

Abstract
Little is known about novel genetic elements that drove the emergence of anthropoid primates. We exploited the sequencing of the marmoset genome to identify 23,849 anthropoid-specific constrained (ASC) regions and confirmed their robust functional signatures. Of the ASC base pairs, 99.7% were noncoding, suggesting that novel anthropoid functional elements were overwhelmingly cis-regulatory. ASCs were highly enriched in loci associated with fetal brain development, motor coordination, neurotransmission, and vision, thus providing a large set of candidate elements for exploring the molecular basis of hallmark primate traits. We validated ASC192 as a primate-specific enhancer in proliferative zones of the developing brain. Unexpectedly, transposable elements (TEs) contributed to >56% of ASCs, and almost all TE families showed functional potential similar to that of nonrepetitive DNA. Three L1PA repeat-derived ASCs displayed coherent eye-enhancer function, thus demonstrating that the “gene-battery” model of TE functionalization applies to enhancers in vivo. Our study provides fundamental insights into genome evolution and the origins of anthropoid phenotypes and supports an elegantly simple new null model of TE exaptation.
Most whole-genome studies of human evolution have focused on the ∼5–7 million years since the human-chimpanzee divergence (Pollard et al. 2006; Prabhakar et al. 2006a, 2008; Haygood et al. 2007; McLean et al. 2011). However, many quintessentially human traits are merely extensions of earlier evolutionary changes that appeared in the ancestors of anthropoid primates (Cartmill 1974; Williams et al. 2010). From a biomedical perspective, these earlier primate-specific changes potentially underlie many of the limitations of nonprimate disease models. Primate-specific changes are also likely to be far more numerous than human-specific alterations, since the former accumulated over a longer timespan: ∼47 million years versus ∼6 million years (Methods). However, with the exception of studies that focus on transcribed genes (Enard et al. 2002; Varki et al. 2008; Tay et al. 2009; Pierron et al. 2012), very little is known about the DNA sequences that drove the emergence of anthropoid primates.
The contribution of transposable elements (TEs) to species evolution is a topic of intense interest. The early view was that TEs, which constitute at least 48% of the human genome, were essentially genomic parasites, although they could occasionally contribute new biological functions purely by chance (Doolittle and Sapienza 1980; Orgel and Crick 1980). As knowledge of genomic function expanded, many notable examples of TE-derived human cis-regulatory elements were identified (Feschotte 2008; Rebollo et al. 2012), but the overall evolutionary impact of TEs was still unclear. Thanks to the advent of whole-genome biochemical assays, we now know that TEs contribute massively to genomic transcription factor (TF) binding, chromatin openness, focal histone modification, and tissue-specific DNA methylation (Johnson et al. 2006; Mariño-Ramírez and Jordan 2006; Wang et al. 2007; Kunarso et al. 2010; Kelley and Rinn 2012; Schmidt et al. 2012; Chuong et al. 2013; Jacques et al. 2013; Xie et al. 2013). However, such studies are indicative only of biochemical activity, i.e., biochemical changes at the molecular level. These molecular processes do not always have an impact on organismal phenotypes or fitness (Eddy 2012; de Souza et al. 2013; Doolittle 2013; Niu and Jiang 2013). In other words, biochemical activity does not necessarily imply biological function. Since transposon-derived sequences are, by definition, bound by host proteins at some point in their life cycle and also actively suppressed by the host genome in most cell types, it is only to be expected that they should be enriched for biochemical activity even in the absence of any impact on species traits (Eddy 2012). Thus, it is important that we analyze the contribution of TEs to human evolution using maps of biological, rather than biochemical, function.
It has been suggested (de Souza et al. 2013) that primate sequence constraint analysis, also known as “phylogenetic shadowing” (Boffelli et al. 2003), could provide a more accurate view of the biological functions of TEs. Another complementary strategy is to characterize allele frequency skews in human populations (de Souza et al. 2013). The advantage of these approaches is that they are based on measures of natural selection, and therefore directly indicative of functions that are biologically relevant. However, since the requisite primate genome sequences were not initially available, multiple studies used mammalian sequence comparisons to identify ancient examples of TE functionalization (Cooper et al. 2005; Kamal et al. 2006). Two such studies estimated that 5%–10% of mammal-specific functional elements were TE-derived (Lowe et al. 2007; Mikkelsen et al. 2007). However, it was acknowledged that these figures based on ancestral mammalian gain of function (>100 Mya) may represent severe underestimates, since ancient TEs are poorly annotated. In contrast, recently functionalized TEs are likely to still be recognizable as repetitive elements. It is therefore imperative that we assess the impact of TEs through whole-genome analysis of sequences that (1) show evidence of natural selection; and (2) became functional only recently (i.e., in the primate lineage). Such a strategy could also be used to examine Britten and Davidson’s “gene battery” hypothesis, which states that multiple genes may be coregulated via insertion of near-identical TEs in their promoter regions (Britten and Davidson 1969).
Here, we exploit the sequencing of the marmoset genome (The Marmoset Genome Sequencing and Analysis Consortium 2014) to address the questions described above. Being a New World monkey, the marmoset, which diverged ∼43 Mya (Hedges et al. 2006), lies on the most distant branch of the anthropoid family tree (Fig. 1A). Consequently, it provides sufficient statistical power to detect anthropoid-specific constrained sequences at the length scale of cis-regulatory modules and large exons (Prabhakar et al. 2006b; Wang et al. 2006). Such sequences are likely to have gained new functions that set anthropoids, or perhaps all primates, apart from distant mammals. Thus, anthropoid-specific constrained sequence analysis constitutes a straightforward method for revealing the molecular drivers, both TE-derived and otherwise, of primate evolution genome-wide.
Figure 1.

Functionality and genomic distribution of ASCs. (A) Phylogenetic tree: anthropoids (blue) and nonprimate mammals (red). (B) Illustrative example: Sequence alignment of ASC19060 shows constraint in anthropoids and unconstrained divergence among nonprimate mammals. Dots represent identical nucleotides. (C) Derived allele frequency spectra of African (AFR) SNPs from the 1000 Genomes Project. SNPs within ASCs are shifted to lower frequencies (<15%), indicating ongoing negative selection in humans. In contrast, SNPs within biochemically defined primate-specific DNase I hypersensitive (HS) sites (Jacques et al. 2013) show only a weak frequency shift. (D) ASCs are enriched for DNase I hypersensitivity in 84 human cell lines. (E) Distribution of constrained elements in the human genome. (Green subplot) Of ASC base pairs, 0.3% are protein-coding relative to 13.1% for ACs.
Results
Identification and validation of anthropoid-specific constrained elements (ASCs) in the human genome
As in our previous locus-specific analyses of primate sequence alignments (Prabhakar et al. 2006b; Wang et al. 2006), we defined ASCs as sequences that show strong constraint among anthropoid primates but little or no constraint among nonprimate mammals (Methods; Boffelli et al. 2003). We used a representative selection of the anthropoid primate genomes that have been sequenced to draft quality or better: human, orangutan, rhesus macaque, and common marmoset. Anthropoid constrained elements (ACs) were detected genome-wide in global alignments of multispecies syntenic regions. At a strict P-value threshold of 1 × 10−3, we discovered 268,000 ACs covering 4.2% of the syntenic genome (FDR<0.1%) (see Methods; Supplemental Note 1). In order to prioritize unambiguous examples of gain of function, we discarded from this set any element that showed even weak evidence of nonprimate mammalian constraint. We then used fastDNAmL (Olsen et al. 1994) to independently validate the lineage-specificity of constraint in the remaining elements and thus defined a final set of 23,849 ACs as ASCs (Methods). ASCs covered 8.7 Mbp (0.34%) of the syntenic genome and had a median length of 276 bp (Supplemental Figs. S1, S2; Supplemental Table S1).
To independently test the effect of ASCs on fitness, we examined the distribution of derived-allele frequencies at single-nucleotide polymorphisms (SNPs) within the ASC set (Fig. 1C). For this analysis, we chose SNPs detected in African populations because they exhibit high genetic diversity (The 1000 Genomes Project Consortium 2010). It has been noted that genomic scans for evolutionary constraint tend to enrich for SNPs at which the reference genome carries the ancestral allele (Ward and Kellis 2013). The constrained element allele frequency spectra shown in Figure 1C were calculated in a manner that corrects for this bias (Supplemental Note 2). Relative to the average in syntenic regions, SNPs within ASCs showed a highly significant shift toward lower frequencies (P = 5 × 10−22) (Fig. 1C; Supplemental Note 2), indicating that mutations in ASCs are more deleterious than mutations at random genomic locations. Thus, ASCs as a group show evidence of natural selection, and therefore biological function, in humans. In order to also test for biochemical functionality, we intersected ASCs with DNase I hypersensitive sites from 84 human cell lines (Thurman et al. 2012). We found twofold enrichment of hypersensitive base pairs at ASCs, which was even slightly greater than the enrichment observed for a widely used whole-genome set of placental-constrained elements (Fig. 1D; Supplemental Note 3; Siepel et al. 2005). These results suggest that, with regard to regulatory and biological function, ASCs are equivalent to constrained elements identified by other means.
We compared our ASCs to other sets of functional elements (Fig. 1C): ACs, placental-constrained sequences, coding exons, and also primate-specific DNase I hypersensitive sites (Supplemental Note 4; Jacques et al. 2013). All five sequence sets showed statistically significant allele frequency shifts (Supplemental Table S2)—clearly, they were all influenced by natural selection. To quantify the per-base-pair impact (“effect size”) of natural selection, we calculated the contribution of low-frequency derived alleles to total heterozygosity (Supplemental Note 2). ASCs showed a substantial frequency shift: 36.20% of heterozygosity derived from SNPs with derived allele frequency <15% versus 33.55% genome-wide (Fig. 1C). In contrast to this excess of 2.65 percentage points in ASCs, primate-specific hypersensitive sites showed only minimal allele frequency suppression (0.14 percentage points) (Fig. 1C). Thus, lineage-specific constrained elements are more relevant to organismal fitness than lineage-specific biochemically active regions.
Interestingly, ASCs were enriched only in distal nonexonic regions (P ≈ 0) relative to the average AC (Fig. 1E; Supplemental Note 5). Only 1.2% of ASC bases overlapped noncoding RNAs, and no enrichment was observed in this category relative to ACs. Untranslated regions (UTRs) were depleted by a factor of 5 (P ≈ 0), suggesting limited contribution of proximal regulatory elements to anthropoid innovation. Most notably, although 13.1% of AC base pairs overlapped coding regions, only 0.28% of ASCs were protein-coding—a 47-fold reduction (P ≈ 0). This bias against gain of functional coding sequence is stronger than previously believed (Mikkelsen et al. 2007). It is possible that the earlier estimate (14-fold) was biased by the fact that it only included noncoding regions showing pan-eutherian constraint. Noncoding functional elements, which tend to evolve more rapidly, would therefore be undercounted (Meader et al. 2010). Since 99.7% of ASC base pairs are noncoding and only 1.2% overlap noncoding RNAs, it is evident that functional blocks gained in ancestral primates were overwhelmingly cis-regulatory (King and Wilson 1975; Carroll 2003).
Overarching functional themes among nonexonic ASCs
In order to examine the in vivo regulatory functions of recently gained elements during human development, we intersected ACs and ASCs with tissue-specific DNase I hypersensitive sites from eight human fetal tissues (Bernstein et al. 2010). Notably, ASCs were significantly more likely than ACs to overlap hypersensitive sites in human fetal brain and fetal thymus (Fig. 2A; Supplemental Note 6). These enriched ASCs thus provide a promising set of candidate regulatory regions for exploring the genetic underpinnings of the profound alterations in primate brain development seen in comparative and fossil studies (Kaas 2006) and also primate-specific immune adaptations.
Figure 2.
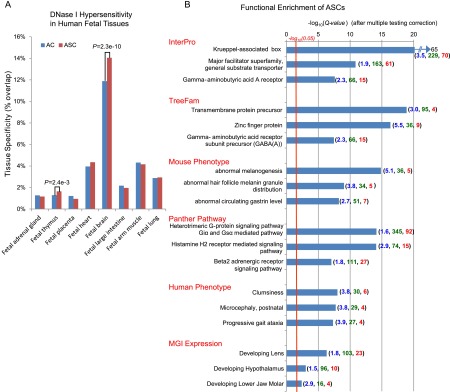
Tissue specificity and functional enrichment of nonexonic ASCs. (A) Red and blue bars indicate the fraction of constrained base pairs overlapping tissue-specific DNase I hypersensitive sites. (B) Top three functional annotations within each ontology that are enriched near nonexonic ASCs (GREAT tool). Numbers indicate fold enrichment (blue), number of contributing ASCs (green), and number of annotated ASC-flanking genes (red).
In order to identify functionally coherent sets of molecular changes in anthropoid evolution on a larger scale, we used the GREAT tool (McLean et al. 2010) to test for enriched annotations in the flanking genes of nonexonic ASCs relative to ACs (Methods). GREAT avoids systematic biases by explicitly controlling for the larger size of genomic loci associated with certain biological functions (for example, neurodevelopment). We filtered enriched functional terms for “jackpot” effects arising from an excess of ASCs near a single gene (Methods). We also avoided cherry-picking of GREAT results by focusing on the top three enriched terms in each of six functional ontologies (Fig. 2B; Supplemental Table S3). Remarkably, almost all the top enriched annotations matched traits known to have evolved uniquely among primates.
We found massive enrichment of nonexonic ASCs near Krüppel-associated box (KRAB) genes (FDR Q-value = 6 × 10−65) and zinc finger genes (Q = 5 × 10−17). Both signals derive from ASCs flanking KRAB-zinc finger (KZNF) genes (Fig. 2B; Supplemental Table S3), which direct repressive chromatin to retroviral sequences and other genomic loci (Rowe et al. 2010). Within the InterPro ontology, we also found a strong excess of ASCs near substrate transporter genes (Q = 1.4 × 10−11). Notably, when these genes were sorted by their individual enrichment for ASCs, seven of the top 14 were either annotated as urate transporters, or adjacent to GWAS SNPs for serum urate level or both (Supplemental Tables S3, S4; Anzai and Endou 2011). Thus, it is possible that, in addition to the known changes in urate homeostasis among apes (Oda et al. 2002), urate levels also evolved through cis-regulatory gain of function in the lineage leading to anthropoids.
Nonexonic ASCs also showed strong enrichment near transmembrane protein precursors (Q = 1 × 10−19), all of which derived from a single locus containing TMEM132B, TMEM132C, and TMEM132D. These genes show robust genetic associations with anxiety, depression, attempted suicide, and panic disorder, and also differential expression in fear-related brain regions (Hindorff et al. 2009; Erhardt et al. 2011). Interestingly, 53/123 ASCs in this locus overlap open-chromatin regions detected by DNase-seq in human fetal brain, adult brain, or neuronal cell lines (Fig. 3A; Supplemental Note 7), suggesting a potential role for ASCs in the evolution of these behavioral traits.
Figure 3.

ASC-enriched genes are associated with disease. Three genomic loci that have accumulated a significant excess of ASCs and also harbor disease-associated SNPs: (A) TMEM132; (B) OCA2; (C) POLN. Numerous ASCs overlap open chromatin regions (vertical red lines). ASC enrichment is computed from a moving window of 10 background ACs. (D) Fourteen of the top 20 genes enriched for flanking nonexonic ASCs are associated with human disease, of which 11 relate to the brain or eye (red).
Gamma-aminobutyric acid A (GABAA) neurotransmitter receptors were among the top three ASC-enriched gene classes within both the InterPro and TreeFam ontologies. Moreover, GABA synthesis genes were also enriched for ASCs (PANTHER Pathway ontology) (Supplemental Table S3). The excess of ASCs near genes expressed in the developing eye lens was also almost entirely attributable to GABAA receptor genes and other GABA-pathway genes. We also observed ASC enrichment near histaminergic, serotonergic, adrenergic, and dopaminergic receptors (TreeFam: TF316350) (Supplemental Table S3). Moreover, ASCs were enriched near heterotrimeric G-protein signaling pathway genes (PANTHER Pathway ontology) (Fig. 2B), which include numerous neurotransmitter receptors and their downstream effectors. Thus, neurotransmission genes in general, and GABAA pathway genes in particular, were preferentially targeted for anthropoid-specific gain of nonexonic function.
Intriguingly, ASCs were strongly enriched near genes related to abnormal melanogenesis. This was initially a surprising result, since anthropoid primates are not known to share any unique external coloration phenotypes. However, upon closer inspection, we noticed that mutations in four of the five ASC-flanked melanogenesis genes (OCA2, LYST, HPS6, and BLOC1S4) result in defective vision and ocular malformations (Oetting and King 1999), suggesting a potential link between these ASCs and primate visual acuity (Martínez-Morales et al. 2004; Nickla and Wallman 2010). We also found ASC enrichment near human genes associated with clumsiness, postnatal microcephaly, and progressive gait ataxia. Notably, three genes were shared across these disease annotations and accounted for most of the enrichment: UBE3A, MECP2, and CDKL5 (Supplemental Table S3). ASCs near these genes constitute promising candidates for exploring the molecular basis of primate-specific motor traits and also primate-specific aspects of human disease (Yasui et al. 2007).
Encouraged by the preceding examples, we hypothesized that top ASC-enriched genes could reveal additional candidates for primate-specific disease biology. The most strongly ASC-enriched gene in the genome was the above-described anxiety-associated gene TMEM132B, followed by the oculocutaneous albinism II gene OCA2 (Fig. 3A,B; Supplemental Note 7). Overall, 14 of the top 20 ASC-enriched genes were associated with human diseases related to behavior, mood, motor coordination, vision, and hearing (Fig. 3D). Moreover, 302 ASCs contained SNPs associated with human diseases (Fig. 3C; Supplemental Table S5; Supplemental Note 8). Thus, we see numerous loci in the genome where primate-specific gene regulation may have altered the biology of specific human diseases.
ASC192 is a primate-specific neurodevelopmental enhancer
It is believed that developmental evolution occurs to a large extent through alterations in gene expression (Carroll 2003). However, experimentally validated examples of this phenomenon are scarce, particularly in the context of primate evolution. We prioritized ASC192 (constraint P = 5.8 × 10−11) (Supplemental Fig. S3) based on chromatin profiling data (Methods) and tested this element for tissue-specific enhancer function in day 11.5 (E11.5) transgenic mouse embryos. We found highly reproducible reporter-gene expression in the central nervous system and eye, with midbrain being the strongest expression domain (Fig. 4; Supplemental Fig. S4). In stark contrast, the mouse and dog orthologs of ASC192 showed no reproducible activity in this assay (Fig. 4A; Supplemental Figs. S5, S6). Thus, ASC192 represents an evolutionarily novel neurodevelopmental enhancer that arose in the ancestral lineage of anthropoid primates.
Figure 4.

Primate-specific enhancer function of ASC192. (A) ASC192 drives consistent primate-specific lacZ expression in the developing central nervous system of E11.5 mouse embryos; four representative embryos are shown for each construct: (fb) forebrain; (mb) midbrain; (hb) hindbrain; (sc) spinal cord. For the entire set of transgenic embryos, see Supplemental Figures S4–S6. (B) Enhancer success rates for ASC192 and its orthologs: ASC192 drove strong lacZ expression in 10/14 transgenic embryos, whereas the mouse and dog orthologs of ASC192 drove strong reporter gene expression in only 1/8 transgenics. (C) Transverse sections of a representative ASC192 embryo; strong expression is visible in forebrain, midbrain, and hindbrain (i, ii). lacZ expression coincides with neuroepithelial zones that spawn neural progenitors (iii, v) and also with neural retina (iv). In the spinal cord, lateral to the roof and floor plates, enhancer activity localizes to regions containing dorsal spinal interneurons and motor neuron progenitors (v).
We examined ASC192 function at greater resolution using transverse embryonic sections (Fig. 4C). In the diencephalon, midbrain, and hindbrain, expression was localized to the alar ventricular, subventricular, marginal zones (VZ, SVZ, and MZ), and the entire roof plate neuroepithelium. Notably, the VZ and SVZ are sites of neurogenesis in the developing brain. ASC192 was also functional in the neural retina and dorsal spinal cord, which are again involved in neurogenesis. In order to uncover the target gene of ASC192, we used the 3C assay, which probes for physical interactions between the enhancer and flanking regions (Hagège et al. 2007). Human brain tissue is inaccessible at this developmental stage. However, human embryonic stem cells (hESCs) provide a convenient alternative because they express all three genes proximal to ASC192 (Supplemental Fig. S7). Furthermore, ASC192 is marked by open chromatin in hESCs (Supplemental Fig. S8). In the 3C assay, ASC192 showed strong long-range interactions with the promoters of both POU2F1 (also known as OCT1) isoforms (Fig. 5). Thus, ASC192 functions as an enhancer of the neurogenesis-related POU2F1 transcription factor (Kiyota et al. 2008; Theodorou et al. 2009) and drives strong primate-specific expression in neurogenic zones during embryonic development.
Figure 5.

3C assay demonstrates looping between ASC192 and POU2F1. (A) Black ticks indicate locations of primers designed to capture long-range DNA interactions with ASC192. Locus-wide relative interaction frequencies reveal that ASC192 interacts most strongly with the two promoters of POU2F1. Error bars represent one standard deviation. (B) DNase-seq signal at ASC192 and the POU2F1promoter regions indicate an open chromatin conformation in human fetal brain.
Massive contribution of TEs to new functional elements
In order to investigate the origins of new anthropoid functional elements, we intersected ASCs with annotated human TEs. Although other estimates are even higher (de Koning et al. 2011), the RepeatMasker track on the UCSC Genome Browser annotates 48% of the syntenic human genome (hg19) as TE-derived (Supplemental Note 9). Unexpectedly, 46% of ASC base pairs were TE-derived, indicating that the earlier estimates of 5.5% and 10% were biased by incomplete annotation of ancestral TEs (Fig. 6A; Lowe et al. 2007; Mikkelsen et al. 2007). As many as 56% of the ASC elements overlapped TEs by at least 50 bp. Note that alignment artifacts are unlikely to contribute significantly to our count of TE-derived ASCs (TE-ASCs) because we detected anthropoid sequence constraint exclusively in quality-filtered multisyntenic global alignments (Methods). Moreover, manual examination of >100 randomly chosen ASCs failed to identify any such artifacts. Nevertheless, in order to independently confirm the functionality of TE-ASCs, we specifically examined their bias-corrected allele frequency distribution (Fig. 6B). Reassuringly, SNPs within TE-derived ASC subregions showed approximately the same effect on human fitness (enrichment for low-frequency alleles) as SNPs within ASCs as a whole. These results indicate that TE-ASCs have massively influenced anthropoid evolution, contributing 3.99 Mbp of newly constrained sequence distributed across 14,546 ASCs genome-wide. Eighty-five percent of ASC base pairs are ancestral, i.e., shared with nonprimate mammals (Supplemental Fig. S9), and 40% of these are TE-derived. The remaining 15% of ASC base pairs have no ortholog beyond primates; 80% of these are TE-derived.
Figure 6.

Massive contribution of transposable elements to ASCs. (A) Percent of human genome annotated as TE-derived compared to percent of lineage-specific constrained base pairs derived from TEs. TE contributions to eutherian- and mammal-specific constrained elements were estimated in previous studies (Lowe et al. 2007; Mikkelsen et al. 2007). (B) Derived allele frequency spectrum of AFR SNPs within TE-derived ASC subregions shows a similar enrichment at low frequencies, indicating that TE-ASCs are not noticeably enriched for false positives. (C) Enrichment was defined as the fraction of constrained (ASC or AC) base pairs attributable to a TE family divided by the fraction of the aligned genome attributable to the same family. TE family names were assigned by RepeatMasker. Family size was defined as the total size in base pairs of all TEs within the family.
Previous studies have highlighted specific ancient TEs, such as MER121 and LF-SINE, that show enrichment for pan-mammalian constraint (Bejerano et al. 2006; Kamal et al. 2006). However, these TEs were inserted over 180 Mya, and therefore their high level of sequence constraint may merely reflect the fact that nonconstrained instances are no longer detectable. In order to systematically examine TE functional enrichment, we tested all RepeatMasker-annotated TE families for enrichment in AC base pairs relative to their overall prevalence in the genome (Fig. 6C, blue bars; Supplemental Note 9). As expected, the older repeat families frequently showed strong overrepresentation in ACs, although this effect was only observed in small, ancient families with relatively few annotated genomic instances, such as “SINE?,” “LTR?,” and “Deu.” The “DNA” TE family was the most recent family showing strong enrichment in ACs. However, even this family predates the mammalian radiation—orthologs are detectable even in the highly diverged platypus genome. The average divergence of “DNA” repeats from their consensus sequence was calculated to be relatively low (0.27 subs/site) only because half the genomic positions assigned to this repeat family were evolutionarily constrained. Overall, the pattern of TE enrichment within ACs is consistent with a model in which all TE families possess approximately the same functional potential. As previously suggested (Lowe at al. 2007; Mikkelsen et al. 2007), the ancient repeat families that do show functional enrichment most likely do so because their nonconstrained instances have decayed so much that they are no longer recognizable as TEs by RepeatMasker.
Due to their relatively recent origin, ASCs provide a unique opportunity to accurately examine the propensity of TEs from various families to become functional. We therefore assessed each TE family for enrichment in ASCs (Fig. 6C, red bars). In contrast to the ∼12-fold maximum enrichment in ACs, we found that only one TE family (piggyBac) showed greater than threefold enrichment in ASCs, and 88% (36/41) showed less than twofold enrichment. Similarly, although 44% (18/41) TE families showed greater than twofold depletion in ACs, only 17% (7/41) showed such a depletion in ASCs. Moreover, three of these seven were of very recent origin (“Other,” ERVK, and Alu), and therefore either largely or entirely too “young” to contribute functional elements to the anthropoid ancestor. Overall, these results suggest that (1) most repeat families are similar in their propensity for contributing new functional elements to the genome; and (2) this propensity is similar to that of unique DNA.
Gene battery model of TE exaptation applies to enhancers in vivo
We sought to determine if ASCs derived from homologous TEs could act as functionally homologous enhancers in vivo as predicted by the gene battery hypothesis (Britten and Davidson 1969). Given the neurodevelopmental themes arising from functional enrichment analysis of ASCs, we prioritized five ASCs derived from three closely related primate-specific subfamilies of the L1 repeat family (L1PA13, L1PA15, L1PA16) (Khan et al. 2006), all of which showed strong DNase I hypersensitivity in human fetal brain (Methods). The mouse developmental stage equivalent to that of the human brain samples is not conducive to the lacZ enhancer assay. We therefore tested the five TE-ASCs for enhancer activity at an earlier developmental stage (E14.5 mouse embryos) (Fig. 7A; Supplemental Figs. S10–S14). Although the TE-ASCs drove lacZ staining in multiple brain regions, these expression domains were highly variable across the embryos. Interestingly however, three/five TE-ASCs drove reproducible lacZ expression in the eye. The other two TE-ASCs also drove eye expression in individual embryos but not consistently enough to be scored as positives. In contrast, embryos transgenic for the empty lacZ vector (negative control) showed no eye staining (Supplemental Fig. S15). Upon further examination, we noticed that all five tested TE-ASCs flanked genes known to be specifically expressed or up-regulated in the eye (Supplemental Table S6).
Figure 7.

Shared enhancer function among three ASCs derived from primate-specific L1PA repeats. (A) Three ASC elements drove consistent lacZ expression in the developing eye at E14.5. Three representative embryos are shown for each construct; the full sets can be found in Supplemental Figures S10–S12. (B) De novo motif discovery in the tested TE-ASCs uncovered binding motifs similar to the known motifs of TCF3 and SIX6. In situ hybridization, sagittal sections: Tcf3 and Six6 show strongest expression in the eye at E14.5.
In order to infer the identities of TFs that may coherently regulate the L1PA-derived ASCs, we performed unbiased motif discovery in the L1PA subregions of the five ASCs using MEME (Bailey et al. 2009). The top-scoring motif thus identified matched the sequence specificity of TCF3 (Fig. 7B). The second- and third-ranked motifs had no database matches (data not shown). However, the fourth-ranked motif matched the Drosophila TFs Optix and sine oculis, and also mammalian SIX6, all of which play crucial roles in eye development (Liu et al. 2006; Bharti et al. 2012). We therefore examined the expression patterns of Tcf3 and Six6 using in situ hybridization at E14.5. Notably, both TFs showed highly specific expression in the eye at this time point (Fig. 7B). It is possible that the shared presence of binding sites for these TFs contributed to the coherent eye-specific expression driven by the TE-ASCs.
In order to explore the gene battery model at the subfamily level, we tested 429 TE-ASC subfamilies for functional coherence of their neighboring genes (Supplemental Table S7; Supplemental Note 10). Note that this analysis has limited statistical power, since ASCs represent only a small subset of newly functionalized sequences, and also because TE subfamilies are narrowly defined to only include very highly homologous transposon relics. Perhaps for this reason, only one TE-ASC subfamily (L1MB5) showed significant functional enrichment relative to the set of all ASCs, after correcting for multiple testing. L1MB5-derived ASCs were enriched near genes containing the Mib-herc2 domain, which mediates Notch signaling. TE-ACs are greater in number than TE-ASCs and therefore provide greater statistical power for functional enrichment analysis. Consequently, we found a larger number of significantly coherent TE-AC subfamilies (33/617 tested) (Supplemental Fig. S16; Supplemental Table S7). In particular, MER121, which has previously been noted for strong mammalian evolutionary constraint (Kamal et al. 2006), was highly enriched near reproductive system development and appendage morphogenesis genes. Other TE-AC subfamilies were enriched near genes related to adaptive immunity (MIRc), susceptibility to seizures (L2b), diverse brain development functions (UCON28b), and synaptic plasticity (MIRb). Note that functional biases in ancestral mammalian exaptation are not the only possible explanation for these results. It is conceivable, for example, that MER121 repeats actually contributed to a broad range of ancestral functions, but the morphogenetic subset showed enrichment in our analysis because morphogenesis gene regulatory elements are more “durable” over evolutionary time.
Discussion
Molecular origins of anthropoid primates
We have exploited the availability of the marmoset genome sequence to perform the first genome-wide screen for anthropoid-specific functional elements. Using a stringent set of filters, we identified a large set of ∼24,000 ASCs covering ∼9 Mbp of the human genome and that shows strong genetic and chromatin-state signatures of functionality. The overwhelmingly nonexonic (97.4%) and noncoding (99.7%) nature of ASCs suggests that, when measured by gain of constraint, anthropoid novelty is overwhelmingly attributable to gene regulatory changes (King and Wilson 1975).
The genomic distribution of ASCs was highly nonrandom, suggesting strong selection for gain of specific functions in the lineage leading to anthropoid primates. The massive excess of ASCs near KZNFs suggests that, in addition to coding sequence evolution (Huntley et al. 2006), novel gene regulation may have played a major role in protecting the ancestral primate genome from retroviral transcription (Thomas and Schneider 2011). The TMEM132 locus showed the strongest ASC enrichment of all loci in the genome, perhaps reflecting altered regulation of TMEM132 genes during primate evolution in response to selection for fear-related behavioral traits. These ASCs are promising candidates for exploring the molecular basis of the known differences in anxiety-related amygdala-prefrontal cortex circuitry between primates and rodents (Kalin et al. 2001).
The broad-based enrichment of ASCs near GABA and other neurotransmitter pathway genes provides a striking molecular correlate to one of the unique features of primate cortical development, namely simultaneous overexpression of multiple neurotransmitter receptors during the early postnatal growth phase (Lidow et al. 1991). GABA genes were also responsible for the enrichment of ASCs in eye lens development loci. GABA signaling influences lens growth (Schwirtlich et al. 2011), and the primate lens is known for its ability to accommodate an exceptionally wide range of focal lengths (Borja et al. 2010). Thus, widespread anthropoid gain of function in the GABA signaling pathway could potentially have played a role in primate visual acuity.
The melanogenesis genes enriched in ASCs also control eye development and visual perception, which is significant in light of the well-known connections between melanin, eye development, and primate evolution (Kirkwood 2009). For example, the choroid layer of the primate eye has evolved higher melanin levels in order to reduce uncontrolled scattering of light behind the retina (Nickla and Wallman 2010). Thus, it is possible that cis-regulatory gain of function in these melanogenesis loci potentially contributed to improved visual acuity in primates (Martínez-Morales et al. 2004). Intriguingly, primates are also known for elevated levels of neuromelanin in dopamine neurons (Marsden 1961).
Finally, ASC enrichment near motor coordination and microcephaly genes (UBE3A, MECP2, CDKL5, and others) suggests that gain of new regulatory elements may have contributed to primate motor adaptation and enlargement of the primate motor cortex (Kaas 2008) and also potentially to primate-specific aspects of neurodevelopmental diseases. Overall, ASCs show a strong and consistent trend of enrichment in gene loci with clear links to known anthropoid-specific phenotypes. Thus, they provide us with the first large set of candidate genomic elements for exploring the cis-regulatory underpinnings of hallmark primate traits.
We have validated ASC192 as the first known neurodevelopmental enhancer specific to primates. ASC192 drives expression in neurogenic zones of the developing brain. Interestingly, POU2F1, the target gene of ASC192, is a driver of developmental neurogenesis (Theodorou et al. 2009) and a vital effector of radial glia formation in the VZ and SVZ (Kiyota et al. 2008). These links between POU2F1 and ASC192 function suggest a possible role for the newly evolved enhancer in primate-specific neuronal proliferation. It is thus possible that ASC192 may have contributed to the increased brain size and unique brain structure of anthropoid primates (Kriegstein et al. 2006).
Origins of new functional elements
Where do new functional elements come from? Many previous whole-genome studies have addressed this question using biochemical technologies such as ChIP-seq, DNase-seq, and DNA methylation profiling (Johnson et al. 2006; Mariño-Ramírez and Jordan 2006; Wang et al. 2007; Kunarso et al. 2010; Kelley and Rinn 2012; Schmidt et al. 2012; Chuong et al. 2013; Jacques et al. 2013; Xie et al. 2013). However, our results indicate that sequences showing lineage-specific biochemical activity have fewer fitness consequences than sequences showing lineage-specific constraint (Fig. 1C). In other words, they are biochemically functional but less enriched for biological function. Other whole-genome studies have taken the approach of mapping ancient (mammalian) constrained elements and intersecting them with TEs (Lowe et al. 2007; Mikkelsen et al. 2007). However, our results indicate that recent functionalization provides a cleaner lens through which to view the process of evolution, most likely because annotation of TEs is more reliable over shorter timescales (Fig. 6A).
The 23,849 ASCs identified in this study constitute the first genome-wide data set that satisfies the two important criteria discussed above: They were recently functionalized, and they are also important for fitness. Remarkably, TEs contributed to >56% of ASCs, and new functional elements arose from repetitive and nonrepetitive DNA without any discrimination between the two categories (Fig. 6A). Just as remarkably, the 41 annotated repeat families all showed approximately the same propensity for contributing new functions once their prevalence in the genome was taken into account (Fig. 6C). Thus, our results support a new, elegantly simple model of molecular evolution in which new functions arise more or less at random in the genome, with little regard to repeat status or repeat family. This is at variance with the conclusions of studies based on biochemical definitions of function, which detect massive enrichment of LTR repeats (Wang et al. 2007; Thurman et al. 2012; Chuong et al. 2013; Jacques et al. 2013). The most parsimonious explanation for these divergent findings is that there exists in the genome a large number of biochemically active LTRs that have little or no biological function. Another possible explanation is that the biochemically based studies examined function in specific cell types, whereas ASCs are cell-type agnostic. However, the enrichment of biochemically functional LTRs was observed across a very broad range of cell types (Jacques et al. 2013), suggesting that the former explanation is more likely.
It has long been hypothesized that the existence of near-identical TE insertions within the upstream regions of functionally related genes could provide a mechanism for their coexpression (Britten and Davidson 1969). This hypothesis has recently found in vitro experimental support: A large set of MER20 LTR-derived sequences was shown to drive expression in mammalian endometrial cells (Lynch et al. 2011). In addition, RLTR13D5 insertions have been shown to drive reporter gene expression in rodent placental cells (Chuong et al. 2013.). However, despite considerable speculation based on individual instances (Bejerano et al. 2006; Tashiro et al. 2011), there has so far been no corresponding evidence for enhancers driving gene batteries in vivo. We have used in vivo enhancer assays of ASCs to show that three highly homologous TE-derived sequences (L1PA subfamilies) drive coherent expression patterns in the developing eye. Experimental dissection of additional TE-ASC subgroups could potentially reveal many additional instances of enhancer-driven gene batteries in primate evolution.
Methods
Duration of primate-specific and human-specific evolution
As defined in this study, anthropoid-specific functional elements arose at some point between the divergence of the nonprimate outgroup species (tree shrew) and the divergence of the marmoset and human lineages (Fig. 1A). These two divergence times have been estimated at ∼90 million years ago and ∼43 million years ago, respectively (Hedges et al. 2006). Consequently, ASCs arose over a time span of ∼47 million years (90 minus 43). In contrast, the evolutionary timespan since the human-chimpanzee divergence is much shorter: ∼6 million years (Hedges et al. 2006).
Global alignment of multisyntenic regions
DNA sequences of human (hg19), three anthropoid primates, Pongo pygmaeus abelii (ponAbe2), Macaca mulatta (rheMac2), and Callithrix jacchus (calJac3), and three nonprimate mammals, Canis familiaris (canFam2), Mus musculus (mm9), and Equus caballus (equCab2), were downloaded from the UCSC Genome Browser (Kent et al. 2002). Their respective pairwise chain and net alignments to human were also downloaded. To identify multispecies syntenic blocks, we divided the human genome into intervals of length ≥50 kbp that were syntenic across all seven species, as evidenced by the existence of “Level-1” net alignments. Level-2 nets >50 kbp were used to fill gaps in Level 1, and Level-3 nets similarly filled gaps in Level 2.
In each of the 3193 multisyntenic regions, we discarded nonhuman sequences to which too small a fraction of human bases was aligned in the “net.” Percent alignment thresholds were selected empirically for each species based on the inflection point in the whole-genome distribution of aligned fractions (orangutan: 60%; rhesus macaque: 55%; marmoset: 45%; dog: 30%; horse: 35%; and mouse: 30%). We discarded syntenic blocks if (1) either marmoset or dog was insufficiently aligned; (2) both rhesus and orangutan were insufficiently aligned; or (3) both mouse and horse were insufficiently aligned, resulting in 1876 filtered multisyntenic regions covering 2.57 Gbp of the human genome.
We used the global aligner MLAGAN (Brudno et al. 2003) to align sequences within each multisyntenic block (see Supplemental Table S8 for scoring matrices). Sequence positions with quality score <30 were masked to “N” in the respective genomes to avoid artifacts from sequencing errors.
Scanning for constraint using Gumby
Multiply aligned syntenic blocks were scanned for constrained segments using Gumby (Prabhakar et al. 2006b). We ran Gumby with a strict P-value threshold when scanning for constraint in anthropoids (P < 0.001) and a loose threshold when scanning for constraint among nonprimate mammals (P < 0.1). We detected constrained elements in multiple species sets: (1) human-marmoset-rhesus-orangutan; (2) human-marmoset-orangutan; (3) human-marmoset-rhesus; (4) dog-horse-mouse; (5) dog-horse; and (6) dog-mouse. For each set, we ran Gumby with two values of the “Ratio” parameter (2 and 5) and merged the resulting sets of constrained regions.
Additional syntenic block filtering using Gumby alignment metrics and GC content
Gumby fits the distribution of column scores of multiple-sequence alignments to a Gumbel distribution with parameters K and λ. We discarded syntenic blocks with outlier K or λ values, i.e., values that deviated from the 25th or 75th percentile for the chromosome by >1.5 times the inter-quartile range (IQR). We also filtered syntenic blocks with respect to GC-adjusted substitution rate. For each nonhuman species and each syntenic block, we linearly regressed the pairwise substitution rate with human against the GC content. If the regression residual of any nonhuman species within a syntenic block was atypical for the chromosome (by the same IQR criterion), the syntenic block was discarded. These filters yielded a final list of 1713 “clean” syntenic blocks covering 2.55 Gbp of the human genome. The 268,521 anthropoid-constrained (AC) sequences cover 4.2% of the syntenic genome.
Scanning for mammal constraint using phastCons on 46-way MULTIZ alignment
In contrast to the ACs, we used an extremely broad definition of nonprimate mammalian constraint (2.1 million elements, 11% of syntenic genome). Our mammalian constraint screen combined two independent algorithms, multiple parameter settings, relatively loose significance thresholds, and four different species sets, one aligned globally and three locally (Supplemental Fig. S1). The details are discussed below.
To increase sensitivity in detecting nonprimate mammal constraint, we used phastCons (Siepel et al. 2005) to scan the subalignmnet of 25 nonprimate mammals (Supplemental Table S9) within the UCSC 46-species whole-genome MULTIZ alignment (Kent et al. 2002; Blanchette et al. 2004). Glire genomes have evolved more rapidly than those of other mammals, resulting in frequent nonfunctionalization of otherwise constrained sequences. Consequently, the inclusion of Glires in the 25-species set occasionally hinders detection of mammalian constraint. Conversely, some functional elements could be specific to Euarchontoglires and therefore constrained only in Glires and primates. For greater sensitivity, we therefore used phastCons to scan only the Glires (seven species) and also only the non-Euarchontoglire mammals (18 species). Again, for completeness, we ran phastCons twice for each of the three mammal subgroups (25, 18, and seven species), using two sets of parameters (“–rho 0.31–target-coverage 0.3–explen 45” and “–rho 0.5–target-coverage 0.2–explen 65”), for a total of six phastCons genome-wide constraint scans. We then filtered the phastCons constrained elements in order to have comparable genomic coverage with the Gumby nonprimate mammal constrained elements. We discarded the rho = 0.5 elements whose phastCons score was less than 50 and used all phastCons constrained elements detected using rho = 0.31. Overlapping constrained elements from the various scans were merged, resulting in a highly sensitive and comprehensive final set of 2.1M nonprimate mammal constrained elements covering 10.96% of the syntenic genome.
Definition of anthropoid-specific constrained (ASC) sequences and validation using fastDNAmL
We defined ASCs as anthropoid-constrained (AC) sequences with at most 10% of their length covered by mammal-constrained elements, resulting in an initial set of 24,999 candidates. We used fastDNAmL (Olsen et al. 1994) to compute branch lengths (substitution rates) within each candidate ASC and also the background substitution rate within nonexonic regions of the same syntenic block. For each candidate element, the total substitution rate within the anthropoid (or nonprimate mammal) phylogenetic tree was calculated by summing over all lineages, and these aggregate substitution rates were compared to the corresponding background rates. We defined the anthropoid constraint factor as
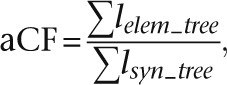 |
where l represents the length of a phylogenetic tree branch, and the subscripts indicate the phylogenetic tree over which summation is performed. We incorporated uncertainty in branch length estimation by defining a “modified anthropoid constraint factor,” which constitutes an upper bound on the constraint factor:
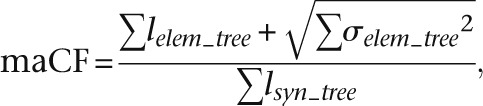 |
where σ is the length uncertainty of an individual tree branch (95% confidence interval reported by fastDNAmL). Note that we have ignored uncertainties in background substitution rates because these are typically small. We similarly computed the nonprimate mammal constraint factors for each candidate ASC and also the constraint factors in the tree-shrew lineage.
We declared an element constrained according to fastDNAmL if it satisfied constraint factor ≤0.7 and modified constraint factor ≤0.85. All 24,999 elements had anthropoid constraint factors below these thresholds, suggesting that Gumby constraint analysis produced no obvious false positives. By the same criterion, 1150 candidate ASCs showed constraint either among nonprimate mammals or in the tree-shrew lineage. We discarded these 1150 elements to obtain a final list of 23,849 validated ASCs, ranging in size from 77–5239 bp, with a median of 276 bp (Supplemental Fig. S2; Supplemental Table S1).
Functional enrichment analysis of ASCs using GREAT
We used GREAT (McLean et al. 2010, version 1.7.0) to determine whether nonexonic ASCs were enriched near genes belonging to specific functional categories. This enrichment analysis was performed using Fisher’s exact test with the entire set of ACs as the “background” set. In other words, we were testing for ASC functional enrichment relative to ACs. We downloaded results for all 20 ontology databases that can be accessed through GREAT. In order to harmonize statistical significance measures across the 20 ontologies, we chose the conservative approach of recalculating the FDR Q-value of each functional category based on the P-values of all tests across all ontologies. In most cases, this had the effect of mildly increasing the Q-value originally calculated by GREAT, thus mildly reducing the estimated statistical significance. We used an FDR Q-value threshold of 0.01 and discarded functional categories in which ASCs were less than 1.2-fold enriched relative to ACs.
Since the GO annotation is hierarchical, we pruned the GREAT results by deleting an enriched functional category if it was a “child” of a “parent” (superset) category that was enriched with a superior P-value. Conversely, if the parent category added nothing to the observed-expected statistic of the child, then the child category was retained. The other 17 ontologies were pruned by defining a functional category as a child of another category if all of the genes within the former were contained within the gene list of the latter. AC-gene assignments made by GREAT were used to ask if any individual genes were enriched in ASCs, using the same Fisher’s exact test approach (Supplemental Table S4).
To avoid “jackpot” effects arising from an abundance of ASCs near a single gene, we recomputed the P-value of each ASC-enriched functional category after removing the single most enriched gene. If the functional category was no longer significant at an uncorrected P-value threshold of 0.05, it was removed from the list. The complete set of GREAT results (FDR Q-value <0.01 and at least three foreground gene hits) are in Supplemental Table S3.
Transgenic enhancer assay
We shortlisted ASC candidates within the top 200 by P-value that showed DNase I hypersensitivity at all available developmental stages in human fetal brain (day 85 to day 142; NHGRI Epigenome Atlas) (Bernstein et al. 2010). We then filtered out all ASCs that overlapped a human mRNA. Among the resulting candidates, ASC192 (constraint P = 5.8 × 10−11) (Supplemental Fig. S3) was chosen because it showed the greatest hypersensitivity at day 85, the earliest time point.
The lacZ reporter constructs for in vivo mouse embryonic enhancer assays were created as previously described (Pennacchio et al. 2006). Embryos were collected and stained for lacZ activity at embryonic day E11.5/E14.5. In brief, all candidate enhancer sequences were PCR-amplified from human, mouse, and dog genomic DNA (Roche) using high fidelity Pfx Polymerase (Invitrogen). PCR fragments were cloned into the pENTR plasmid (Invitrogen), transferred into the Gateway-compatible hsp68-lacZ reporter vector using LR recombination, and validated by Sanger sequencing. NotI digestion was used to excise the vector backbone from the reporter construct. The gel extracted fragment was further diluted and used for pronuclear microinjection of mouse zygotes. Pronuclear microinjection of the DNA was performed by Cyagen Biosciences (USA) using standard procedures. Two rounds of injection were performed for each construct. Embryos were collected and stained for lacZ activity. The stained embryos were fixed with 4% paraformaldehyde overnight at 4°C, washed with phosphate buffer saline, dehydrated, and embedded in paraffin. Ten-micron-thick paraffin sections were obtained using a microtome (Leica RM2165). The air-dried sections were counterstained with eosin (pink stain) and mounted with di-n-butyl phthalate in xylene (DPX, Fischer Scientific). Zeiss Axio Imager Z1 and Leica M205FA were used for imaging and documenting sectioned and whole embryos, respectively.
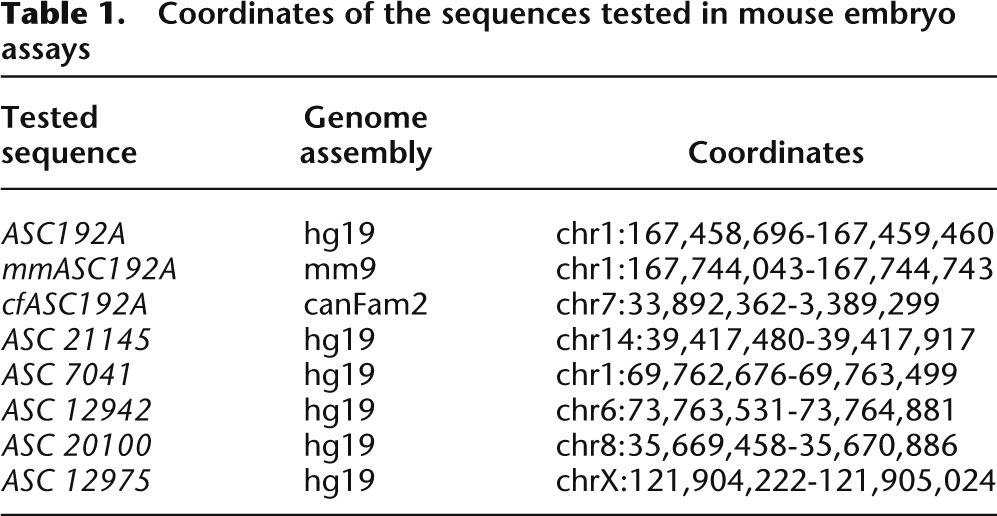
The coordinates of the sequences tested in mouse embryo assays are given in Table 1.
Table 1.
Coordinates of the sequences tested in mouse embryo assays
Motif discovery in L1PA-derived ASC bases
The subregions of ASC21145, ASC7041, ASC12942, ASC20100, and ASC12975 that intersect L1PA13,15,16 repeats were scanned for motifs using MEME (Bailey et al. 2009) with the following parameter settings: “zoops” model of motif occurrence; minw = 6; maxw = 14; nmotifs = 20; and -revcomp. The discovered motifs were matched to motifs in the TRANSFAC, UniPROBE, and JASPAR databases using TOMTOM (Gupta et al. 2007).
The top motif discovered by MEME had an E-value of 0.067. This motif was matched by TOMTOM to the TCF3 UniPROBE motif (P = 0.00037). The fourth MEME motif had an E-value of 6 × 102, which exceeds the conventional E-value threshold of 0.1. We nevertheless followed up on this motif since it had a statistically significant match to a UniPROBE motif (SIX6; P = 0.001).
Chromosome conformation capture (3C)
We performed the 3C assay on ASC192 and neighboring gene promoter regions according to a previously described protocol (Hagège et al. 2007). Briefly, ∼1.5 × 107 H1 human embryonic stem cells were crosslinked at a final concentration of 1% formaldehyde. The cells were lysed and their nuclei isolated. HindIII was used as the primary restriction endonuclease to digest the nuclear samples, and the regions of chromosome contact were ligated using the T4 DNA ligase. Following protein digestion, reverse crosslinking, and purification, physical interactions between genomic regions were detected by quantitative real-time PCR (qPCR) using PCR primers specific for each of the ligated interaction products. Primers were designed using Primer 3 and are listed in Supplemental Table S10. To quantitatively compare signal intensities obtained from different primer sets, a control template containing all possible ligation products in equimolar amounts was used to correct for the PCR efficiency of each primer set. For this purpose, two BACs (CH17-214G02 and CH17-46K14) with minimal overlap spanning the locus of interest were digested and ligated as before. The ligated control fragments were diluted to appropriate concentrations to obtain a final working concentration of 50 ng/mL total DNA, similar to that of the 3C templates. The control templates were also used to draw standard curves for the quantitative real-time PCR as described in the protocol.
Histology and RNA in situ hybridization
Embryos were fixed, dehydrated in graded ethanol, and stored at −20°C. The cDNA clones of Tcf3 (IMAGE clone:2631291) and Six6 (Riken clone: 4832418K20) were linearized with EcoRI and NaeI, respectively, and used as templates for synthesizing antisense DIG-labeled Tcf3 and Six6 RNA probes (DIG RNA labeling kit, Roche). RNA in situ hybridization was performed essentially as previously described (Tribioli et al. 1997) on 10-μm paraffin-embedded embryonic sections (Leica RM 2165 microtome). Following hybridization and washing, sections were stained with NBT/BCIP and exposed overnight at 4°C in the dark according to the manufacturer’s instructions. Sections were washed in PBS, mounted with glycerol/gelatin, and subjected to imaging.
Acknowledgments
We thank Kim Worley and the Marmoset Genome Sequencing and Analysis Consortium for providing the genome assembly; Petra Kraus, Thomas Lufkin, and Axel Vissel for reagents and technical guidance; James Noonan, Nadav Ahituv, and Qianfei Wang for helpful feedback; Meet Singhal for bioinformatic protocols; and Xin Lixia and Toh Jun Hong for assistance with the 3C assay. This work was funded by the Agency for Science, Technology and Research (A*STAR), Singapore.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.168963.113.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzai N, Endou H. 2011. Urate transporters: an evolving field. Semin Nephrol 31: 400–409 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, Kent WJ, Haussler D. 2006. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 441: 87–90 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. . 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28: 1045–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti K, Gasper M, Ou J, Brucato M, Clore-Gronenborn K, Pickel J, Arnheiter H. 2012. A regulatory loop involving PAX6, MITF, and WNT signaling controls retinal pigment epithelium development. PLoS Genet 8: e1002757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. . 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391–1394 [DOI] [PubMed] [Google Scholar]
- Borja D, Manns F, Ho A, Ziebarth NM, Acosta AC, Arrieta-Quintera E, Augusteyn RC, Parel JM. 2010. Refractive power and biometric properties of the nonhuman primate isolated crystalline lens. Invest Ophthalmol Vis Sci 51: 2118–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ, Davidson EH. 1969. Gene regulation for higher cells: a theory. Science 165: 349–357 [DOI] [PubMed] [Google Scholar]
- Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S. 2003. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res 13: 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB 2003. Genetics and the making of Homo sapiens. Nature 422: 849–857 [DOI] [PubMed] [Google Scholar]
- Cartmill M 1974. Rethinking primate origins. Science 184: 436–443 [DOI] [PubMed] [Google Scholar]
- Chuong EB, Rumi MA, Soares MJ, Baker JC. 2013. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat Genet 45: 325–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Program NCS, Green ED, Batzoglou S, Sidow A. 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15: 901–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7: e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza FS, Franchini LF, Rubinstein M. 2013. Exaptation of transposable elements into novel cis-regulatory elements: Is the evidence always strong? Mol Biol Evol 30: 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF 2013. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci 110: 5294–5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF, Sapienza C. 1980. Selfish genes, the phenotype paradigm and genome evolution. Nature 284: 601–603 [DOI] [PubMed] [Google Scholar]
- Eddy SR 2012. The C-value paradox, junk DNA and ENCODE. Curr Biol 22: R898–R899 [DOI] [PubMed] [Google Scholar]
- Enard W, Khaitovich P, Klose J, Zollner S, Heissig F, Giavalisco P, Nieselt-Struwe K, Muchmore E, Varki A, Ravid R, et al. . 2002. Intra- and interspecific variation in primate gene expression patterns. Science 296: 340–343 [DOI] [PubMed] [Google Scholar]
- Erhardt A, Czibere L, Roeske D, Lucae S, Unschuld PG, Ripke S, Specht M, Kohli MA, Kloiber S, Ising M, et al. . 2011. TMEM132D, a new candidate for anxiety phenotypes: evidence from human and mouse studies. Mol Psychiatry 16: 647–663 [DOI] [PubMed] [Google Scholar]
- Feschotte C 2008. Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9: 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. 2007. Quantifying similarity between motifs. Genome Biol 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagège H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, Forné T. 2007. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc 2: 1722–1733 [DOI] [PubMed] [Google Scholar]
- Haygood R, Fedrigo O, Hanson B, Yokoyama KD, Wray GA. 2007. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat Genet 39: 1140–1144 [DOI] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. 2006. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22: 2971–2972 [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci 106: 9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley S, Baggott DM, Hamilton AT, Tran-Gyamfi M, Yang S, Kim J, Gordon L, Branscomb E, Stubbs L. 2006. A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressors. Genome Res 16: 669–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques PÉ, Jeyakani J, Bourque G. 2013. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet 9: e1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, Gamblin RJ, Ooi L, Bruce AW, Donaldson IJ, Westhead DR, Wood IC, Jackson RM, Buckley NJ. 2006. Identification of the REST regulon reveals extensive transposable element-mediated binding site duplication. Nucleic Acids Res 34: 3862–3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas JH 2006. Evolution of the neocortex. Curr Biol 16: R910–R914 [DOI] [PubMed] [Google Scholar]
- Kaas JH 2008. The evolution of the complex sensory and motor systems of the human brain. Brain Res Bull 75: 384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin NH, Shelton SE, Davidson RJ, Kelley AE. 2001. The primate amygdala mediates acute fear but not the behavioral and physiological components of anxious temperament. J Neurosci 21: 2067–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal M, Xie X, Lander ES. 2006. A large family of ancient repeat elements in the human genome is under strong selection. Proc Natl Acad Sci 103: 2740–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D, Rinn J. 2012. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 13: R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan H, Smit A, Boissinot S. 2006. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res 16: 78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116 [DOI] [PubMed] [Google Scholar]
- Kirkwood BJ 2009. Albinism and its implications with vision. Insight 34: 13–16 [PubMed] [Google Scholar]
- Kiyota T, Kato A, Altmann CR, Kato Y. 2008. The POU homeobox protein Oct-1 regulates radial glia formation downstream of Notch signaling. Dev Biol 315: 579–592 [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, Martínez-Cerdeño V. 2006. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci 7: 883–890 [DOI] [PubMed] [Google Scholar]
- Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, Ng HH, Bourque G. 2010. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42: 631–634 [DOI] [PubMed] [Google Scholar]
- Lidow MS, Goldman-Rakic PS, Rakic P. 1991. Synchronized overproduction of neurotransmitter receptors in diverse regions of the primate cerebral cortex. Proc Natl Acad Sci 88: 10218–10221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Thurig S, Mohamed O, Dufort D, Wallace VA. 2006. Mapping canonical Wnt signaling in the developing and adult retina. Invest Ophthalmol Vis Sci 47: 5088–5097 [DOI] [PubMed] [Google Scholar]
- Lowe CB, Bejerano G, Haussler D. 2007. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc Natl Acad Sci 104: 8005–8010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch VJ, Leclerc RD, May G, Wagner GP. 2011. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat Genet 43: 1154–1159 [DOI] [PubMed] [Google Scholar]
- Mariño-Ramírez L, Jordan IK. 2006. Transposable element derived DNase I-hypersensitive sites in the human genome. Biol Direct 1: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Marmoset Genome Sequencing and Analysis Consortium 2014 The common marmoset genome provides insight into primate biology and evolution. Nat Genet doi: 10.1038/ng.3042.
- Marsden CD 1961. Pigmentation in the nucleus substantiae nigrae of mammals. J Anat 95: 256–261 [PMC free article] [PubMed] [Google Scholar]
- Martínez-Morales JR, Rodrigo I, Bovolenta P. 2004. Eye development: a view from the retina pigmented epithelium. Bioessays 26: 766–777 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, et al. . 2011. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471: 216–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meader S, Ponting CP, Lunter G. 2010. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res 20: 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, et al. . 2007. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447: 167–177 [DOI] [PubMed] [Google Scholar]
- Nickla DL, Wallman J. 2010. The multifunctional choroid. Prog Retin Eye Res 29: 144–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu DK, Jiang L. 2013. Can ENCODE tell us how much junk DNA we carry in our genome? Biochem Biophys Res Commun 430: 1340–1343 [DOI] [PubMed] [Google Scholar]
- Oda M, Satta Y, Takenaka O, Takahata N. 2002. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 19: 640–653 [DOI] [PubMed] [Google Scholar]
- Oetting WS, King RA. 1999. Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hum Mutat 13: 99–115 [DOI] [PubMed] [Google Scholar]
- Olsen GJ, Matsuda H, Hagstrom R, Overbeek R. 1994. fastDNAmL: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput Appl Biosci 10: 41–48 [DOI] [PubMed] [Google Scholar]
- Orgel LE, Crick FH. 1980. Selfish DNA: the ultimate parasite. Nature 284: 604–607 [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. . 2006. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444: 499–502 [DOI] [PubMed] [Google Scholar]
- Pierron D, Wildman DE, Hüttemann M, Letellier T, Grossman LI. 2012. Evolution of the couple cytochrome c and cytochrome c oxidase in primates. Adv Exp Med Biol 748: 185–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, et al. . 2006. Forces shaping the fastest evolving regions in the human genome. PLoS Genet 2: e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Noonan JP, Paabo S, Rubin EM. 2006a. Accelerated evolution of conserved noncoding sequences in humans. Science 314: 786. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin EM, Couronne O, Pennacchio LA. 2006b. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res 16: 855–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, Fitzpatrick DR, Afzal V, et al. . 2008. Human-specific gain of function in a developmental enhancer. Science 321: 1346–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R, Romanish MT, Mager DL. 2012. Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet 46: 21–42 [DOI] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. . 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463: 237–240 [DOI] [PubMed] [Google Scholar]
- Schmidt D, Schwalie PC, Wilson MD, Ballester B, Goncalves A, Kutter C, Brown GD, Marshall A, Flicek P, Odom DT. 2012. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148: 335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwirtlich M, Kwakowsky A, Emri Z, Antal K, Lacza Z, Cselenyák A, Katarova Z, Szabó G. 2011. GABAergic signaling in primary lens epithelial and lentoid cells and its involvement in intracellular Ca2+ modulation. Cell Calcium 50: 381–392 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. . 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro K, Teissier A, Kobayashi N, Nakanishi A, Sasaki T, Yan K, Tarabykin V, Vigier L, Sumiyama K, Hirakawa M, et al. . 2011. A mammalian conserved element derived from SINE displays enhancer properties recapitulating Satb2 expression in early-born callosal projection neurons. PLoS ONE 6: e28497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay SK, Blythe J, Lipovich L. 2009. Global discovery of primate-specific genes in the human genome. Proc Natl Acad Sci 106: 12019–12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorou E, Dalembert G, Heffelfinger C, White E, Weissman S, Corcoran L, Snyder M. 2009. A high throughput embryonic stem cell screen identifies Oct-2 as a bifunctional regulator of neuronal differentiation. Genes Dev 23: 575–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH, Schneider S. 2011. Coevolution of retroelements and tandem zinc finger genes. Genome Res 21: 1800–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. . 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribioli C, Frasch M, Lufkin T. 1997. Bapx1: An evolutionary conserved homologue of the Drosophila bagpipe homeobox gene is expressed in splanchnic mesoderm and the embryonic skeleton. Mech Dev 65: 145–162 [DOI] [PubMed] [Google Scholar]
- Varki A, Geschwind DH, Eichler EE. 2008. Explaining human uniqueness: genome interactions with environment, behaviour and culture. Nat Rev Genet 9: 749–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QF, Prabhakar S, Wang Q, Moses AM, Chanan S, Brown M, Eisen MB, Cheng JF, Rubin EM, Boffelli D. 2006. Primate-specific evolution of an LDLR enhancer. Genome Biol 7: R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, Burgess SM, Brachmann RK, Haussler D. 2007. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci 104: 18613–18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M. 2013. Response to comment on “Evidence of abundant purifying selection in humans for recently acquired regulatory functions”. Science 340: 682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BA, Kay RF, Kirk EC. 2010. New perspectives on anthropoid origins. Proc Natl Acad Sci 107: 4797–4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL, et al. . 2013. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat Genet 45: 836–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. 2007. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci 104: 19416–19421 [DOI] [PMC free article] [PubMed] [Google Scholar]