10 air objective and standard epifluorescence microscope. Hemocytometer grids are shown in light grey. Only DAPI-positive nuclei are shown in this image, as GFP fluorescence was not visible under this objective on the epifluorescence microscope. (C) Representative image from a repo-GAL4, UAS-EGFP::Msp-300KASHpost-isolation sample analyzed as described for panel A. The isolated EGFP::KASH tagged glial nuclei (marked by arrows) stain positively for DAPI, and are surrounded by the beads, which autofluoresce at low levels in the GFP channel. Scale bar, 50 μm. (D) Higher magnification image of a typical isolated EGFP::KASH tagged glial nuclei (marked by arrow) surrounded by beads. Scale bar, 5 μm. Please click here to view a larger version of this figure.
Figure 2. Characterization of nuclei in “pre-isolation” and “post-isolation” samples. (A) Representative image from a repo-GAL4, UAS-EGFP::Msp-300KASHpre-isolation sample analyzed using a X 20 / 0.75 oil immersion objective and standard confocal microscope. DNA is stained with DAPI (blue), and EGFP::KASH tagged glial nuclei are shown in green (marked by arrows). Scale bar, 50 μm. (B) Representative image of DAPI-stained nuclei from a pre-isolation sample on a hemocytometer using a X 10 air objective and standard epifluorescence microscope. Hemocytometer grids are shown in light grey. Only DAPI-positive nuclei are shown in this image, as GFP fluorescence was not visible under this objective on the epifluorescence microscope. (C) Representative image from a repo-GAL4, UAS-EGFP::Msp-300KASHpost-isolation sample analyzed as described for panel A. The isolated EGFP::KASH tagged glial nuclei (marked by arrows) stain positively for DAPI, and are surrounded by the beads, which autofluoresce at low levels in the GFP channel. Scale bar, 50 μm. (D) Higher magnification image of a typical isolated EGFP::KASH tagged glial nuclei (marked by arrow) surrounded by beads. Scale bar, 5 μm. Please click here to view a larger version of this figure.
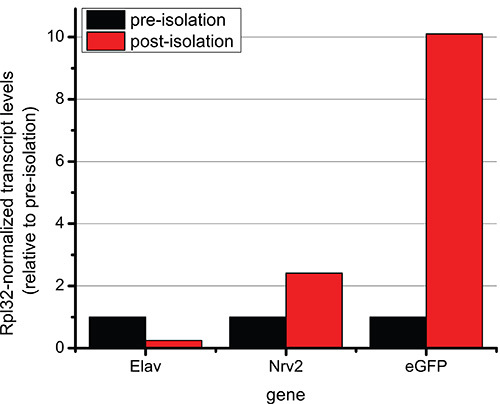 Figure 3. Representative qRT-PCR data from “pre-isolation” and “post-isolation” samples. Transcript levels of target genes were determined by qRT-PCR of pre-isolation and post-isolation samples obtained from repo-GAL4, UAS-EGFP::Msp-300KASHoptic lobes and eye imaginal discs. All transcript levels are normalized to Rpl32 transcript levels, and the pre-isolation sample for each gene target is set to one. Representative results from one biological experiment are shown for the following genes: elav, nrv2 and eGFP.
Figure 3. Representative qRT-PCR data from “pre-isolation” and “post-isolation” samples. Transcript levels of target genes were determined by qRT-PCR of pre-isolation and post-isolation samples obtained from repo-GAL4, UAS-EGFP::Msp-300KASHoptic lobes and eye imaginal discs. All transcript levels are normalized to Rpl32 transcript levels, and the pre-isolation sample for each gene target is set to one. Representative results from one biological experiment are shown for the following genes: elav, nrv2 and eGFP.
| Gene | Primer sequence (5' -3') | Size PCR product (bp) |
| eGFP | GAGGGATACGTGCAGGAGAG | 102 |
| GATCCTGTTGACGAGGGTGT | ||
| nrv2 | GGCTGGTTGTAGTCGCAGTT | 125 |
| GGCACCCAACACGAGAACTA | ||
| elav | GCACCATTCGGAGCAATAAT | 153 |
| TGTGCAGCTGGATGGTGTAG | ||
| Rpl32 | GCTAAGCTGTCGCACAAATG | 160 |
| CGTTGTGCACCAGGAACTT |
Table 1. Primers used for qRT-PCR analysis.
| Total number of nuclei in pre-isolation sample | Estimated total number of nuclei in post-isolation sample | RNA yield from post-isolation sample (ng) |
| 0.8 - 1.2 x 107 | 1.3 - 2.7 x 104 | 160-220 ng |
Table 2. Representative nuclei and RNA yields obtained. Typical ranges of nuclei and RNA yields obtained from pre-isolation and post-isolation samples from 100 dissected optic lobe and eye imaginal discs from the repo-GAL4, UAS-EGFP::Msp-300KASH genotype, in which glial cells are labeled with EGFP::KASH. Nuclei were quantified as described in 2.6.3 and RNA was quantified as described in 3.1.2.
Discussion
This protocol can be used to isolate or highly enrich specifically tagged nuclei from mixed cell populations in Drosophila embryonic or larval tissues. Using this protocol, nuclei can be isolated from dissected tissues in approximately 1 hr. The purity and yield of the isolated nuclei must be experimentally determined for each cell type. It can be difficult to quantify the bead-bound nuclei at the post-isolation stage of the protocol using a hemocytometer because of the beads present in the sample. Thus, if exact numbers of nuclei are required for a downstream application, it will be necessary to estimate nuclei yield based on an alternative method such as DNA content. Exact calculations of nuclei yield are not necessary for gene expression analysis if RNA can be successfully isolated and quantified. It is recommended to use a fluorescence-based method for quantifying RNA yield, as we have found that spectrophotometric determination of RNA concentration in these low concentration samples is highly variable.
There are two important aspects of the protocol that will influence its success: the choice of magnetic beads and antibody. Magnetic beads that are coupled to GFP or protein G are commercially available from several different sources. However, it is strongly recommended to use the protein G magnetic beads available from Invitrogen for the procedure described in this protocol for two major reasons. First, the size of the Invitrogen beads at 2.8 μm is slightly smaller than the average size of the nuclei in the cell types we have examined, which is approximately 5 μm (Figure 2D). Magnetic beads that are smaller (e.g. 50 nm, μMACS α-GFP, Miltenyi Biotech) do not bind rapidly to batch-wash style magnets, and the columns provided for these beads can clog with larval tissue extracts. Larger magnetic beads (e.g. 10 μm, PureProteome protein G magnetic beads, Millipore) did not bind well to the nuclei in our preliminary tests, and these also exhibit strong autofluorescence in the same channels as both DAPI and GFP, making it difficult to identify nuclei in the bead-bound sample by standard microscopy techniques. In addition to the choice of magnetic beads, it is important to use a GFP antibody that works well for immunoprecipitation but that does not have high nonspecific background. To aid in the reproducibility of this protocol, we have used a monoclonal mouse antibody against GFP. Polyclonal rabbit antibodies against GFP were also successfully trialed in our preliminary studies, but these tended to have higher nonspecific background. Other monoclonal antibodies were also tested (e.g. Developmental Studies Hybridoma Bank), with varying levels of success. Thus, antibody choice is an important factor in the successful isolation of labeled nuclei.
Although in this protocol we describe methods to determine gene transcript levels in isolated nuclei, it is anticipated that the approach described in steps 2.1-2.11 will also be suitable for isolating nuclei that can be used for subsequent chromatin immunoprecipitation analysis. If tissue is fixed in 1% formaldehyde prior to homogenization (step 2.3), then dissected tissues can be collected over several days and sufficient material isolated to perform chromatin immunoprecipitation analysis. Based on counts obtained using the hemocytometer, which provide only rough estimates as discussed above, we typically obtained 1.3 x 104 and 2.7 x 104 glial nuclei from the eye imaginal disc and optic lobe of 100 larvae (Table 2). Thus, using our approach, it should be feasible to obtain sufficient material to perform chromatin immunoprecipitation analysis, depending on the abundance of the targeted cell type, and the epitope of interest.
One useful genetic application of this technique is that it can be used to positively label nuclei in specific cell types in embryos or larvae that are homozygous for a recessive lethal mutant allele. To accomplish this, two separate fly stocks must be generated in which the mutant allele of interest is recombined with a specific Gal4 driver, or with the UAS-EGFP::Msp-300KASH transgene on the third chromosome. If flies carrying the mutant allele and Gal4 driver are crossed to flies carrying the mutant alleles and the UAS-EGFP::Msp-300KASH transgene, only progeny that have both copies of the mutant allele will be labeled with GFP in the tissue in which the Gal4 driver is expressed. Thus, cell-specific effects of recessive lethal alleles can be examined in the embryonic or larval stages, prior to the stage of lethality. This genetic technique has been previously used to positively label cells in embryonic muscle that were homozygous for mutations in the SAGA subunit, sgf111.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Janice Fischer for providing the UAS-EGFP::Msp-300KASH plasmid. The mAB24B10 antibody was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology. The repo-GAL4 stock (BL7415) was obtained from the Bloomington Stock Center at Indiana University. Support from the American Cancer Society Institutional Research Grant (IRG #58-006-53) to the Purdue University Center for Cancer Research is gratefully acknowledged. Jingqun Ma is supported by an Agricultural Research at Purdue Assistantship in Food and Agriculture from Purdue University.
References
- Weake VM, et al. Post-transcription initiation function of the ubiquitous SAGA complex in tissue-specific gene activation. Genes Dev. 2011;25:1499–1509. doi: 10.1101/gad.2046211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn S, et al. Cell type-specific chromatin immunoprecipitation from multicellular complex samples using BiTS-ChIP. Nat. Protoc. 2012;7:978–994. doi: 10.1038/nprot.2012.049. [DOI] [PubMed] [Google Scholar]
- Haenni S, et al. Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3'-end-seq. Nucleic Acids Res. 2012;40:6304–6318. doi: 10.1093/nar/gks282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelson RA, Lambert GM, Vanier C, Lynch RM, Galbraith DW. Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells. BMC Genom. 2007;8 doi: 10.1186/1471-2164-8-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Barthelson RA, Lambert GM, Galbraith DW. Global characterization of cell-specific gene expression through fluorescence-activated sorting of nuclei. Plant Physiol. 2008;147:30–40. doi: 10.1104/pp.107.115246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff S. The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011;6:56–68. doi: 10.1038/nprot.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell. 2010;18:1030–1040. doi: 10.1016/j.devcel.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner FA, Talbert PB, Kasinathan S, Deal RB, Henikoff S. Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genom. Res. 2012;22:766–777. doi: 10.1101/gr.131748.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Henry GL, Davis FP, Picard S, Eddy SR. Cell type-specific genomics of Drosophila neurons. Nucleic Acids res. 2012;40:9691–9704. doi: 10.1093/nar/gks671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr DA. KASH and SUN proteins. Curr Biol. 2011;21:414–415. doi: 10.1016/j.cub.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer JA, et al. Drosophila klarsicht has distinct subcellular localization domains for nuclear envelope and microtubule localization in the eye. Genetics. 2004;168:1385–1393. doi: 10.1534/genetics.104.028662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson K, et al. The functions of Klarsicht and nuclear lamin in developmentally regulated nuclear migrations of photoreceptor cells in the Drosophila eye. Mol. Biol. Cell. 2004;15:600–610. doi: 10.1091/mbc.E03-06-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, et al. The KASH domain protein MSP-300 plays an essential role in nuclear anchoring during Drosophila oogenesis. Dev. Biol. 2006;289:336–345. doi: 10.1016/j.ydbio.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. U.S.A. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracklauer MP, Banks SM, Xie X, Wu Y, Fischer JA. Drosophila klaroid encodes a SUN domain protein required for Klarsicht localization to the nuclear envelope and nuclear migration in the eye. Fly. 2007;1:75–85. doi: 10.4161/fly.4254. [DOI] [PubMed] [Google Scholar]
- Markstein M, Pitsouli C, Villalta C, Celniker SE, Perrimon N. Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet. 2008;40:476–483. doi: 10.1038/ng.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp KJ, Schulte J, Auld VJ. Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev. Biol. 2001;238:47–63. doi: 10.1006/dbio.2001.0411. [DOI] [PubMed] [Google Scholar]
- Fujita SC, Zipursky SL, Benzer S, Ferrus A, Shotwell SL. Monoclonal antibodies against the Drosophila nervous system. Proc. Natl. Acad. Sci. U.S.A. 1982;79:7929–7933. doi: 10.1073/pnas.79.24.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]