

Abstract
Historically, the midbrain and hindbrain (MBHB) have been considered “support staff” for the cerebrum, which has typically been acknowledged as the most important part of the brain. Radiologists and pathologists did not regularly examine these structures, also known as the brainstem and cerebellum, because they are small and difficult to remove without damage. With recent improvements in neuroimaging, neuropathology and neurogenetics, many developmental disorders of the MBHB have emerged as significant causes of neurodevelopmental dysfunction. This review provides an overview of MBHB disorders important to clinicians and developmental biologists. A basic understanding of MBHB embryology is essential to understanding the malformations that occur in MBHB structures; therefore, a brief embryology review is provided, as is a review of MBHB anatomy as assessed by MRI, and an approach to MRI analysis of the individual structures. Clinical features common to many MBHB disorders are presented, followed by a more in depth summary of the clinical presentations, MRI features and genetic causes of many common, and some less common, malformations. Research advances that may change how we treat these patients in the future are briefly discussed. The information provided in this review will improve the clinical acumen of the practicing neurologist in regard to malformations of the MBHB, while at the same time adding to their understanding of brainstem and cerebellar development, genetics, and function.
Introduction
Recent advances in genetic and neuroimaging technology have promoted enormous progress in the understanding of midbrain and hindbrain (MBHB) malformations.1 Combining modern imaging with cutting edge genetic techniques has provided heretofore unimaginable clarity to the wide spectrum of developmental disorders affecting the MBHB. While many of these disorders result from inherited mutations, a number are caused by mutations that occur de novo in the affected individual, and are therefore not inherited. Great strides have also been made in understanding the mechanisms underlying normal and abnormal MBHB development using animal models such as mouse, chicken and zebrafish. These combined advances are poised to yield better diagnostic testing, and hopefully, more specific and effective treatments in the future.
The MBHB develops abnormally in many disorders, so this review focuses on congenital conditions in which the MBHB is predominantly affected and where progress has been made in the diagnosis and/or genetics. We omit MBHB disorders due to in utero insults, forebrain disorders secondarily affecting the MBHB, progressive disorders with onset after birth, and neural tube defects. In addition, later onset and acquired disorders that result in cerebellar atrophy have been reviewed elsewhere2, 3 and are not covered. For comprehensive classification of MBHB malformations, see Barkovich et al. 20074 and Barkovich et al. 2009.5
This review begins with a summary of basic MBHB embryology and developmental genetics that provides a framework for descriptions of the clinical features and causes of human MBHB malformations. A neuroimaging approach to the MBHB is then suggested to aid in the detection of subtle abnormalities. Finally, the clinico-radiologic features of a series of conceptually and clinically important MBHB malformations are described.
Basic embryology of the MBHB
After neural tube formation and segmentation into brain regions (fore-, mid- and hind-brain) and spinal cord, neurons of the MBHB are mostly generated in the neuroepithelium that lines the walls of the 4th ventricle (Fig 1). In the developing midbrain, neurons are generated from the ventricular zone and primarily migrate radially to form the tectum dorsally, and substantia nigra, red nuclei and cranial motor nerves 3 and 4, ventrally.
Figure 1. MBHB development and genes associated with human MBHB malformations.

The diagrams depict representative stages of mouse MBHB development, the model in which MBHB development is best understood. The upper diagrams depict views from the dorsal aspect at E9.5, E12.5 and E18.5 (days after conception in the mouse embryo). Dotted lines in the upper diagrams indicate the position of the axial (A) or sagittal (B and C) cross sections depicted in the lower diagrams. (A) The lower figure represents an axial cut through most rostral level of the hindbrain. HOXA1 is involved in Anterior/Posterior (A/P) patterning of the MBHB, while rhombencephalosynapsis may be due to defects in Dorsal/Ventral (D/V) patterning at the most dorsal and rostral aspect of the hindbrain (red area), where the cerebellar vermis is formed. (B) The upper diagram shows the location of early vermis formation at the most dorsal and anterior portion of the hindbrain (red circle). Early vermis formation may require the genes involved in Joubert/Meckel syndromes. Lower image (sagittal view) shows PTF1A expressed in the cerebellar ventricular zone is required for GABAergic Purkinje cell precursor fate, FOXC1 is required for induction between mesenchyme and rhombic lip, and the RELN pathway, O-glycosylation genes, and tubulins are likely required for migration of precursors out of the rhombic lip and ventricular zone. (C) By E18.5, the mouse cerebellar hemispheres (blue in upper figure) and vermis (yellow in upper figure) have partially formed. The midline sagittal (lower) diagram depicts early foliation and cortical lamination. Multiple pathways are likely involved in proliferation, migration, and survival of neuronal precursors and other cell types. ROBO3, Joubert/Meckel and tubulin genes are required for axon pathfinding to establish connections with cerebellar and brainstem nuclei. Fore=Forebrain, Mid=Midbrain, Hind=Hindbrain
The cerebellum arises from the dorsal anterior hindbrain (rhombomere 1), and most neurons are generated in two distinct germinal matrices: 1) the dorsal- and rostral-most portion of the rhombic lip, which generates neuronal precursors that develop into glutamatergic neurons (cerebellar granule neurons and others), and 2) the adjacent dorsal ventricular zone, which generates cerebellar GABAergic neurons (Purkinje cells (PCs) and others). Although ATOH1 (formerly called MATH1) and PTF1A are critical for defining these germinal zones, most genes required for cerebellar development remain to be identified.6, 7 After generation in the rhombic lip, granule cells migrate tangentially to form a transient external granular layer (EGL) on the outside of the developing cerebellar hemisphere.8 The EGL is a secondary germinal zone, as immature granule cells undergo many cycles of mitosis (under the influence of sonic hedgehog, a mitogen secreted by underlying developing PCs), while migrating over the enlarging cerebellum. As EGL progenitors differentiate into granule neurons, they migrate inwards to form the internal granular layer (IGL) located under the developing PCs. In humans, this process continues into the second postnatal year. Concurrently, GABAergic PCs are generated in the ventricular zone and migrate along radial glial processes into the cerebellar anlage, where they form an array of PC clusters under the EGL.9 Reelin, secreted by the EGL, binds to receptors on the PC processes, reducing PC-PC adhesion and allowing the cell clusters to string out into long parasagittal stripes, which form the skeleton of the mature cerebellum.10-12
In the posterior hindbrain, the lower rhombic lip is a germinal zone distinct from the upper rhombic lip. It does not contribute to the cerebellum, instead producing the neurons that migrate to form the precerebellar nuclei (pontine, pontine reticulotegmental nuclei, inferior olivary nuclei, external cuneate, and lateral reticular), all of which are directly connected to the cerebellum at an early developmental stage and maintain connections as the structures grow and mature.13 Development of the upper and lower rhombic lips is regulated by overlapping sets of genes, disruption of which can cause both cerebellar and brainstem defects, reinforcing the developmental interdependence of these structures.
Clinical features of MBHB malformations
Little population-based data on the prevalence of MBHB malformations exists, due to the uncommonness of these disorders and substantial under-recognition on brain imaging studies. The postnatal prevalence of Dandy-Walker malformation has been estimated to be ∼1/30,00014 and 1/5,00015. A prenatal prevalence of ∼1/10,000 was estimated from a large birth defects registry in the United Kingdom.16
Clinical classification of MBHB disorders is important to provide accurate prognostic and recurrence risk information, as well as guide further evaluation and medical management. In addition, a clinical diagnosis can lead to identification of a genetic cause that can then be used for prenatal diagnosis and carrier testing. The utility of a given diagnostic category rests on whether it can be used to predict outcome, response to treatment and/or underlying cause. Given the overlapping clinical and imaging features of MBHB malformations, correct classification requires a holistic approach that integrates history, physical examination, imaging and laboratory testing.
Due to the widespread use of prenatal ultrasound and MRI to evaluate the fetal brain, MBHB malformations are frequently diagnosed before birth, although the sensitivity and specificity of prenatal imaging for MBHB malformations are unknown.17-20 Prenatal ultrasound can identify cerebellar hypoplasia, abnormal fluid collections in the posterior fossa or poor delineation of posterior fossa landmarks. Further evaluation during the pregnancy can involve fetal MRI, genetic amniocentesis, cell free fetal DNA testing and evaluation for in utero infection. If work-up during the pregnancy does not identify a specific etiology, postnatal examination, imaging and laboratory testing are often revealing.
Postnatal presentation of patients with MBHB malformations is typically non-specific; features include hypotonia, motor delay, nystagmus, and decreased visual attention (Table 1). More severely affected patients can present with apnea, feeding difficulties, aspiration, spasticity, lack of developmental progress or seizures. Signs of cranial neuropathy, such as abnormal eye movements, ptosis, facial palsy, hearing impairment (with consequent speech delay) and facial/corneal anesthesia, may also be observed. Ophthalmologic evaluation may reveal chorioretinal coloboma or retinal dystrophy in a subset of patients with Joubert syndrome (JS), a variety of structural eye abnormalities in patients with cobblestone malformations, or eye movement abnormalities. Cognitive impairment is common, but not universal, and autistic features are also observed.21 The range of neurodevelopmental outcome is broad. Mildly affected patients may have relatively isolated cranial nerve dysfunction, as in Duane retraction syndrome and horizontal gaze palsy with progressive scoliosis.
Table 1. Common clinical features in patients with MBHB malformations.
| Hypotonia |
| Ataxia |
| Abnormal eye movements (nystagmus, oculomotor apraxia, abnormal smooth pursuit, saccades, optokinetic nystagmus and/or vestibulo-ocular reflex) |
| Motor delay |
| Vestibular issues (e.g. poor balance, head shaking, disequilibrium) |
| Dysarthria |
| Cranial nerve deficits |
| Dysphagia |
| Trigeminal anesthesia |
| Hearing loss |
| Dysarthria |
| Cognitive-affective signs and symptoms |
| Cognitive impairment |
| Hyperactivity/Impulsivity |
| Emotional lability |
| Autistic behaviors |
While these common features related to cerebellar and brainstem dysfunction help identify patients with MBHB malformations and prompt brain imaging, idiosyncratic features can help differentiate between specific diagnoses (Table 2 and sections below). For instance, alternating tachypnea/apnea, polydactyly, coloboma, and/or progressive retinal, kidney or liver disease can be seen in some, but not all, patients with JS. Most patients with rhombencephalosynapsis have persistent head-shaking, often in a figure-of-eight pattern; others have scalp alopecia, trigeminal anesthesia and hyperactivity. Extreme prematurity or intrauterine infection can point to a non-genetic cause; however, these are usually diagnoses of exclusion. Laboratory evaluation can also help differentiate patients: elevated creatine kinase in patients with cobblestone malformations, abnormal transferrin glycosylation in CDGS, elevated glucose and absent insulin in patients with PTF1A-related cerebellar and pancreatic agenesis.
Table 2. Typical clinical, imaging and genetic characteristics of specific MBHB malformations involving the cerebellum*.
| Disorder | Clinical features | Imaging features | Causes/testing | Inheritance |
|---|---|---|---|---|
| Dandy Walker malformation | Macrocephaly common Variable cognitive, NDV impairment Axenfield-Rieger syndrome (FOXC1) |
Hypoplastic vermis, variable hemisphere hypoplasia, large posterior fossa, upwardly rotated vermis | FOXC1 (rare), ZIC1/ZIC4 deletions (rare), most unknown | Sporadic (recurrence risk <5%) |
| Lissencephaly-related MBHB malformations | Spasticity, seizures Severe NDV impairment in patients with RELN mutations Broad range of NDV outcome in patients with tubulinopathies and ARX mutations |
Heterogeneous RELN: pachygyria with mildly thickened cortex ARX: variable spectrum, lissencephaly with moderately thick cortex (more severe posteriorly), small to absent basal ganglia, ACC, small pons with normal cerebellum Tubulinopathies: highly variable spectrum of PMG to lissencephaly, enlarged tectum, dysmorphic basal ganglia, thin/absent CC |
RELN, ARX, TUBA1A | AR (RELN), X-linked (ARX), de novo AD (TUBA1A) |
| VLDLR-related cerebellar hypoplasia | Dysequilibrium, quadripedal locomotion reported in some, substantial ataxia Moderate to severe NDV impairment |
Cerebellar hypoplasia with decreased foliation (vermis worse than hemispheres), simplified cortical gyral pattern | VLDLR | AR |
| X-linked intellectual disability with cerebellar hypoplasia | Moderate NDV impairment | Mild vermis hypoplasia, mild cerebellar hemisphere hypoplasia, variable ventriculomegaly | OPHN1 | X-linked |
| PHACE syndrome | Segmental hemangioma (usually head and neck), intracranial and great vessel abnormalities Variable NDV impairment in <50% (usually mild) |
Unilateral cerebellar hypoplasia with or without vermis involvement, DWM Dysmorphic or absent major vessels Occasional heterotopia, PMG |
Unknown | Sporadic |
| Oculocerebrocutaneous syndrome | Orbital cysts, micro/anophthalmia, focal skin defects and appendages | Severely hypoplastic vermis, normal or hypoplastic cerebellar hemispheres, enlarged dysplastic tectum, thick, vertical SCPs, ACC, frontal PMG | Unknown | Sporadic |
| RES all types | Alopecia, trigeminal anesthesia (GLH syndrome), head shaking (usually “figure of eight” pattern), hyperactivity/impulsivity, VACTERL features (<50%), variable ataxia Full range of NDV outcome |
Absent septum pellucidum, aqueductal stenosis, fused colliculi, posterior holoprosencephaly (rare), absent olfactory bulbs (<50%) | Unknown | Sporadic (one recurrence reported) |
| Cerebellar hyperplasia | Highly variable features of Alexander disease, fucosidosis, Sotos, Williams, Costello, MCAP/MPPH syndromes | Large cerebellum, cerebellar ectopia, cerebellum wrapped around brainstem | Chromosomal anomalies, genes for associated disorders | Various depending on specific disorder |
| CMS | Hearing loss, mild or no ataxia Mild NDV impairment |
Cerebellar hemisphere dysplasia, inferior cerebellar vermis hypoplasia, partial agenesis of the corpus callosum, frontal subcortical heterotopia, frontal PMG, arachnoid cysts | GPSM2 | AR |
| PCH Type 1 | Spinal muscular atrophy | Hypoplastic pons (often mild), proportional vermis and hemispheres hypoplasia | EXOSC3, VRK1 | AR |
| TSEN-related PCH | Neonatal encephalopathy, severe progressive microcephaly, increased tone, dyskinesia, seizures, cortical visual impairment NDV impairment usually profound |
Postmigrational microcephaly, small pons and cerebellum, atrophic appearing cortex, thin corpus callosum, vermis less hypoplastic than hemispheres | TSEN54, TSEN2, TSEN34 | AR |
| PCH Type 6 | Elevated cerebrospinal fluid lactate | Small pons and cerebellum, vermis more severely hypoplastic than hemispheres | RARS2 | AR |
| PTF1A-related PCH | Congenital diabetes, intrauterine growth retardation, microcephaly Severe NDV impairment |
Small to absent cerebellum, small pons | PTF1A | AR |
| CASK-related PCH | Typically females (rare males), progressive microcephaly, variable hearing loss Severe NDV impairment |
Small pons and cerebellum (can be mild), proportionate cerebellar vermis and hemisphere hypoplasia | CASK | X-linked |
| CHMP1A-related PCH | Acquired microcephaly, increased extremity tone and contractures Moderate to severe NDV impairment |
Small pons and cerebellum, proportionate vermis and hemisphere hypoplasia | CHMP1A | AR |
| PTCD | Hearing loss, trigeminal anesthesia/corneal scarring, dysphagia, variable cardiac and vertebral/rib defects, substantial ataxia Moderate to severe NDV impairment |
Hypoplastic pons, mildly hypoplastic cerebellum, “cap” of white matter on dorsum of pons, markedly hypoplastic middle and inferior cerebellar peduncles | No specific testing | Sporadic |
| CDGS Type 1a | Abnormal fat distribution, retinitis pigmentosa, other diverse features: failure to thrive, elevated transaminases, coagulopathy, hypothyroidism, hypogonadism, seizures, stroke-like episodes, substantial ataxia Mild to severe NDV impairment |
Pontocerebellar hypoplasia with progressive atrophy, cortical atrophy in some patients | PMM2, transferrin glycosylation analysis | AR |
| Joubert syndrome | Hypotonia, alternating apnea and tachypnea (improves with age), other diverse features: Retinal dystrophy, coloboma, liver fibrosis, nephronophthisis, polydactyly, substantial ataxia Moderate-severe NDV impairment |
Vermis hypoplasia, long/thick, horizontal SCPs, encephalocele (rare), foramen magnum cephalocele (occasional), anterior midbrain and dorsal medulla heterotopia (occasional), PMG and ACC (rare) | AHI1, ARL13B, C5ORF42, CC2D2A, CEP41, CEP290, INPP5E, KIF7, MKS1, NPHP1, OFD1, RPGRIP1L, TCTN1, TCTN2, TCTN3, TMEM67, TMEM138, TMEM216, TMEM237 | AR, X-linked (OFD1 only) |
| Dystroglycanopathies (Walker-Warburg syndrome, Muscle-eye-brain disease, Fukuyama muscular dystrophy) | Eye involvement (cataracts, coloboma, high myopia), muscle involvement (variably increased CK) Severe NDV impairment |
Kinked brainstem, large tectum, cerebellar hypoplasia, subcortical cerebellar cysts, cobblestone cortex, abnormal myelination | POMT, POMGNT1, FKRP, POMT1, POMT2, FKTN, LARGE, GPR56, ISPD, GTDC2 | AR |
Note that information published for many of the disorders is quite limited, so the full spectrum of characteristics is likely broader than depicted in this table.
Imaging Approach to MBHB malformations
Many physicians are comfortable with looking at images of the cerebral hemispheres and have worked out an approach to analyzing CT and MRI scans that allows them to detect significant abnormalities without missing any important features. The MBHB has always been more difficult to image than supratentorial structures, requiring a modified approach to evaluating the images. The MBHB is composed of the brainstem and the cerebellum. The brainstem is composed of the midbrain, derived from the embryonic mesencephalon, the pons, formed from the rostral part of the rhombencephalon (the metencephalon), and the medulla, formed from the caudal part of the rhombencephalon (myelencephalon). The cerebellum, which is a dorsal extension of the rostral rhombencephalon, is composed of the medial/midline vermis and the bilateral hemispheres. It is best to start by looking at the midline sagittal image (Fig 2A), which allows an overall estimate of the size of the posterior fossa in addition to detailed views of the brainstem, the fourth ventricle and the cerebellar vermis. The posterior margin of the brainstem from the distal aqueduct to the obex should be nearly straight. The fastigium of the fourth ventricle should be just below the mid-point of the ventral pons. As an approximation, the cranio-caudal size (height) of the vermis on this image should be equal to the distance from the intercollicular sulcus of the midbrain tectum to the obex. The vermis is divided into three sections by the primary and prepyramidal fissures (Fig 2A, arrows). These sections are approximately equal in size, with the anterior vermis (the portion anterior to the primary fissure) being largest and the middle section typically the smallest. The rostrocaudal length of the ventral pons at its largest point should be approximately double the size of the midbrain from the anteromedial most point of the midbrain on this image to the third ventricle; the latter is equal to the distance from the anterior-posterior level of the obex to the inferior margin of the ventral pons. The cerebellar hemispheres are characterized on coronal images by fissures that radiate towards the cerebellar nuclei; this is best seen on images at the level of the vermian nodulus (Fig 2B). The cerebellar peduncles should be evaluated for size, contour, and location (Fig 2C-D). On axial images, the hemispheric folia of the inferior half of the cerebellum are seen to course parallel to the calvarium (Fig 2D).
Figure 2. Normal brain MRI images.
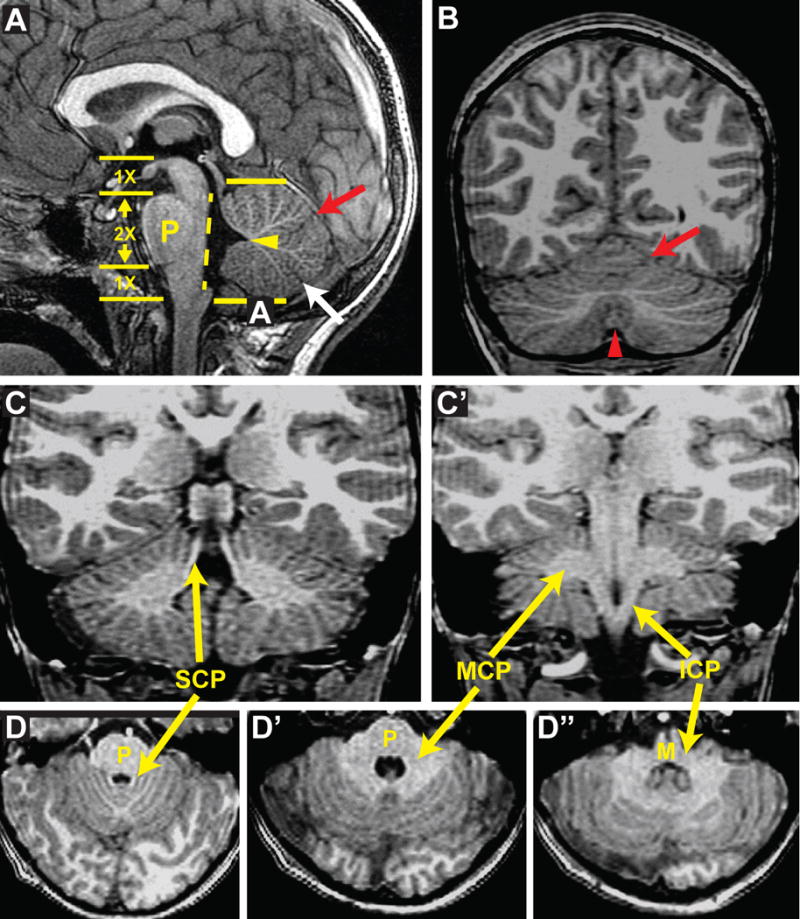
(A) Sagittal image demonstrates patent aqueduct, normal tectum, normal sized posterior fossa, vermis and pons (P), straight brainstem with flat dorsal surface (dotted line), appropriately positioned fastigium (arrowhead) just below the mid-point of the ventral pons, primary (red arrow) and pre-pyramidal (white arrow) fissures dividing vermis into 3 segments. Yellow arrows and numbers give approximate proportions of brain stem. (B) Coronal image demonstrating normal-sized cerebellar hemispheres and vermis (red arrowhead) with folia radiating toward the deep cerebellar nuclei; Note primary fissure (red arrow). (C and C′) Coronal images demonstrating superior (SCP), middle (MCP) and inferior (ICP) cerebellar peduncles. (D-D″) Axial images demonstrating SCP, MCP and ICP; note the normal-sized cerebellar hemispheres and vermis.
Specific Malformations
Predominantly Cerebellar Malformations
Cerebellar hypoplasia has many causes including chromosomal disorders, specific genetic syndromes, and prenatal disruptions. The vermis and both hemispheres may be equally small or may be hypoplastic in any combination. Pontine hypoplasia and midbrain malformation are typically, but not invariably, associated.
Dandy-Walker Malformation
Perhaps the most common MBHB malformation is Dandy-Walker malformation (DWM MIM 220200). Although variably defined in the literature since the term was first coined by Benda in 1954,22 this heterogeneous disorder is defined by a hypoplastic (and commonly counterclockwise rotated) vermis, an enlarged fourth ventricle, and an enlarged posterior fossa with an elevated torcular (Fig 3B). Typically, the cerebellar hemispheres are less affected than the vermis, and the brainstem is normal to moderately hypoplastic. Subsets of patients have other brain findings such as agenesis of the corpus callosum and hydrocephalus. DWM can be isolated, part of a defined syndrome or associated with extra-CNS malformations that do not fit with a known condition (summarized in Poretti et al. 2012).23 Detailed evaluation of brain imaging is important to 1) distinguish DWM from more benign entities such as persistent Blake pouch cyst, in which a normal vermis is rotated counterclockwise by the cyst, and 2) identify mass-effect that might result in obstructive hydrocephalus. Midsagittal thin slices (0.5 mm) using high-resolution steady state MRI sequences (CISS, FIESTA) best demonstrate the level of obstructed CSF flow, if present; axial or coronal images can demonstrate the abnormal membranes of arachnoid cysts (although most cysts cause no clinically significant abnormality).
Figure 3. Sagittal views of MBHB malformations.
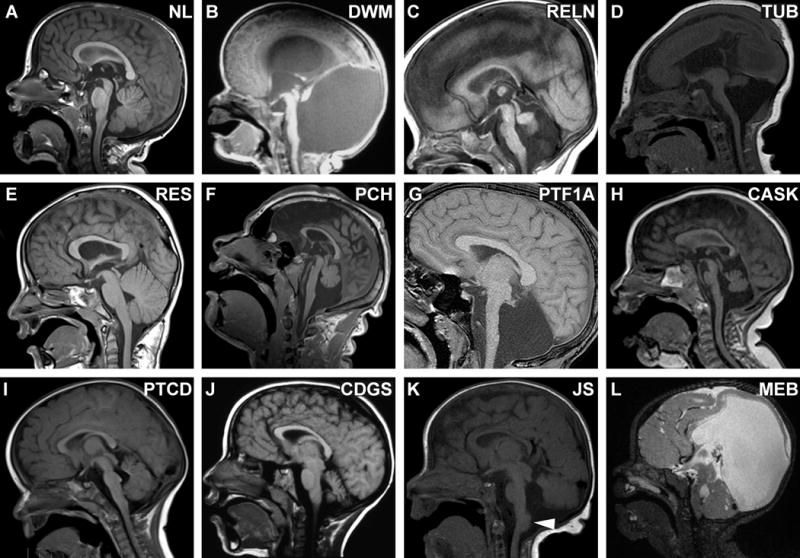
(A) Normal sagittal image for comparison. (B) Dandy-Walker malformation (unknown cause) with hypoplastic, rotated vermis and marked enlargement of 4th ventricle and posterior fossa. (C) Cerebellar hypoplasia in a patient with biallelic RELN mutations, demonstrating hypoplastic brainstem and characteristic absent folia of the vermis; note the normal tectum. (D) Tubulinopathy (TUBA1A mutation) with brainstem hypoplasia, vermis hypoplasia, lissencephaly and microcephaly; note the large, dysplastic tectum. (E) Rhombencephalosynapsis (unknown cause) with hemisphere-like vermis morphology (the three segments described in Fig 2A are not seen); note the normal size and configuration of the pons and vermis. (F) Pontocerebellar hypoplasia (homozygous TSEN54 mutation) with hypoplastic brainstem and vermis (which is less affected than hemispheres), note the normal tectum. (G) Pontocerebellar hypoplasia in a patient with congenital diabetes; note the extremely small vermis and flat pons with preserved tectum. (H) CASK-related PCH; note that the pons is not severely affected in this patient. (I) Pontine tegmental cap dysplasia (unknown cause) with ventral pons hypoplasia and an ectopic “cap” of white matter on the dorsal pons (arrowhead); the vermis is mildly hypoplastic with prominent folia. (J) Congenital disorders of glycosylation Type 1a due to biallelic PMM2 mutations. (K) TCTN2-related Joubert syndrome with vermis hypoplasia (obscured by hemispheres in this image), horizontal superior cerebellar peduncles, large dysplastic tectum and heterotopia at the dorsal cervicomedullary junction (arrowhead). With kind permission from Springer Science and Business Media: Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol 2012; 123: 695–709. (L) Muscle-eye-brain disease (MEB) due to POMTGN1 mutations; note the markedly hypoplastic and dysplastic brainstem, cerebellar cysts, abnormal tectum and hydrocephalus.
Developmental outcome spans a wide range, with additional malformations and/or cerebellar vermis dysplasia likely to be associated with more severe impairment.24 A substantial subset of patients requires shunting for hydrocephalus, and a smaller subset has seizures. Empirically, the recurrence risk is low.25 Although few genetic causes have been identified, one cause, loss of FOXC1 function, has led to the hypothesis that cerebellar and posterior skull development are linked through inductive interactions between the mesenchyme and rhombic lip.26
Lissencephaly and cerebellar hypoplasia
Cerebellar hypoplasia may be found in patients with cerebral malformations in the lissencephaly spectrum, caused by defects in the RELN pathway or microtubule function. The RELN pathway is incompletely understood and it is likely that some, perhaps many, patients with cerebellar hypoplasia have defects in this pathway. Reported patients with RELN mutations have pachygyria and minimal foliation of a very hypoplastic cerebellum, vermis worse than hemispheres (Fig 3C and 4A).27-30 Profound developmental disability, microcephaly, sloping forehead, seizures, and congenital lymphedema have also been reported in these patients. In contrast, patients with mutations in VLDLR have a simplified, mildly pachygyric cerebral cortex and milder cerebellar hypoplasia with some degree of cerebellar foliation.31, 32 RELN signaling, via the VLDLR and APOER2 receptors and DAB1 effector, is important for dispersion of Purkinje cells from their clumped configuration during early cerebellar formation.12
Figure 4. Other imaging features of MBHB malformations.
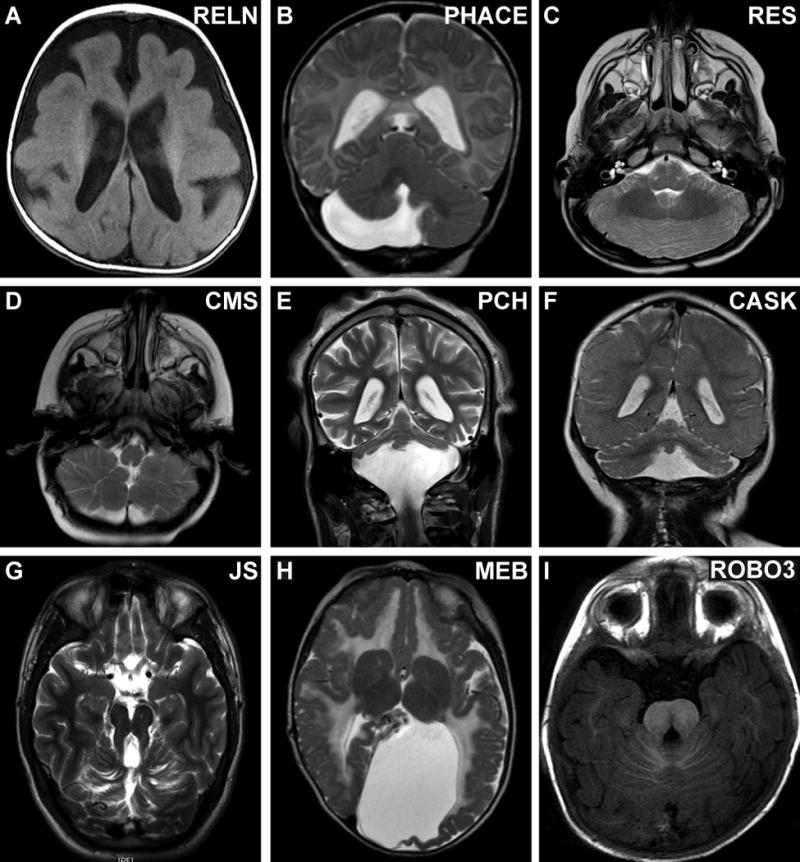
(A) Incomplete lissencephaly (pachygyria) in a patient with biallelic RELN mutations. (B) Unilateral cerebellar hemisphere hypoplasia with milder vermis hypoplasia in a patient with PHACE syndrome (unknown cause). (C) Deficient vermis and fused cerebellar white matter tracts in a patient with rhombencephalosynapsis (unknown cause). (D) Inferior cerebellar hemisphere dysplasia in a patient with Chudley-McCullough syndrome (CMS) due to a homozygous GPSM2 mutation. (E) Cerebellar hypoplasia with relatively preserved vermis in a patient with PCH due to biallelic TSEN54 mutations. (F) Cerebellar hypoplasia with proportionally affected hemispheres and vermis in a patient with PCH due to a CASK mutation. (G) Molar tooth sign in a patient with Joubert syndrome due to a homozygous TMEM216 mutation. (H) Cobblestone cortical malformation and abnormal white matter signal in a patient with Muscle-eye-brain disease due to POMTGN1 mutations. (I) Dysplastic pons in a patient with HGPPS due to biallelic ROBO3 mutations; compare to Figure 2D.
The “tubulinopathies” are caused by mutations in genes involved in microtubule formation and function. The spectrum of phenotypes associated with mutations in TUBA1A, TUBA8, TUBB2B, TUBB3 and TUBB5 ranges from isolated congenital fibrosis of the extraocular muscles to severe intellectual disability, quadriplegic cerebral palsy, seizures, cranial neuropathies and hydrocephalus.33-38 Imaging features include cortical dysgenesis (lissencephaly or polymicrogyria), malformation of cranial nerves and basal ganglia, often with cerebellar and pontine hypoplasia, as well as defects in the corpus callosum, anterior commissure and internal capsule (Fig 3D). Dysmorphic features are infrequently reported,39 and remarkably, other organ systems are not affected. While most occurrences are sporadic and due to de novo mutations, recurrences have been reported due to germline mosaicism and autosomal recessive inheritance.36
Other cerebellar hypoplasias
While the cause of cerebellar hypoplasia remains unknown in many patients, a number of other rare cerebellar hypoplasia disorders deserve mention. Cerebellar hypoplasia, ventriculomegaly, intellectual disability, seizures and mildly dysmorphic facial features are characteristic in males and occasional females with mutations in the X-linked OPHN1 gene.40-42 OPHN1 is a Rho GTPase-activating protein involved in clathrin-mediated endocytosis and dendritic spine formation; however, its role in cerebellar development is unknown. PHACE syndrome is characterized by Posterior fossa brain malformations, Hemangioma of the head and/or neck, Arterial lesions of the head and/or neck, Cardiac defects including aortic coarctation, and Eye abnormalities.43, 44 Unilateral cerebellar hypoplasia, ipsilateral to the hemangioma, and cerebrovascular lesions are the most common brain imaging findings with occasional cerebellar vermis and supratentorial malformations (corpus callosum dysgenesis, polymicrogyria, and heterotopia).45, 46 Given the lack of recurrences and typically unilateral involvement, PHACE syndrome is likely to be caused by de novo somatic mutations, similar to Proteus, Megalencephaly-Capillary Malformation-Polymicrogyria (MCAP) and Megalencephaly-Polydactyly-Polymicrogyria-Hydrocephalus (MPPH) syndromes.47-50
Rhombencephalosynapsis
Rhombencephalosynapsis (RES) consists of partial or complete absence of the cerebellar vermis with continuity of the hemispheres across the midline (Fig 4C), thought to be due to aberrant dorsal-ventral patterning.51-53 The severity of RES on imaging correlates with clinical outcome.54 When severe, the fused cerebellar nuclei arch in a horseshoe shape across the midline, with a narrow fourth ventricle; the primary and prepyramidal fissures of the vermis cannot be identified (Fig 3E). Directionally encoded FA maps (from DTI data) showed absent transverse fibers of the vermis; instead, a bundle of fibers runs rostrocaudally in the midline of the fused cerebellar hemispheres and the superior cerebellar peduncles fail to decussate.55 RES is often associated with midbrain abnormalities (aqueductal stenosis and midline fusion of the colliculi), and supratentorial abnormalities such as absent septum pellucidum and corpus callosum dysmorphisms.
In addition the typical clinical features of MBHB disorders, patients with RES display persistent headshaking behavior, high activity level and impulsiveness. Affected patients can be divided into at least three recognizable categories: 1) Gómez-López-Hernández syndrome (MIM 601853) combining RES with scalp alopecia and trigeminal anesthesia, 2) RES plus features of VACTERL association (Vertebral defects, Anal atresia, Cardiac defects, Tracheo-Esophageal fistula, Renal defects and Limb defects), and 3) RES with atypical holoprosencephaly, most severely affecting the occipital lobes.54, 56 Developmental outcomes vary widely; patients with isolated RES can function independently in adulthood, while patients with VACTERL features or HPE are more likely to manifest significant impairments. No genetic defects have been linked to RES and no animal models recapitulate the phenotype, so the mechanism remains unknown.
Cerebellar hyperplasia
Cerebellar hyperplasia (macrocerebellum) is a rare imaging finding that can be seen in isolation, or in a variety of disorders including chromosomal abnormalities, Alexander disease and fucosidosis, as well as Sotos, Williams, Costello, MCAP/MPPH syndromes (reviewed in Poretti et al., 2012).57 While the developmental mechanisms underlying cerebellar hyperplasia in most of these disorders remains unknown, substantial progress has been made in the understanding of brain overgrowth in MCAP/MPPH. MCAP/MPPH are typically diagnosed due to supratentorial overgrowth and polymicrogyria. 58 Although cerebellar size is normal at birth, many patients develop macrocerebellum with normal posterior fossa size, resulting in cerebellar ectopia/Chiari I malformation and associated symptoms (posterior headache, dysphagia, stridor) and hydrocephalus. Other features include seizures, capillary malformations, macrocephaly, and polydactyly. Most patients have pathway activating de novo mutations in components of the PI3K-AKT-mTOR pathway that result in increased cell growth.48-50 Mutations affecting this pathway are found in a variety of cancers, opening the possibility of using drugs in development for cancer treatment to try to reduce brain overgrowth and neurological issues in patients with MCAP/MPPH.59, 60
Cerebellar dysplasia
Any part of the cerebellum can be dysplastic, from small focal portions of a single hemisphere to abnormal foliation of the entire cerebellum.61 Very often, such as in JS and cobblestone malformations, hypoplastic cerebella are also dysmorphic. Outside of these well-recognized conditions, little is known about the causes of cerebellar cortical dysgenesis. One exception is Chudley-McCullough syndrome (CMS – MIM 604213), in which patients have striking disorganization of the inferior cerebellar hemisphere folia (Fig 4D). Clinically, these patients present with severe sensorineural hearing loss and mild delays, but are not usually dysmorphic and do not have other malformations.62 Brain imaging reveals additional abnormalities including frontal polymicrogyria with subcortical heterotopia, corpus callosum hypogenesis and arachnoid cysts. All patients to date have biallelic truncating mutations in the GPSM2 gene that encodes a GTPase regulator required for correct orientation of stem cell divisions in multiple tissues.63 It is likely that aberrant cell division underlies the cerebellar dysplasia, but the mechanism remains unknown.
Cerebellar and Brainstem malformations
Pontocerebellar hypoplasia (PCH)
PCH occurs in a number of disorders that can be distinguished, in part, by imaging findings and associated features (Fig 3F-J). At least 6 clinical types of PCH were described before genetic causes were identified. PCH type 1(MIM 607596 and 614678), associated with mutations in EXOSC364 and VRK165, is characterized by moderate PCH on imaging in combination with spinal muscular atrophy, resulting in substantial global weakness and decreased or absent reflexes. PCH types 2 (MIM 277470, 612389 and 612390), 4 (MIM 225753) and 5 are now known to be caused predominantly by mutations in genes that encode tRNA splicing endonucleases (TSEN54, TSEN34 and TSEN2).66-68 Patients with PCH type 2 represent the less severe end of the spectrum with early hyperreflexia, developmental delay, and feeding problems, eventually developing spasticity and involuntary movements in childhood, while patients with PCH type 4 represent the severe end of the spectrum characterized by polyhydramnios, severe hyperreflexia, contractures and early death due to central respiratory failure. Microcephaly is present at birth in patients with PCH4, while microcephaly develops over time in PCH2. Seizures are common in both groups. A typical feature of TSEN-related PCH is more severe involvement of the cerebellar hemispheres compared to the vermis (Fig 4E).69 Described in only one report, PCH type 3 (MIM 608027) is associated with optic atrophy and the genetic cause is unknown.70 PCH type 6 (MIM 611523) is associated with elevated CSF lactate and caused by mutations in the RARS2 gene.71
Other disorders can mimic the TSEN-related PCHs. Patients with PTF1A-related cerebellar hypoplasia present with diabetes in infancy due to agenesis of the pancreas Fig 3G).72 While quite rare, this disorder highlights the essential role for PTF1A in the development of ventricular zone-derived precursors. The severe end of the spectrum of CASK-related disease also presents with progressive microcephaly and PCH that proportionately affects the vermis and hemispheres (Fig 3H).73, 74 Patients may also display dysmorphic features of the nose, eyes, and ears. The CASK gene is on the X chromosome, so CASK-related PCH is more common in females, presumably due to lethality in males. Most recently, recessive loss of function mutations in CHMP1A have been shown to cause severe PCH with proportionate involvement of the cerebellar vermis and hemispheres and preserved foliation pattern.75 Described patients had moderate to severe developmental delay, acquired microcephaly, increased extremity tone and contractures. CHMP1A appears facilitate the repression of CDKN2A expression by the transcriptional repressor BMI1, resulting in decreased proliferation and stem cell renewal.
Pontine Tegmental Cap Dysplasia (PTCD, MIM 614688) is characterized by multiple cranial neuropathies, particularly hearing loss, trigeminal anesthesia, facial paralysis and swallowing dysfunction. 76, 77 PTCD can be distinguished from other PCHs by the characteristic “cap” of tissue on the dorsal pons and severe hypoplasia of the inferior and middle cerebellar peduncles (Fig 3I). Congenital heart, kidney, vertebral and rib defects are present in subsets of patients. Developmental disability is typically severe, and the degree of impairment may correlate with the severity of brainstem dysplasia, with mildly affected patients having a rounded bump (“cap”) and those more severely affected having a more angular brainstem kink (“beak”).77-79 Neither familial recurrence nor genetic causes have been reported.
Hypoplasia of the pons and cerebellum with progressive volume loss is characteristic of Congenital Disorders of Glycosylation Type 1a (CDGS, MIM 212065, Fig 3J).80 Patients present with hypotonia and developmental delay, but can also display a striking variety of other features including abnormal fat distribution, coagulopathy, retinal degeneration, peripheral neuropathy, stroke-like episodes, and seizures. The range of developmental outcome is broad. CDG Type1a is autosomal recessive, caused by biallelic mutations in the PMM2 gene, required for N-glycosylation of proteins.81 The identity and role of N-glycosylated proteins during hindbrain development remain under investigation.
Many causes of PCH remain undefined, although atypical presentations of disorders such as VLDLR-related disequilibrium syndrome82 or pre- and post-natal insults (most commonly complications of extreme prematurity) may explain additional subsets of patients.83
Joubert syndrome/Molar Tooth Malformation
In patients with neonatal hypotonia, abnormal eye movements and alternating apnea and tachypnea, JS should be considered. Polydactyly is present in a minority of patients and subsets of patients also develop retinal dystrophy, nephronophthisis and liver fibrosis. Brain MRI is usually diagnostic, revealing cerebellar vermis hypoplasia/dysplasia, long, thick, elevated superior cerebellar peduncles (SCPs), and a thin MBHB junction with a deep interpeduncular fossa resulting in the “molar tooth sign” on axial MRI (Fig 3K and 4G). Other brain malformations can be present, including polymicrogyria, brainstem and cortical heterotopia, agenesis of the corpus callosum and/or cephalocele.84 DTI can be used to generate color FA maps and tractography, showing laterally displaced and dysmorphic deep cerebellar nuclei, hypoplastic medial lemnisci, and absent transverse fibers in the central vermis and deficient SCP decussation.85
Diagnosing JS is important, since it is recessive and carries a 25% recurrence risk. In addition, close monitoring for the progressive retinal, kidney and liver disease is required to reduce complications. Mutations in 20 genes can cause JS and related disorders.86 The gene products function in the primary cilium, the cellular antenna that mediates a variety of signaling processes, making these disorders part of an expanding group of disorders called “ciliopathies.”87, 88 The brain malformations may result from defects in midline fusion of the developing vermis89 or defects in sonic hedgehog-mediated granule cell proliferation.90, 91
Cobblestone malformations
Cobblestone malformations comprise a broad spectrum of clinically and genetically overlapping disorders resulting from defects in the pial limiting membrane and the attachment of radial glial fibers thereto.92-98 The Walker-Warburg phenotype represents the severe end of the spectrum with profoundly small and dysmorphic cerebellar hemispheres often with cysts, absent vermis and very small brainstem, as well as other major brain malformations: cerebral cobblestone cortex, abnormal white matter, anomalous corpus callosum, enlarged dysplastic tectum, and hypoplastic/dysplastic brainstem (Fig 3L and 4H). The cerebellar cortical and subcortical cysts represent small areas of pia/subarachnoid space herniating inward through gaps in the pial limiting membrane. Patients frequently have hydrocephalus, eye abnormalities (microphthalmia, optic nerve hypoplasia, chorioretinal coloboma, cataract, glaucoma and/or high myopia), seizures, hypotonia and/or muscular dystrophy (creatine kinase levels 2-15X normal). The muscle-eye-brain phenotype represents more moderately affected patients with milder cerebellar and brainstem hypoplasia, and Fukuyama muscular dystrophy represents the mild end of the spectrum that still includes brain malformation. Recessive mutations in multiple genes (POMT1 MIM 607423, POMT2 MIM 607439, POMGNT1 MIM 606822, FKTN MIM 607440, FKRP MIM 606596, LARGE MIM 603590, ISPD MIM 614631, and GTDC2 MIM 614830) can cause overlapping phenotypes across the entire spectrum of disease, but a substantial proportion of patients remain unexplained.
Predominantly Brainstem Malformations
Congenital Cranial Dysinnervation Disorders
The term Congenital Cranial Dysinnervation Disorders was coined in 2002 to refer to abnormalities of cranial nerve development that result in abnormal movement of the face and eyes,99 most of which do not have obvious brainstem abnormalities on clinical imaging studies. The exception is Horizontal Gaze Palsy with Progressive Scoliosis (HGPPS MIM 617313), an autosomal recessive disorder associated with a “butterfly shaped pons and medulla” (Fig 4I).100 Patients typically present with progressive scoliosis during early childhood and congenital eye movement abnormalities and defects in midline axon crossing demonstrated by somatosensory and motor evoked potentials. HGPPS is due to biallelic mutations in ROBO3 which encodes a receptor required for axon guidance.101
Although not apparent on clinical MRI, Athabaskan brainstem dysgenesis102 and Bosley-Salih-Alorainy syndromes (MIM 601536)103 represent brainstem patterning disorders due to HOXA1 loss of function in individuals of Athabaskan and Saudi descent respectively.103 In mouse models, HOXA1 is required for correct specification of rhombomeres 4 and 5 in the developing hindbrain,104, 105 as well as inner ear16, 17 and heart106 development, so it is not surprising that patients have cranial nerve palsies, sensorineural hearing loss, abnormal intracranial blood vessels and cardiac outflow tract defects. Similarly, a number of other cranial dysinnervation disorders have facilitated the identification of genes required for cranial nerve formation/survival and axon guidance such as CHN1-related Duane retraction syndrome (MIM 604356)107, SALL4-related Duane radial ray syndrome (MIM 607323),108 and Congenital Fibrosis of the Extraocular Muscles Types 1-3 (MIM 135700, 602078, 600638, 609384) due to mutations in KIF21A, PHOX2A, TUBB3 and TUBB2B.35, 109-111 Finally, the term Moebius syndrome (MIM 157900) is used to describe patients with cranial nerve dysfunction, most commonly facial palsy with impaired ocular abduction; however, the term is used quite variably, limiting its utility for guiding work-up, providing prognostic information, predicting recurrence risk or directing treatment and identifying the underlying cause(s).
Evaluation of patients with cranial nerve disorders should include 3-dimensional, heavily T2-weighted, steady state (CISS, FIESTA) MRI sequences that are able to visualize cranial nerves as they course through the cisterns at the skull base. In addition, high resolution diffusion tensor imaging of the brainstem holds some promise for further delineating axonal pathfinding disorders of the brainstem in the absence of obvious structural defects.
Predominantly Midbrain Malformations
Relatively few disorders that predominantly affect the midbrain have been described. Zaki at al. (2012) recently reported similar midbrain dysplasia in several consanguineous Egyptian families.112 On imaging, the patients had rostral-caudal shortening and dorsal-ventral lengthening of the midbrain, with a deep interpeduncular cistern. In two patients, the corticospinal tracks were not detectable at the level of the pons by diffusion tensor imaging. Variable features included cortical calcifications, agenesis of the corpus callosum, ventriculomegaly, brainstem dysplasia and cerebellar vermis hypoplasia. Clinically, the patients had progressive microcephaly, spasticity, intellectual disability and seizures. Inheritance appeared to be autosomal recessive, but the genetic cause(s) remain unknown. Although the midbrain is abnormal in a number of other disorders, further delineation of midbrain malformations is required.3
Conclusions
Despite developments in genetics and neuroimaging that have facilitated the diagnosis of malformations of the MBHB in fetuses and children, these disorders remain underdiagnosed and poorly understood because of historical difficulties in assessing these areas of the brain. It is hoped that the information contained in this review in regard to the range of these disorders, the methodological clinical and radiologic approaches, the characteristic clinical and imaging findings that aid in their initial diagnoses, and the genetic studies that confirm the diagnoses will help neurologists and radiologists in this regard. Increased cognizance of these disorders among physicians, along with careful analyses of clinical and imaging studies, will raise the general awareness of them in the medical community.
Defining MBHB malformations and identifying their causes provides direct benefits to patients and their families: 1) diagnostic, carrier and prenatal testing; 2) more accurate prognostic and recurrence risk information; 3) avoidance of additional, unnecessary diagnostic testing; 4) early diagnosis of associated complications through medical monitoring; 5) reduction in stress caused by diagnostic uncertainty; 6) relief of parental guilt and anxiety for causing their child's disability. Defining the molecular pathways underlying normal and abnormal human MBHB development (Fig 1) is the first step toward developing specific therapies, although it will likely be many years before they are available to our patients. It is uncertain whether early developmental brain anomalies can be corrected, but many gene products required for brain development are later required for ongoing brain function, so intervention targeting the affected pathways may be possible. For example, loss of function in the genes associated with JS disrupts protein localization to the primary cilium113 where several neurotransmitter receptors are normally localized,114, 115 likely contributing to the intellectual disability, behavior problems and mental health issues experienced by these patients. In the future, pharmacological treatments may be able to address ongoing brain dysfunction caused by ciliary receptor mislocalization.
Beyond direct benefits to patients with MBHB malformations, understanding these disorders may inform other areas of human health and disease. Developing treatments for neurological dysfunction caused by trauma, infection, and neurodegenerative conditions depends on a solid understanding of MBHB development and function in humans. Similarly, successful treatment of oculomotor, vestibular, and respiratory control dysfunction will benefit from basic knowledge about how these neurological systems develop in humans. In addition to functions typically assigned to the MBHB, mounting evidence indicates that the MBHB plays a role in autism spectrum, mental health and cognitive disorders.110-112 Although recent progress in understanding normal and abnormal human MBHB development has been remarkable, further advances in brain imaging, genetics and animal modeling foretell continued progress in the future.
Table 3. Typical clinical, imaging and genetic characteristics of specific MBHB malformations primarily involving the brainstem*.
| Disorder | Clinical features | Imaging features | Causes/testing | Inheritance |
|---|---|---|---|---|
| HGPPS | Scoliosis, horizontal gaze palsy Normal NDV outcome |
Brain: Dorsal midline cleft leads to “butterfly” appearance of pons, medulla Spine: early and progressive scoliosis |
ROBO3 | AR |
| Duane retraction syndrome, Duane radial ray syndrome |
CHN1 mutations Limited eye adduction with globe retraction and palpebral fissure narrowing SALL4 mutations Radial ray abnormalities (thenar hypoplasia, thumb hypoplasia/aplasia, triphalangeal thumb, shortened forearm, preaxial polydactyly), kidney malformations, coloboma, cardiac malformations (rare), hearing loss Normal NDV outcome |
Hypoplastic/absent abducens nerve, likely other oculomotor nerve defects | CHN1, SALL4 | AD (SALL4, ∼50% de novo) |
| Athabaskan brainstem dysgenesis and Bosley-Salih-Alorainy syndromes | Cranial nerve palsies, sensorineural hearing loss, abnormal intracranial blood vessels and cardiac outflow tract defects | No published brain imaging abnormalities | HOXA1A | AR |
| Moebius syndrome | Facial palsy with impaired ocular abduction (variable involvement of other cranial nerves) Variable somatic malformations Variable NDV outcome |
Heterogeneous: hypoplastic brainstem with calcifications, hypoplastic or absent cranial nerves | Misoprostol exposure in utero, TUBB3, HOXA1A and HOXB1 mutations in patients with Moebius features | Sporadic (rarely AD and AR) |
| Diencephalic–mesencephalic junction dysplasia | Progressive microcephaly, spasticity, intellectual disability, seizures | rostral-caudal shortening and dorsal-ventral lengthening of the midbrain, deep interpeduncular cistern Variable features: cortical calcifications, agenesis of the corpus callosum, ventriculomegaly, brainstem dysplasia and cerebellar vermis hypoplasia |
Unknown | AR |
Note that information published for many of the disorders is quite limited, so the full spectrum of characteristics is likely broader than depicted in this table.
Acknowledgments
Funding: We acknowledge funding support for brain malformation research from NIH to D.D. (R01NS064077), K.J.M. (R01NS072441), and A.J.B. (R01NS35129 and R01NS058721). No funding source played a role in writing this manuscript.
Abbreviations
- ACC
Agenesis of the Corpus Callosum
- AD
Autosomal Dominant
- AR
Autosomal Recessive
- CDGS
Congenital Disorders of Glycosylation
- CMS
Chudley-McCullough Syndrome
- DTI
Diffusion Tensor Imaging
- DWM
Dandy-Walker Malformation
- EGL
External Granule cell Layer
- IGL
Internal Granule cell Layer
- GLH
Gómez-López-Hernández syndrome
- HGPPS
Horizontal Gaze Palsy and Progressive Scoliosis
- ICP
Inferior Cerebellar Peduncle
- MCAP
Megalencephaly-Capillary Malformation-Polymicrogyria
- MCP
Middle Cerebellar Peduncle
- MPPH
Megalencephaly-Polydactyly-Polymicrogyria-Hydrocephalus
- NDV
NeuroDeVeopmental
- OCCS
Oculocerebrocutaneous syndrome
- PC
Purkinje Cell
- PCH
PontoCerebellar Hypoplasia
- PHACE
Posterior fossa brain malformations, Hemangioma, Arterial lesions, Cardiac defects, and Eye abnormalities
- PMG
PolyMicroGyria
- PTCD
Pontine Tegmental Cap Dysplasia
- RES
RhombEncephaloSynapsis
- SCP
Superior Cerebellar Peduncle
- VACTERL
Vertebral defects, Anal atresia, Cardiac defects, Tracheo-Esophageal fistula, Renal defects and Limb defects
Footnotes
Conflict of interest statement: None of the authors has a conflict of interest.
Author contributions: D.D. and A.J.B. reviewed the literature and drafted the initial manuscript. D.D. and K.J.M. drafted the initial figures. All three authors edited the final text and figures.
Search Strategy: We searched all PubMed records through December 2012 with keywords for MBHB malformation anatomic features (cerebellar hypoplasia, cerebellar dysplasia, pontocerebellar hypoplasia, brainstem dysgenesis, molar tooth sign, rhombencephalosynapsis, pontine tegmental cap dysplasia), syndromes (PHACE, PHACES, Gómez-López-Hernández, Chudley-McCullough, Joubert, Walker-Warburg, Duane retraction, Duane radial ray, Athabaskan brainstem dysgenesis, Bosley-Salih-Alorainy, Moebius), disorders (Dandy-Walker, congenital disorders of glycosylation, dystroglycanopathy, Muscle-eye-brain disease, Fukuyama muscular dystrophy, Horizontal Gaze Palsy and Progressive Scoliosis) and selected genes (RELN, VLDLR, TSEN*, PTF1A, CASK, CHMP1A, tubulins, OPHN1). Due to the breadth of this review, we did not perform comprehensive literature searches, and selected articles that made substantial contributions to the clinical description (including natural history), identified genetic causes, and/or described treatment. We apologize for not being able to cite all publications relating to MBHB malformations due to space constraints.
Additional resources: GeneReviews http://www.ncbi.nlm.nih.gov/sites/GeneTests/review?db=GeneTests
Online Mendelian Inheritance in Man www.omim.org
References
- 1.Dandy WE, Blackfan KD. Internal hydrocephalus: an experimental, clinical, and pathological study. Am J Dis Child. 1914;8:406–82. [Google Scholar]
- 2.Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet neurology. 2007;6(3):245–57. doi: 10.1016/S1474-4422(07)70054-6. [DOI] [PubMed] [Google Scholar]
- 3.Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. The New England journal of medicine. 2012;366(7):636–46. doi: 10.1056/NEJMra1006610. [DOI] [PubMed] [Google Scholar]
- 4.Barkovich AJ, Millen KJ, Dobyns WB. A developmental classification of malformations of the brainstem. Ann Neurol. 2007;62(6):625–39. doi: 10.1002/ana.21239. [DOI] [PubMed] [Google Scholar]
- 5.Barkovich AJ, Millen KJ, Dobyns WB. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132(Pt 12):3199–230. doi: 10.1093/brain/awp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pascual M, Abasolo I, Mingorance-Le Meur A, Martinez A, Del Rio JA, Wright CV, et al. Cerebellar GABAergic progenitors adopt an external granule cell-like phenotype in the absence of Ptf1a transcription factor expression. Proc Natl Acad Sci U S A. 2007;104(12):5193–8. doi: 10.1073/pnas.0605699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390(6656):169–72. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- 8.Rakic P, Sidman RL. Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. The Journal of comparative neurology. 1970;139(4):473–500. doi: 10.1002/cne.901390407. [DOI] [PubMed] [Google Scholar]
- 9.Miyata T, Ono Y, Okamoto M, Masaoka M, Sakakibara A, Kawaguchi A, et al. Migration, early axonogenesis, and Reelin-dependent layer-forming behavior of early/posterior-born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev. 2010;5:23. doi: 10.1186/1749-8104-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374(6524):719–23. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 11.D'Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17(1):23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyata T, Nakajima K, Mikoshiba K, Ogawa M. Regulation of Purkinje cell alignment by reelin as revealed with CR-50 antibody. J Neurosci. 1997;17(10):3599–609. doi: 10.1523/JNEUROSCI.17-10-03599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bloch-Gallego E, Causeret F, Ezan F, Backer S, Hidalgo-Sanchez M. Development of precerebellar nuclei: instructive factors and intracellular mediators in neuronal migration, survival and axon pathfinding. Brain research Brain research reviews. 2005;49(2):253–66. doi: 10.1016/j.brainresrev.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch JF, Pierre-Kahn A, Renier D, Sainte-Rose C, Hoppe-Hirsch E. The Dandy-Walker malformation. A review of 40 cases. J Neurosurg. 1984;61(3):515–22. doi: 10.3171/jns.1984.61.3.0515. [DOI] [PubMed] [Google Scholar]
- 15.Parisi MA, Dobyns WB. Human malformations of the midbrain and hindbrain: review and proposed classification scheme. Mol Genet Metab. 2003;80(1-2):36–53. doi: 10.1016/j.ymgme.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 16.Long A, Moran P, Robson S. Outcome of fetal cerebral posterior fossa anomalies. Prenatal diagnosis. 2006;26(8):707–10. doi: 10.1002/pd.1485. [DOI] [PubMed] [Google Scholar]
- 17.Saleem SN, Zaki MS. Role of MR imaging in prenatal diagnosis of pregnancies at risk for Joubert syndrome and related cerebellar disorders. Ajnr. 2010;31(3):424–9. doi: 10.3174/ajnr.A1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patek KJ, Kline-Fath BM, Hopkin RJ, Pilipenko VV, Crombleholme TM, Spaeth CG. Posterior fossa anomalies diagnosed with fetal MRI: associated anomalies and neurodevelopmental outcomes. Prenatal diagnosis. 2012;32(1):75–82. doi: 10.1002/pd.2911. [DOI] [PubMed] [Google Scholar]
- 19.Scott JA, Hamzelou KS, Rajagopalan V, Habas PA, Kim K, Barkovich AJ, et al. 3D morphometric analysis of human fetal cerebellar development. Cerebellum (London, England) 2012;11(3):761–70. doi: 10.1007/s12311-011-0338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garel C, Fallet-Bianco C, Guibaud L. The fetal cerebellum: development and common malformations. Journal of child neurology. 2011;26(12):1483–92. doi: 10.1177/0883073811420148. [DOI] [PubMed] [Google Scholar]
- 21.Bolduc ME, Du Plessis AJ, Sullivan N, Khwaja OS, Zhang X, Barnes K, et al. Spectrum of neurodevelopmental disabilities in children with cerebellar malformations. Developmental medicine and child neurology. 2011;53(5):409–16. doi: 10.1111/j.1469-8749.2011.03929.x. [DOI] [PubMed] [Google Scholar]
- 22.Benda CE. The Dandy-Walker syndrome or the so-called atresia of the foramen Magendie. Journal of neuropathology and experimental neurology. 1954;13(1):14–29. doi: 10.1093/jnen/13.1.14. [DOI] [PubMed] [Google Scholar]
- 23.Poretti A, Millen KJ, Boltshauser E. Dandy-Walker Malformation. In: Boltshauser E, Schmahmann JD, editors. Cerebellar Disorders in Children. London: Mac Keith Press; 2012. pp. 140–8. [Google Scholar]
- 24.Boddaert N, Klein O, Ferguson N, Sonigo P, Parisot D, Hertz-Pannier L, et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiol. 2003;45:320–4. doi: 10.1007/s00234-003-0980-6. [DOI] [PubMed] [Google Scholar]
- 25.Murray JC, Johnson JA, Bird TD. Dandy-Walker malformation: etiologic heterogeneity and empiric recurrence risks. Clin Genet. 1985;28:272–83. doi: 10.1111/j.1399-0004.1985.tb00401.x. [DOI] [PubMed] [Google Scholar]
- 26.Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009;41(9):1037–42. doi: 10.1038/ng.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26(1):93–6. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- 28.Zaki M, Shehab M, El-Aleem AA, Abdel-Salam G, Koeller HB, Ilkin Y, et al. Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am J Med Genet A. 2007;143A(9):939–44. doi: 10.1002/ajmg.a.31667. [DOI] [PubMed] [Google Scholar]
- 29.Jissendi-Tchofo P, Kara S, Barkovich AJ. Midbrain-hindbrain involvement in lissencephalies. Neurology. 2009;72(5):410–8. doi: 10.1212/01.wnl.0000333256.74903.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hourihane JO, Bennett CP, Chaudhuri R, Robb SA, Martin ND. A sibship with a neuronal migration defect, cerebellar hypoplasia and congenital lymphedema. Neuropediatrics. 1993;24(1):43–6. doi: 10.1055/s-2008-1071511. [DOI] [PubMed] [Google Scholar]
- 31.Boycott KM, Parboosingh JS. VLDLR-Associated Cerebellar Hypoplasia. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. Seattle (WA): 1993. [Google Scholar]
- 32.Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77(3):477–83. doi: 10.1086/444400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, Cleak J, et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128(1):45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaglin XH, Poirier K, Saillour Y, Buhler E, Tian G, Bahi-Buisson N, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009;41(6):746–52. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140(1):74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poirier K, Saillour Y, Bahi-Buisson N, Jaglin XH, Fallet-Bianco C, Nabbout R, et al. Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet. 2010;19(22):4462–73. doi: 10.1093/hmg/ddq377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdollahi MR, Morrison E, Sirey T, Molnar Z, Hayward BE, Carr IM, et al. Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am J Hum Genet. 2009;85(5):737–44. doi: 10.1016/j.ajhg.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breuss M, Heng JI, Poirier K, Tian G, Jaglin XH, Qu Z, et al. Mutations in the beta-Tubulin Gene TUBB5 Cause Microcephaly with Structural Brain Abnormalities. Cell Rep. 2012;2(6):1554–62. doi: 10.1016/j.celrep.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sohal AP, Montgomery T, Mitra D, Ramesh V. TUBA1A mutation-associated lissencephaly: case report and review of the literature. Pediatric neurology. 2012;46(2):127–31. doi: 10.1016/j.pediatrneurol.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 40.Billuart P, Bienvenu T, Ronce N, des Portes V, Vinet MC, Zemni R, et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392(6679):923–6. doi: 10.1038/31940. [DOI] [PubMed] [Google Scholar]
- 41.Zanni G, Saillour Y, Nagara M, Billuart P, Castelnau L, Moraine C, et al. Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology. 2005;65(9):1364–9. doi: 10.1212/01.wnl.0000182813.94713.ee. [DOI] [PubMed] [Google Scholar]
- 42.Philip N, Chabrol B, Lossi AM, Cardoso C, Guerrini R, Dobyns WB, et al. Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J Med Genet. 2003;40:441–6. doi: 10.1136/jmg.40.6.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frieden IJ, Reese V, Cohen D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Archives of dermatology. 1996;132(3):307–11. doi: 10.1001/archderm.132.3.307. [DOI] [PubMed] [Google Scholar]
- 44.Metry D, Heyer G, Hess C, Garzon M, Haggstrom A, Frommelt P, et al. Consensus Statement on Diagnostic Criteria for PHACE Syndrome. Pediatrics. 2009;124(5):1447–56. doi: 10.1542/peds.2009-0082. [DOI] [PubMed] [Google Scholar]
- 45.Oza VS, Wang E, Berenstein A, Waner M, Lefton D, Wells J, et al. PHACES association: a neuroradiologic review of 17 patients. Ajnr. 2008;29(4):807–13. doi: 10.3174/ajnr.A0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tangtiphaiboontana J, Hess CP, Bayer M, Drolet BA, Nassif LM, Metry DW, et al. Neurodevelopmental Abnormalities in Children With PHACE Syndrome. Journal of child neurology. 2012 doi: 10.1177/0883073812450073. [DOI] [PubMed] [Google Scholar]
- 47.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. The New England journal of medicine. 2011;365(7):611–9. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44(8):934–40. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44(8):941–5. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74(1):41–8. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obersteiner H. Ein Kleinhirn ohne Wurm. Arbeit Neurol Inst Univ Wien. 1916;21:124–36. [Google Scholar]
- 52.Sarnat HB. Molecular genetic classification of central nervous system malformations. Journal of child neurology. 2000;15(10):675–87. doi: 10.1177/088307380001501007. [DOI] [PubMed] [Google Scholar]
- 53.Yachnis AT. Rhombencephalosynapsis with massive hydrocephalus: case report and pathogenetic considerations. Acta Neuropathol. 2002;103(3):301–4. doi: 10.1007/s004010100454. [DOI] [PubMed] [Google Scholar]
- 54.Ishak GE, Dempsey JC, Shaw DW, Tully H, Adam MP, Sanchez-Lara PA, et al. Rhombencephalosynapsis: a hindbrain malformation associated with incomplete separation of midbrain and forebrain, hydrocephalus and a broad spectrum of severity. Brain. 2012;135(Pt 5):1370–86. doi: 10.1093/brain/aws065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Widjaja E, Blaser S, Raybaud C. Diffusion tensor imaging of midline posterior fossa malformations. Pediatric radiology. 2006;36(6):510–7. doi: 10.1007/s00247-006-0146-x. [DOI] [PubMed] [Google Scholar]
- 56.Tully HM, Dempsey JC, Ishak GE, Adam MP, Curry CJ, Sanchez-Lara P, et al. Beyond Gomez-Lopez-Hernandez syndrome: Recurring phenotypic themes in rhombencephalosynapsis. Am J Med Genet A. 2012;158A(10):2393–406. doi: 10.1002/ajmg.a.35561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poretti A, Mall V, Smitka M, Grunt S, Risen S, Toelle SP, et al. Macrocerebellum: Significance and Pathogenic Considerations. Cerebellum (London, England) 2012 doi: 10.1007/s12311-012-0379-1. [DOI] [PubMed] [Google Scholar]
- 58.Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A(2):269–91. doi: 10.1002/ajmg.a.34402. [DOI] [PubMed] [Google Scholar]
- 59.Holmes D. PI3K pathway inhibitors approach junction. Nature reviews Drug discovery. 2011;10(8):563–4. doi: 10.1038/nrd3527. [DOI] [PubMed] [Google Scholar]
- 60.Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. The New England journal of medicine. 2012;367(17):1596–606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Demaerel P. Abnormalities of cerebellar foliation and fissuration: classification, neurogenetics and clinicoradiological correlations. Neuroradiol. 2002;44:639–46. doi: 10.1007/s00234-002-0783-1. [DOI] [PubMed] [Google Scholar]
- 62.Chudley AE, McCullough C, McCullough DW. Bilateral sensorineural deafness and hydrocephalus due to foramen of Monro obstruction in sibs: a newly described autosomal recessive disorder. American journal of medical genetics. 1997;68(3):350–6. doi: 10.1002/(sici)1096-8628(19970131)68:3<350::aid-ajmg19>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 63.Doherty D, Chudley AE, Coghlan G, Ishak GE, Innes AM, Lemire EG, et al. GPSM2 Mutations Cause the Brain Malformations and Hearing Loss in Chudley-McCullough Syndrome. Am J Hum Genet. 2012;90(6):1088–93. doi: 10.1016/j.ajhg.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wan J, Yourshaw M, Mamsa H, Rudnik-Schoneborn S, Menezes MP, Hong JE, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet. 2012;44(6):704–8. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Renbaum P, Kellerman E, Jaron R, Geiger D, Segel R, Lee M, et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet. 2009;85(2):281–9. doi: 10.1016/j.ajhg.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Budde BS, Namavar Y, Barth PG, Poll-The BT, Nurnberg G, Becker C, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008;40(9):1113–8. doi: 10.1038/ng.204. [DOI] [PubMed] [Google Scholar]
- 67.Namavar Y, Barth PG, Baas F. Pontocerebellar Hypoplasia Type 2 and Type 4. GeneReviews 2009. 2009 Sep 22; [cited 2012 November 16]; Available from: http://www.ncbi.nlm.nih.gov/books/NBK9673/
- 68.Namavar Y, Chitayat D, Barth PG, van Ruissen F, de Wissel MB, Poll-The BT, et al. TSEN54 mutations cause pontocerebellar hypoplasia type 5. Eur J Hum Genet. 2011;19(6):724–6. doi: 10.1038/ejhg.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Namavar Y, Barth PG, Kasher PR, van Ruissen F, Brockmann K, Bernert G, et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain. 2011;134(Pt 1):143–56. doi: 10.1093/brain/awq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rajab A, Mochida GH, Hill A, Ganesh V, Bodell A, Riaz A, et al. A novel form of pontocerebellar hypoplasia maps to chromosome 7q11-21. Neurology. 2003;60(10):1664–7. doi: 10.1212/01.wnl.0000068548.58498.41. [DOI] [PubMed] [Google Scholar]
- 71.Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81(4):857–62. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36(12):1301–5. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
- 73.Moog U, Kutsche K, Kortum F, Chilian B, Bierhals T, Apeshiotis N, et al. Phenotypic spectrum associated with CASK loss-of-function mutations. J Med Genet. 2011;48(11):741–51. doi: 10.1136/jmedgenet-2011-100218. [DOI] [PubMed] [Google Scholar]
- 74.Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J, et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet. 2008;40(9):1065–7. doi: 10.1038/ng.194. [DOI] [PubMed] [Google Scholar]
- 75.Mochida GH, Ganesh VS, de Michelena MI, Dias H, Atabay KD, Kathrein KL, et al. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat Genet. 2012;44(11):1260–4. doi: 10.1038/ng.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barth PG, Majoie CB, Caan MW, Weterman MA, Kyllerman M, Smit LM, et al. Pontine tegmental cap dysplasia: a novel brain malformation with a defect in axonal guidance. Brain. 2007;130(Pt 9):2258–66. doi: 10.1093/brain/awm188. [DOI] [PubMed] [Google Scholar]
- 77.Jissendi-Tchofo P, Doherty D, McGillivray G, Hevner R, Shaw D, Ishak G, et al. Pontine tegmental cap dysplasia: MR imaging and diffusion tensor imaging features of impaired axonal navigation. Ajnr. 2009;30(1):113–9. doi: 10.3174/ajnr.A1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rauscher C, Poretti A, Neuhann TM, Forstner R, Hahn G, Koch J, et al. Pontine tegmental cap dysplasia: the severe end of the clinical spectrum. Neuropediatrics. 2009;40(1):43–6. doi: 10.1055/s-0029-1224100. [DOI] [PubMed] [Google Scholar]
- 79.Briguglio M, Pinelli L, Giordano L, Ferraris A, Germano E, Micheletti S, et al. Pontine Tegmental Cap Dysplasia: developmental and cognitive outcome in three adolescent patients. Orphanet journal of rare diseases. 2011;6:36. doi: 10.1186/1750-1172-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feraco P, Mirabelli-Badenier M, Severino M, Alpigiani MG, Di Rocco M, Biancheri R, et al. The shrunken, bright cerebellum: a characteristic MRI finding in congenital disorders of glycosylation type 1a. Ajnr. 2012;33(11):2062–7. doi: 10.3174/ajnr.A3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) Nat Genet. 1997;16(1):88–92. doi: 10.1038/ng0597-88. [DOI] [PubMed] [Google Scholar]
- 82.Kolb LE, Arlier Z, Yalcinkaya C, Ozturk AK, Moliterno JA, Erturk O, et al. Novel VLDLR microdeletion identified in two Turkish siblings with pachygyria and pontocerebellar atrophy. Neurogenetics. 2010;11(3):319–25. doi: 10.1007/s10048-009-0232-y. [DOI] [PubMed] [Google Scholar]
- 83.Messerschmidt A, Brugger PC, Boltshauser E, Zoder G, Sterniste W, Birnbacher R, et al. Disruption of cerebellar development: potential complication of extreme prematurity. Ajnr. 2005;26(7):1659–67. [PMC free article] [PubMed] [Google Scholar]
- 84.Poretti A, Huisman TA, Scheer I, Boltshauser E. Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. Ajnr. 2011;32(8):1459–63. doi: 10.3174/ajnr.A2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poretti A, Boltshauser E, Loenneker T, Valente EM, Brancati F, Il'yasov K, et al. Diffusion tensor imaging in Joubert syndrome. Ajnr. 2007;28(10):1929–33. doi: 10.3174/ajnr.A0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parisi MA, Glass IA. Joubert syndrome. GeneReviews 2007. 2012 Sep 12; [cited 2012 November 16]; Available from: [Google Scholar]
- 87.Badano JL, Mitsuma N, Beales PL, Katsanis N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu Rev Genomics Hum Genet. 2006;7:125–48. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 88.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. The New England journal of medicine. 2011;364(16):1533–43. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, et al. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert Syndrome. Nat Med. 2011 doi: 10.1038/nm.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, et al. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci. 2007;27(36):9780–9. doi: 10.1523/JNEUROSCI.5586-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spassky N, Han YG, Aguilar A, Strehl L, Besse L, Laclef C, et al. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Developmental biology. 2008;317(1):246–59. doi: 10.1016/j.ydbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saito Y, Yamamoto T, Mizuguchi M, Kobayashi M, Saito K, Ohno K, et al. Altered glycosylation of alpha-dystroglycan in neurons of Fukuyama congenital muscular dystrophy brains. Brain research. 2006;1075(1):223–8. doi: 10.1016/j.brainres.2005.12.108. [DOI] [PubMed] [Google Scholar]
- 93.Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130(Pt 10):2725–35. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 94.Clement E, Mercuri E, Godfrey C, Smith J, Robb S, Kinali M, et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol. 2008;64(5):573–82. doi: 10.1002/ana.21482. [DOI] [PubMed] [Google Scholar]
- 95.Hewitt JE. Abnormal glycosylation of dystroglycan in human genetic disease. Biochimica et biophysica acta. 2009;1792(9):853–61. doi: 10.1016/j.bbadis.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 96.Chan YM, Keramaris-Vrantsis E, Lidov HG, Norton JH, Zinchenko N, Gruber HE, et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet. 2010;19(20):3995–4006. doi: 10.1093/hmg/ddq314. [DOI] [PubMed] [Google Scholar]
- 97.Luo R, Jin Z, Deng Y, Strokes N, Piao X. Disease-associated mutations prevent GPR56-collagen III interaction. PloS one. 2012;7(1):e29818. doi: 10.1371/journal.pone.0029818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chiang NY, Hsiao CC, Huang YS, Chen HY, Hsieh IJ, Chang GW, et al. Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. The Journal of biological chemistry. 2011;286(16):14215–25. doi: 10.1074/jbc.M110.183830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gutowski NJ, Bosley TM, Engle EC. 110th ENMC International Workshop: the congenital cranial dysinnervation disorders (CCDDs). Naarden, The Netherlands, 25-27 October, 2002. Neuromuscular disorders : NMD. 2003;13(7-8):573–8. doi: 10.1016/s0960-8966(03)00043-9. [DOI] [PubMed] [Google Scholar]
- 100.Jen JC, Chan WM, Bosley TM, Wan J, Carr JR, Rub U, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304(5676):1509–13. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bosley TM, Salih MA, Jen JC, Lin DD, Oystreck D, Abu-Amero KK, et al. Neurologic features of horizontal gaze palsy and progressive scoliosis with mutations in ROBO3. Neurology. 2005;64(7):1196–203. doi: 10.1212/01.WNL.0000156349.01765.2B. [DOI] [PubMed] [Google Scholar]
- 102.Holve S, Friedman B, Hoyme HE, Tarby TJ, Johnstone SJ, Erickson RP, et al. Athabascan brainstem dysgenesis syndrome. Am J Med Genet A. 2003;120A(2):169–73. doi: 10.1002/ajmg.a.20087. [DOI] [PubMed] [Google Scholar]
- 103.Tischfield MA, Bosley TM, Salih MA, Alorainy IA, Sener EC, Nester MJ, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet. 2005;37(10):1035–7. doi: 10.1038/ng1636. [DOI] [PubMed] [Google Scholar]
- 104.Mark M, Lufkin T, Vonesch JL, Ruberte E, Olivo JC, Dolle P, et al. Two rhombomeres are altered in Hoxa-1 mutant mice. Development (Cambridge, England) 1993;119(2):319–38. doi: 10.1242/dev.119.2.319. [DOI] [PubMed] [Google Scholar]
- 105.Chisaka O, Musci TS, Capecchi MR. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene Hox-1.6. Nature. 1992;355(6360):516–20. doi: 10.1038/355516a0. [DOI] [PubMed] [Google Scholar]
- 106.Makki N, Capecchi MR. Cardiovascular defects in a mouse model of HOXA1 syndrome. Hum Mol Genet. 2012;21(1):26–31. doi: 10.1093/hmg/ddr434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miyake N, Chilton J, Psatha M, Cheng L, Andrews C, Chan WM, et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane's retraction syndrome. Science. 2008;321(5890):839–43. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Al-Baradie R, Yamada K, St Hilaire C, Chan WM, Andrews C, McIntosh N, et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet. 2002;71(5):1195–9. doi: 10.1086/343821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamada K, Andrews C, Chan WM, McKeown CA, Magli A, de Berardinis T, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1) Nat Genet. 2003;35(4):318–21. doi: 10.1038/ng1261. [DOI] [PubMed] [Google Scholar]
- 110.Nakano M, Yamada K, Fain J, Sener EC, Selleck CJ, Awad AH, et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet. 2001;29(3):315–20. doi: 10.1038/ng744. [DOI] [PubMed] [Google Scholar]
- 111.Cederquist GY, Luchniak A, Tischfield MA, Peeva M, Song Y, Menezes MP, et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum Mol Genet. 2012;21(26):5484–99. doi: 10.1093/hmg/dds393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zaki MS, Saleem SN, Dobyns WB, Barkovich AJ, Bartsch H, Dale AM, et al. Diencephalic-mesencephalic junction dysplasia: a novel recessive brain malformation. Brain. 2012;135(Pt 8):2416–27. doi: 10.1093/brain/aws162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43(8):776–84. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Verge D. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain research. 2000;872(1-2):271–5. doi: 10.1016/s0006-8993(00)02519-1. [DOI] [PubMed] [Google Scholar]
- 115.Handel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, et al. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience. 1999;89(3):909–26. doi: 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]