

Abstract
Olfactory receptors (ORs) detect odors in the environment, and OR genes constitute the largest multigene family in mammals. Numbers of OR genes vary greatly among species—reflecting the respective species' lifestyles—and this variation is caused by frequent gene gains and losses during evolution. However, whether the extent of gene gains/losses varies among individual gene lineages and what might generate such variation is unknown. To answer these questions, we used a newly developed phylogeny-based method to classify >10,000 intact OR genes from 13 placental mammal species into 781 orthologous gene groups (OGGs); we then compared the OGGs. Interestingly, African elephants had a surprisingly large repertoire (∼2000) of functional OR genes encoded in enlarged gene clusters. Additionally, OR gene lineages that experienced more gene duplication had weaker purifying selection, and Class II OR genes have evolved more dynamically than those in Class I. Some OGGs were highly expanded in a lineage-specific manner, while only three OGGs showed complete one-to-one orthology among the 13 species without any gene gains/losses. These three OGGs also exhibited highly conserved amino acid sequences; therefore, ORs in these OGGs may have physiologically important functions common to every placental mammal. This study provides a basis for inferring OR functions from evolutionary trajectory.
Olfaction is essential for the survival of most mammals. It is used for finding foods, avoiding dangers, identifying mates and offspring, and identifying marked territory (Buck and Axel 1991; Nei et al. 2008; Touhara and Vosshall 2009; Niimura 2012). Various odor molecules in the environment are detected by olfactory receptors (ORs) expressed in the olfactory epithelium of the nasal cavity. Using rats, Buck and Axel were the first to identify mammalian OR genes (Buck and Axel 1991). They estimated that the murine genome may contain as many as ∼1000 OR genes, and further studies confirmed that OR genes constitute the largest multigene family in mammals. ORs are G-protein coupled receptors (GPCRs) and have seven α-helical transmembrane regions. Mammalian ORs can be clearly classified into two groups, Class I or Class II, based on amino acid sequence. The functional difference between the two classes is not well understood, but apparently Class I ORs tend to bind hydrophilic odorants, and Class II ORs hydrophobic odorants (Saito et al. 2009).
It is generally thought that the olfactory system utilizes “combinatorial coding” (Malnic et al. 1999). In this model, each OR does not have a one-to-one relationship with an odorant; instead, one odorant may be recognized by multiple ORs, and one OR may recognize multiple odorants. Ultimately, different odorants are represented as different combinations of activated ORs. Researchers have worked intensively for >15 yr to identify ligands for ORs (Krautwurst et al. 1998; Touhara et al. 1999; Yoshikawa et al. 2013; Shirasu et al. 2014). Saito et al. (2009) performed the most comprehensive of these studies; in that study, they screened 93 odorants against 464 ORs and successfully deorphanized 10 human and 52 mouse ORs. Their results indicate that the combinatorial coding scheme is correct (Saito et al. 2009). They also demonstrated that some ORs are “generalists” that are broadly tuned and bind to a wide variety of odorants, while others are “specialist” ORs that are narrowly tuned and bind to only a limited number of structurally related odorants. However, most ORs remain orphans, and our knowledge and understanding of OR–odorant relationships is still quite limited.
Bioinformatic analyses of diverse mammalian genome sequences revealed that the numbers of OR genes vary greatly among species (Niimura 2012). Apparently, the number of OR genes is affected by each species’ environment (Hayden et al. 2010). Mice and rats have 1000–1200 functional OR genes in their respective genomes, as Buck and Axel (1991) have correctly estimated, and cows and opossums have similar numbers (Niimura and Nei 2007). However, higher primates generally have much fewer OR genes. The human genome harbors ∼400 functional OR genes, and interestingly, it also contains >400 OR pseudogenes (Niimura and Nei 2003; Matsui et al. 2010). Chimpanzees have nearly the same number of functional OR genes as do humans; orangutans and macaques have even fewer OR genes (Matsui et al. 2010). These observations are thought to reflect that higher primates heavily rely on vision and less on olfaction, although the timing and reason(s) for OR gene losses in primate evolution are unclear (Matsui et al. 2010). Platypuses are semi-aquatic egg-laying mammals, and they too have a relatively small repertoire of functional OR genes (∼350) (Niimura and Nei 2007). They have electroreceptors in their bills and can sense subtle changes in electric fields. Therefore, it may be that different modalities of senses affect each other, and development of one sense can coincide with retrogression of another sense.
In addition to the variation in OR gene number among species, OR genes are characterized by frequent gene gains and losses in the course of evolution. A previous study of genome sequences from seven mammals indicates that hundreds of gains and losses of OR genes have occurred in an order-specific manner (Niimura and Nei 2007). Consequently, the OR gene family is recognized as an extreme example of a gene family subject to birth-and-death evolution (Nei and Rooney 2005), in which new genes are created by repeated gene duplication, while others are lost by pseudogenization. As a whole, the repertoire of OR genes has changed dynamically during mammalian evolution. However, there may be some variation among individual OR genes in the extent of gene gains and losses. That is, some ancestral OR genes may have yielded flourishing lineages with large numbers of descendants, while others may have become extinct soon after birth. Other genes may be evolutionarily stable without any gene duplications or losses.
We cannot foresee the evolutionary fate of extant genes; however, we can trace back the evolutionary history of genes by comparing genes among species. In this study, we define an orthologous gene group (OGG) as all extant descendant genes originated from a single gene in the most recent common ancestor (MRCA) of a given set of species (Gabaldon and Koonin 2013). By comparing among OGGs, therefore, we can investigate differences in the extent of gene gains and losses among different gene lineages derived from individual founding genes in the ancestral species.
To identify orthologous relationships, pairwise methods based on reciprocal best hits (RBHs) are often used, but RBH-based methods give erroneous results when lineage-specific gene duplications and/or gene losses have taken place (Gabaldon 2008). Therefore, we adopted a phylogeny-based method. However, identification of OGGs of OR genes among multiple mammalian species is not straightforward for three main reasons. (1) The number of genes for inferring orthology and paralogy is very large. (2) Orthologous relationships between genes from two species are usually not one-to-one; they can be multiple-to-multiple or zero-to-multiple relationships, among others, because of frequent lineage-specific gene duplications and losses. (3) The radiation of most orders of placental mammals occurred during a relatively short time period around the Cretaceous-Paleogene boundary (Meredith et al. 2011; O’Leary et al. 2013); therefore, gene trees are often incongruent with species trees due to incomplete lineage sorting, statistical noises, or both. To overcome these difficulties, we invented a novel method for OGG identification that is semi-automated and based on phylogeny.
We sought to understand the extent of variation in evolutionary dynamics among individual OR genes and to identify factors that generate such variation. For these purposes, we first comprehensively identified the OR gene repertoires in 13 species of placental mammals for which deep-coverage genome sequences are available. We then used a phylogeny-based method to identify OGGs among these 13 species and finally compared among OGGs. Our findings demonstrate that evolutionary fates varied greatly among OGGs and that this variation was associated with OR class, extent of functional constraints, ligand specificity, and OR expression patterns.
Results
OR genes in 13 species of placental mammals
We examined genome sequences from 13 species of placental mammals for which deep-coverage genome sequences are available. These 13 species belong to seven different mammalian orders, and African elephants are located at the most basal position in the phylogeny of these species (Supplemental Fig. 1). In all, >20,000 OR genes were identified in the genomes of these 13 species (Fig. 1A). Among these OR genes, those from African elephant, horse, rabbit, and guinea pig were newly identified in this study, and those from cow or mouse were updated because we used the latest genome data. Nucleotide and predicted amino acid sequences for OR genes that were newly identified or updated in this study are provided in Supplemental Data Sets S1 and S2, respectively. Each OR gene sequence was classified into one of three categories: intact gene, truncated gene, or pseudogene (Niimura and Nei 2007). For each species examined, the number of truncated genes is relatively small (Fig. 1A); therefore, the number of intact genes is expected to be a good estimate of the number of functional OR genes.
Figure 1.
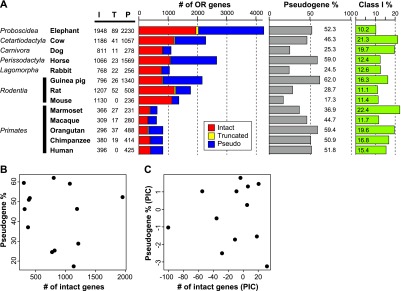
Numbers of OR genes in the genome sequence from 13 placental mammal species. (A) “I,” “T,” and “P” represent the number of intact genes, truncated genes, and pseudogenes, respectively. An intact gene was defined as a sequence starting from an initiation codon and ending with a stop codon that did not contain any disrupting mutations. A pseudogene was defined as a sequence with a nonsense mutation, frameshift, deletion within conserved regions, or some combination thereof. A truncated gene was defined as a partial, intact sequence located at a contig end. An intact gene was assumed to be functional, while a truncated gene was presumed to be either a functional gene or a pseudogene. The fraction of OR pseudogenes was calculated as the number of OR pseudogenes divided by the total number of OR genes. The fraction of Class I genes was calculated as the number of intact Class I genes divided by the total number of intact OR genes. Dog and rat data were taken from Niimura and Nei (2007), and the data for the five primates were from Matsui et al. (2010). (B) There was no significant correlation between the number of intact OR genes within a genome and the fraction of OR pseudogenes within that same genome among these 13 species (r = −0.137; P = 0.655); (C) again, there was no significant correlation (r = −0.003; P = 0.992) after the comparative method of phylogenetically independent constants (PICs) was used to remove phylogenetic dependence (Felsenstein 1985).
The total number of OR genes vary widely among species (Fig. 1A). Notably, African elephants had by far the largest repertoire (1948) of intact OR genes ever identified within a single species (Fig. 1A) and more than rats, which have the largest previously identified repertoire (Niimura 2012). African elephants had an even larger number of OR pseudogenes (2230), and this genome contained, in all, >4200 OR genes.
The fraction of OR pseudogenes, like the total number of OR genes, varies widely among species (Fig. 1A). Guinea pigs had the largest fraction of OR pseudogenes (∼62%). The fraction of OR pseudogenes within a genome did not correlate with the number of intact OR genes (r = −0.137; P = 0.655) (Fig. 1B). Once phylogenetic dependence was removed (Felsenstein 1985), any correlation completely disappeared (r = −0.003; P = 0.992) (Fig. 1C). Therefore, the fraction of OR pseudogenes could not be used to predict the number of functional OR genes in a given genome. In contrast, the absolute number of pseudogenes, unlike the fraction of pseudogenes, did correlate with the number of intact genes even after removing phylogenetic dependence (r = 0.59; P = 0.042) (Supplemental Fig. S2).
Identification of orthologous gene groups (OGGs)
Using OR genes from these 13 species, we identified OGGs using a novel phylogeny-based method (see Methods; Supplemental Fig. S3). We classified 10,659 intact OR genes from these 13 species into 781 OGGs. Truncated genes and pseudogenes were also assigned to the 781 OGGs based on sequence similarity to constituent intact genes (see Methods). By definition, all genes in a given OGG are supposed to have originated from a single ancestral gene in the MRCA of placental mammals. Therefore, we assert that the MRCA of placental mammals may have had ∼781 functional OR genes. This estimate was in good agreement with the previous estimate (800) (Niimura and Nei 2007), which was obtained with the reconciled-tree method.
Each OR gene was clearly classified into Class I or Class II, and 145 OGGs (18.6%) contained Class I genes and 636 (81.4%) contained Class II genes. Therefore, ∼19% of all OR genes in the MRCA of placental mammals were presumed to be in Class I; this value was within the range of the percentages of Class I OR genes in the extant species (11%–22%) (Fig. 1A). Each Class I and each Class II OGG was named separately in a descending order based on the number of constituent intact genes. For example, OGG2-1 represents the OGG containing the largest number of intact Class II genes. Gene names within each OGG and the number of genes from each species within each OGG are provided in Supplemental Data Set S3 and Supplemental Table S1, respectively.
Most OGGs contained a relatively small number of OR genes. The mean number and median number of intact OR genes per OGG were 13.6 and 11, respectively (Fig. 2A), and for pseudogenes, the mean and median were 11.9 and 7, respectively (Fig. 2B). For all OGGs, the number of intact genes and the number of pseudogenes within an OGG were correlated to each other (r = 0.731) (Fig. 2C); therefore, OGGs with many intact genes also tended to contain many pseudogenes.
Figure 2.

Distribution of the total number of OR genes—(A) intact genes and (B) pseudogenes—belonging to each of the 781 OGGs found among 13 placental mammal species. Red and blue indicate Class I and Class II genes, respectively (A–F). (C) The number of intact genes was positively correlated with the number of pseudogenes belonging to the respective OGG (r = 0.731); the dashed line indicates the regression line. (D,E) Boxplots of comparison between Class I (“1”; red) and Class II (“2”; blue) OGGs for estimated numbers of gene gains and losses (D) and estimated ω values (E). (*) P < 0.05, (**) P < 0.01, and (***) P < 0.001. (F) The ω value for an OGG was positively correlated with the number of intact genes in the respective OGG (r = 0.346; P < 2.2 × 10−16); the dashed line indicates the regression line.
Comparison between Class I and Class II genes and extent of purifying selection
Class II OGGs generally contained more genes than did Class I OGGs (Fig. 2A–C), but the difference between the two classes was not significant for the number of intact genes (P = 0.14, Wilcoxon test) or of pseudogenes (P = 0.099). For each OGG separately, we then used the reconciled-tree method (Niimura and Nei 2007) to estimate the numbers of gene gains and losses during the evolution of placental mammals (see Methods). The number of gene gains (P = 0.031, Wilcoxon test) and the total number of gene gains and losses (P = 0.0085) were significantly larger for Class II genes than for Class I genes (Fig. 2D).
Next, we used the maximum likelihood method implemented in PAML (Yang and Nielsen 2000) to calculate ω, the ratio of nonsynonymous to synonymous change rates, for each OGG separately. The ω value reflected the extent of purifying selection. Notably, the ω values were significantly smaller for Class I genes (median ω = 0.196) than for Class II genes (median ω = 0.230; P = 0.00016, Wilcoxon test) (Fig. 2E). These observations indicated that evolution of Class II genes was more dynamic than that of Class I genes. The ω value for each OGG was positively correlated with the number of gene gains in the respective OGG (r = 0.35; P < 2.2 × 10−16) (Fig. 2F). The ω value was also correlated with the number of intact genes (r = 0.27; P = 3.8 × 10−13) (Supplemental Fig. S4). In contrast, the correlation between ω and the number of gene losses was not significant (r = 0.06; P = 0.095) (Supplemental Fig. S4). These observations indicated that gene lineages that had experienced more duplication tended to be under weaker evolutionary constraints.
Expanded OGGs
Some OGGs were very large, indicating that some individual ancestral OR genes had each generated >100 descendant genes in these 13 species (see Fig. 2A,C; Supplemental Table S1). OGG2-1 contained the largest number (159) of intact OR genes. Moreover, OGG2-1 included 25 dog, 43 cow, 28 rabbit, and nine orangutan genes; this OGG also contained the largest number of genes from each of these species (Supplemental Table S2). Phylogenetic analyses suggested that gene expansion has occurred independently in each lineage (Fig. 3A,B; Supplemental Fig. S5A). The second largest OGG, OGG2-2, contained 84 intact elephant genes; therefore, this gene lineage has expanded drastically in African elephants (Fig. 3C,D; Supplemental Fig. S5B). OGG2-3, the third largest OGG, contained the largest number (388) of pseudogenes. Reportedly, the one human intact gene belonging to OGG2-3, named OR7E24, has generated numerous pseudogenes that are scattered throughout the human genome (collectively called H* OR genes) (Newman and Trask 2003; Niimura and Nei 2003; Go and Niimura 2008). Interestingly, this group of pseudogenes is also highly expanded in nonhuman primates and even in nonprimate mammals (Supplemental Table S1).
Figure 3.

Expanded OGGs. Neighbor-joining (NJ) phylogenetic trees were constructed from all intact OR genes in OGG2-1 (A) and OGG2-2 (C). In each tree (A,C), a colored symbol indicates a gene from the species depicted in B and D. Each scale bar indicates the number of amino acid substitutions per site. Gene names and bootstrap values are shown in Supplemental Figure S5A,B. (B,D) Number of gene gains and losses in each branch and the number of genes at ancestral nodes (shown in cyan circles) calculated from the OGG2-1 (A) and OGG2-2 (C) trees, respectively, by the reconciled-tree method (Niimura and Nei 2007). A number in a yellow box indicates the number of intact OR genes in each species belonging to each OGG. (E) For each OGG, the total number of intact genes in elephant and mouse within respective OGGs was negatively correlated with amino acid sequence identity between elephant and mouse among intact OR genes within the respective OGGs (rs = −0.52; P < 2.2 × 10−16). In all, 414 OGGs that contained at least one intact gene from both elephant and mouse were considered. When an OGG included two or more genes from either or both species, the mean of the amino acid sequence identities for all possible interspecies combinations of genes was used.
We calculated an OGG-specific and species-specific “expansion rate” for each OGG within a species; each rate represented the extent of lineage-specific gene expansion with accounting for phylogenetic relatedness among the 13 species (Supplemental Table S3). The results demonstrated that elephant-specific expansions have occurred frequently. The OGG with the largest expansion rate was OGG2-22; it contained 46 intact genes from elephant and just one intact gene each from cow, dog, rabbit, rat, and mouse, and none from all other species (Supplemental Fig. S5C). OGG2-2 ranked second.
We also examined the relationships between lineage-specific gene expansion and amino acid sequence similarity. In an analysis of 414 OGGs that were found in both elephant and mouse, mean between-species within-OGG amino acid sequence identities were negatively correlated with the total number of intact genes within the respective OGGs of both species (Spearman’s rank correlation coefficient rs = −0.52; P < 2.2 × 10−16) (Fig. 3E). Similar negative correlations were observed with each comparison between elephant and another species (Supplemental Fig. S6). Therefore, the genes that experienced more lineage-specific gene duplications tended to show lower amino acid sequence similarities.
OGGs showing one-to-one orthology
Of the 781 OGGs, only 28 contained genes from each of the 13 species. Among these 28 OGGs, only three—OGG1-44, OGG1-45, and OGG2-256—showed complete one-to-one orthologous relationships among all 13 species; each of these OGGs contained only one intact OR gene from each of the 13 species, and none contained any truncated genes or any pseudogenes. Phylogenetic analyses showed that each gene tree (OGG1-44, OGG1-45, and OGG2-256) was consistent with the species tree, suggesting that, for each of these OGGs, all member genes were truly orthologous to one another (Fig. 4A). The gene lineages in these OGGs may not have undergone any gene duplication or loss events.
Figure 4.

Conserved OGGs showing complete one-to-one orthology. (A) NJ phylogenetic trees for OGG1-44, OGG1-45, and OGG2-256. A colored symbol of a gene name indicates a species depicted in Figure 3B,D. Each scale bar indicates the number of amino acid substitutions per site. Bootstrap values obtained from 500 resamplings are shown only for the nodes with bootstrap values >70%. (B) Distribution of amino acid sequence identities between intact human and mouse OR genes for 252 OGGs containing at least one intact gene from both human and mouse. Note that pseudogenes and truncated genes were not used for the calculation of amino acid sequence identity. When an OGG included two or more genes from either or both species, the mean of the amino acid sequence identities for all possible interspecies combinations of genes was used. The OGGs with the three highest amino acid sequence identity values and that with the lowest value are shown with the respective percent identity. The mean and the median amino acid sequence identity among the 252 OGGs are 81.3% and 82.1%, respectively.
The distribution of amino acid sequence identities between human and mouse intact ORs for OGGs containing at least one intact gene from both human and mouse is shown in Figure 4B. Remarkably, the three OGGs that showed the highest identity in amino acid sequence were the exact same three OGGs that showed complete one-to-one orthology. Therefore, these three OGGs are conservative not only in gene number during evolution, but also in amino acid sequences. These observations indicated that the function of each of these ORs is important and common to every placental mammal (see Discussion). Conversely, OGG2-3, which contained the largest number of pseudogenes (Fig. 2C), showed the least identity in amino acid sequence between human and mouse ORs (Fig. 4B).
OR gene clusters in African elephants
Because the African elephant genome contained the largest OR gene repertoire yet reported, we investigated the organization of elephant OR gene clusters in greater detail. Humans and mice have similar numbers of OR gene clusters, although the mouse OR gene repertoire is much larger than the human repertoire (Niimura and Nei 2005; Aloni et al. 2006). Here, we compared OR gene clusters in African elephant with those in mouse to determine whether the elephant genome contained a larger number of OR gene clusters than the mouse genome.
We defined an OR gene cluster using the criterion that any distances between two neighboring OR genes in a cluster are <500 kb (Niimura and Nei 2003). We denote a cluster containing five or more OR genes as a “5+ cluster.” The elephant genome contained 148 5+ clusters, which is many more than the mouse (34) or human (35) genomes (Supplemental Table S4). However, because the elephant genome sequence data are not assembled into chromosomes but comprise many (>2000) scaffolds, individual OR gene clusters were often fragmented into multiple scaffolds; therefore, we may have greatly overestimated the number of clusters. When an OR gene cluster was located near the end of a scaffold, the cluster may have been a part of a larger OR gene cluster. For this reason, we designated OR gene clusters that were located near the end of a scaffold as “truncated clusters” and distinguished them from “intact clusters”; moreover, we further classified truncated clusters into “one-end” and “both-end” clusters (Supplemental Table S4). Among the 148 5+ OR gene clusters in elephant, there were 22 intact, 24 one-end truncated, and 102 both-end truncated clusters (Supplemental Table S4). Therefore, most OR gene clusters were fragmented in the current data set of the elephant genome. Under the assumption that each of the truncated clusters was embedded within a larger cluster, we roughly estimated that the number of 5+ clusters in the African elephant genome was 22 + 24/2 = 34. This number was very close to the number of 5+ clusters in mouse (34) or human (35).
Among the 34 5+ clusters in mouse and the 148 in elephant, we could identify 5+ cluster counterparts in the other species for 33 of the mouse and 144 of the elephant clusters (Supplemental Table S5). Therefore, species-specific clusters were very rare. The order of OR genes in each cluster was generally well conserved between elephant and mouse (Fig. 5). An OR gene cluster on mouse chromosome 7 contained 158 Class I OR genes and occupied a 2.89-Mb genomic region (Fig. 5A; Supplemental Table S5). This mouse cluster corresponded to nine 5+ clusters in elephant; among the nine clusters, two clusters on scaffold79 and scaffold21 were one-end truncated, while the other seven clusters were both-end truncated (Fig. 5A). Therefore, these nine elephant clusters may have constituted one large cluster. In total, the nine clusters contained 353 OR genes and spanned a 5.42-Mb region in the elephant genome. Similarly, a 2.48-Mb mouse OR gene cluster on chromosome 9 contained 118 Class II OR genes and corresponded to seven 5+ clusters in elephant; two of the elephant clusters (on scaffold50 and scaffold58) were one-end truncated, while the others were both-end truncated (Fig. 5B; Supplemental Table S5). These seven clusters encompassed 286 OR genes and were 7.93 Mb long in total. Therefore, again, apparently one large cluster comprised these seven clusters in the elephant genome. All these lines of evidence indicated that (1) the number of OR gene clusters in African elephant was similar to that in mouse in spite of the difference in repertoire size between the two species, and (2) each OR gene cluster tended to be larger in elephant than in mouse.
Figure 5.

Comparison of OR gene clusters between mouse and African elephant. (A) A mouse cluster on chromosome 7 (Mm7.6) corresponded to nine elephant clusters, and (B) a mouse cluster on chromosome 9 (Mm9.3) corresponded to seven elephant clusters (see Supplemental Table S5). Each horizontal line represents a mouse chromosome (top) or a scaffold of the African elephant genome (bottom). The position of each OR gene is represented by a colored vertical bar above or below a horizontal line, the latter indicating the opposite transcriptional direction to the former. Long, medium, and short vertical bars depict an intact gene, a truncated gene, or a pseudogene, respectively. Each bar is colored according to the OGG to which the OR gene belongs; the color code chart is at the bottom of the figure. Class I OGGs are colored between red and yellow in the color chart in order of OGG numbers, while Class II OGGs are colored between yellow and red. When a mouse gene and an elephant gene belong to the same OGG, the two genes are connected by a gray line. The scaffold number for each elephant cluster is shown below the diagram; for example, “s79” indicates scaffold79. A scaffold with an asterisk (e.g., s277*) indicates that the respective scaffold is drawn in reverse orientation. A black vertical bar on a horizontal line is shown at intervals of 1 Mb. (A,B) The rightmost and the leftmost elephant scaffolds (s79 and s21 in A and s50 and s58 in B) contain one-end truncated clusters, while the others contain both-end truncated clusters. A dashed horizontal line indicates that DNA sequences are omitted. The entire length is drawn for a scaffold containing a both-end truncated cluster.
Gains and losses of OR genes during evolution
The numbers of putative OR gene gains and losses among the 781 OGGs were summed to estimate the total numbers of gene gains and losses in each branch during the evolution of placental mammals. In agreement with the previous study (Niimura and Nei 2007), the results showed that hundreds of gains and losses of OR genes have occurred in each taxonomic lineage (Fig. 6). Consequently, two species with similar numbers of OR genes may have very different OR gene repertoires. For example, dogs and guinea pigs each had ∼800 OR genes, but only ∼51% of the OR genes in these two species were shared in common (Supplemental Fig. S7). Moreover, each of the 13 species has apparently lost hundreds of the functional OR genes that were present in the MRCA of placental mammals (Fig. 6). Primates have lost more than half of the putative functional OR genes in the MRCA, and notably orangutans have lost ∼70% (= 547/781) of them.
Figure 6.

Changes in the number of OR genes during the evolution of placental mammals. Each number in a yellow box indicates the number of intact OR genes in an extant species. Each number in a cyan oval represents the number of functional OR genes in an ancestral node estimated by the reconciled-tree method (Niimura and Nei 2007). Estimated numbers of gene gains and gene losses in each branch are also shown. Black and orange bars to the right of a species name indicate the number of gene gains and that of gene losses, respectively, compared with the 781 ancestral OR genes that were present in the MRCA of placental mammals. For example, 462 out of the 781 OR genes in the MRCA were lost in the human lineage, but 77 gene gains also occurred and resulted in the current human repertoire of 396 intact OR genes. Note that the number of gene losses in a black bar is not equal to the sum of gene losses in the branches from the MRCA to a given species, because the number of gene losses at each branch includes that of gene losses that occurred after gene duplication. For the same reason, the number of gene gains in an orange bar is not the same as the total number of gene gains in the branches from the MRCA to the species considered. The divergence time at each node was obtained from TimeTree (http://www.timetree.org/) (see Supplemental Fig. S1; Hedges et al. 2006).
We also estimated the rate of gene birth β and the rate of gene death δ in each taxonomic branch. β and δ are defined as the number of gene gains or gene losses, respectively, per million years per gene; both were assumed to be constant along each branch. We developed a novel method for calculating β and δ from the number of gene gains and losses by solving simultaneous differential equations (see Methods for details). The results showed that β became faster in the elephant and murine lineages, while δ accelerated in the primate lineages (Supplemental Fig. S8). The weighted means of the birth and death rates during the evolution of placental mammals were calculated to be 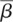 = 0.0062 and
= 0.0062 and 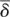 = 0.0059 (per gene per million years), respectively. These values were considerably larger than the previously reported value of the average genomic turnover (birth and death) rate among all mammalian gene families, 0.0016 per gene per million years (Demuth et al. 2006).
= 0.0059 (per gene per million years), respectively. These values were considerably larger than the previously reported value of the average genomic turnover (birth and death) rate among all mammalian gene families, 0.0016 per gene per million years (Demuth et al. 2006).
Discussion
In this study, we found that African elephants have a surprisingly large repertoire of OR genes. The African elephant genome contained ∼2000 functional genes and >2200 pseudogenes, which is by far the largest OR gene repertoire among the genomes examined. The large repertoire of elephant OR genes might be attributed to elephants’ heavy reliance on olfaction in various contexts, including foraging, social communication, and reproduction (Langbauer 2000; Rasmussen and Krishnamurthy 2000). In fact, the Asian elephant (Elephas maximus) is among the few mammalian species for which a sex pheromone has been chemically identified (Rasmussen et al. 1996, 1997). African and Asian elephants possess a specific scent gland, called the temporal gland, behind each eye, and male elephants exude an oily odoriferous secretion from the temporal gland annually during musth, which is characterized by increased aggressiveness and elevated levels of testosterone (Rajaram and Krishnamurthy 2003). Neuroanatomical studies also indicate that elephants have well-developed olfactory systems that include large olfactory bulbs and large olfactory areas in the brain (Shoshani et al. 2006).
Recently, some behavioral tests have been conducted to assess the olfactory ability of Asian elephants. Rizvanovic and colleagues showed that Asian elephants successfully discriminated between 12 enantiomeric odor pairs and between 12 other odor pairs of aliphatic alcohols, aldehydes, ketones, and carboxylic acids, each of them having only a one-carbon difference between pair members (Rizvanovic et al. 2013). These findings indicate that elephants perform at least as well as mice and clearly better than humans, pigtail macaques, or squirrel monkeys in olfaction tests. As yet, no systematic studies assessing olfactory capabilities in African elephants have been published. However, African elephants can reportedly distinguish between two Kenyan ethnic groups—the Maasai, whose young men demonstrate virility by spearing elephants, and the Kamba, who are agricultural people that pose little threat to elephants—by using olfactory cues (Bates et al. 2007). Additionally, African elephants can recognize possibly up to 30 individual family members from olfactory cues in mixtures of urine and earth (Bates et al. 2008).
Together, this evidence supports that elephants have a superior sense of smell, and their large repertoires of OR genes are consistent with this conclusion. It is unclear which aspects of olfactory ability the number of OR genes reflects, but it is reasonable to assume that a species with a larger number of OR genes can discriminate among more subtle differences in structurally related odorants, and that the number of OR genes determines the resolution of the olfactory world rather than the sensitivity to a given odor.
We found that an ancestral gene of OGG2-2 had been specifically expanded in the African elephant lineage. Humans, chimpanzees, and macaques each have only one intact gene belonging to OGG2-2. The human gene (OR8K3) and its chimpanzee and macaque orthologs each reportedly bind (+)-menthol (Adipietro et al. 2012). Mice have 17 OR genes belonging to OGG2-2, and all are located in one big cluster on chromosome 2. Among the 17 mouse ORs, one (OLFR1079) is activated by some enantiomeric pairs—including (+)- and (−)-camphor and (+)- and (−)-carvone—with a different sensitivity between enantiomers (Saito et al. 2009). The functions of each of the 84 African elephant ORs in OGG2-2 are unknown, but the discrimination of the ligands of these ORs may be important for adaptation to the environmental and social conditions of elephants.
We showed that the fraction of OR pseudogenes within a genome did not correlate with the number of intact OR genes in that genome (Fig. 1B,C), indicating that the fraction of OR pseudogenes is a very poor indicator of the olfactory ability of a given species. If (1) no gene duplication has occurred and (2) genes that have undergone pseudogenization remain in the genome during mammalian evolution, then the fractions of OR pseudogenes would be negatively correlated with the number of functional genes. However, this scenario is unlikely because the fraction of OR pseudogenes could easily change during evolution due to frequent gene losses and elimination of pseudogenes from the genome. In fact, the African elephant and human genomes show nearly the same fraction of OR pseudogenes, but the number of functional genes is approximately fivefold larger in African elephant than in human (Fig. 1A).
Nevertheless, some studies have compared the fractions of OR pseudogenes among several species in order to compare their olfactory abilities (Gilad et al. 2004; Kishida et al. 2007; Hayden et al. 2010; Kishida and Hikida 2010). For example, Gilad and colleagues examined the fraction of pseudogenes among 100 randomly sequenced OR genes from each of the 19 primate species. They found that the fractions of OR pseudogenes are significantly higher in primate species with full trichromatic vision (catarrhines and howler monkey) than in other primates and mammals (Gilad et al. 2004). Based on this observation, they concluded that loss of functional OR genes coincided with the acquisition of full trichromatic vision during primate evolution (“color vision priority hypothesis”). However, this logic is correct only when the fraction of OR pseudogenes is negatively correlated with the number of functional OR genes. In fact, Matsui and colleagues showed that the color vision priority hypothesis is not supported by an analysis of whole-genome sequences from five primate species (Matsui et al. 2010). Therefore, whole-genome sequences should be used whenever comparing OR gene repertoires from different species.
We found that OR gene lineages that had undergone fewer gene duplications tended to be under stronger purifying selection (Fig. 2F). This observation is reasonable because the evolutionary rate is expected to accelerate following gene duplication due to the relaxation of functional constraints. Ohno proposed that gene duplication creates two functionally redundant copies, and consequently, one copy is free from constraints and can acquire a new function, as the other copy retains the original function (Ohno 1970). Several studies of many gene families in genomes have shown that evolutionary rates accelerated following gene duplication (Lynch and Conery 2000; Kondrashov et al. 2002; Jordan et al. 2004), but here, our analysis of a single gene family clearly demonstrated the acceleration of evolutionary rates within this family.
We used a phylogeny-based method to identify OGGs among >10,000 intact OR genes from these 13 species (see Methods). In this method, we first computationally identified potential pairwise orthology between two species on the basis of phylogenetic trees. We then merged candidate orthologous pairs into potential OGGs. Visual inspection of the phylogenetic gene trees constructed from genes in candidate OGGs indicated that these candidate OGGs often contained paralogous genes; consequently, we used semi-automated methods to separate the genes in such “entangled” candidate OGGs into true OGGs. African elephants occupy the most basal position in the phylogeny of 13 species. The basic idea for identifying true OGGs was to identify monophyletic clades that contained both elephant and non-elephant genes in a phylogenetic gene tree of a candidate OGG. If a clade contained elephant genes and genes from other species, those elephant and non-elephant genes were likely to have originated from a single ancestral gene in the MRCA of placental mammals, and therefore, they were distinguishable from the genes descended from other MRCA genes. Notably, this strategy should work effectively only when a large number of elephant genes exist. Therefore, the key to the successful identification of OGGs of ORs was the usage of elephant genes.
Among the 781 OGGs identified, there were only three OGGs that showed complete one-to-one orthology. The human genes contained in the three OGGs—OGG1-44, OGG1-45, and OGG2-256—are OR51E1, OR51E2, and OR6B1, respectively. Both OR51E1 and OR51E2 are located in the Class I OR gene cluster on human chromosome 11, and they are juxtaposed to each other with a pseudogene in between. These two ORs share 57% amino acid sequence identity. Remarkably, both of them are ubiquitously expressed in various tissues. Recently, Flegel and colleagues investigated the expression of human OR genes in 16 different nonolfactory tissues (Flegel et al. 2013). They found that OR51E1 and OR51E2 are expressed in 13 and 12, respectively, of the 16 nonolfactory tissues and they are the two most broadly expressed human OR genes among those examined.
Both OR51E1 and OR51E2 have been deorphanized; known ligands of OR51E1 are 3- and 4-methyl-valeric acid (Fujita et al. 2007) and nonanoic acid (nonanoic acid is also a ligand of its mouse ortholog, Olfr558) (Saito et al. 2009), and those of OR51E2 are β-ionone and androstenone derivatives (Neuhaus et al. 2009). In fact, OR51E2, also known as prostate-specific GPCR (PSGR) (Xu et al. 2000), is one of the most well-characterized ORs. It is highly expressed in the human prostate and is strongly up-regulated in prostate cancer; therefore, it can be used as a marker of prostate cancer (Xu et al. 2000). Activation of this OR inhibits the proliferation of prostate cancer cells (Neuhaus et al. 2009). OR51E1, also known as PSGR2, is overexpressed in human prostate cancer as well (Weng et al. 2006), and OR51E1 overexpression in neuroendocrine carcinomas was reported recently (Leja et al. 2009). These results indicate that both of these ORs are involved in some physiological process(es) other than olfaction, and the conservation of these two ORs among placental mammals may indicate that the process(es) is(are) essential and common among many mammalian species. To our knowledge, OR6B1, the only Class II OR among these three human ORs, has not been deorphanized. OR6B1 is therefore an interesting target for deorphanization and further investigation of physiological function.
To reveal possible connections between OR function and OR evolution, we preliminarily examined the correlation between the number of ligands that bind to a given OR and the size of the OGG containing that OR. We used the entire data set from Saito et al. (2009) regarding OR–odorant pairs for 52 mouse ORs and 10 human ORs, which is currently the largest data set obtained under the same condition. We found that the number of ligands per OR was positively correlated with OGG size (Spearman’s correlation coefficient rs = 0.470; P = 0.00020) (Supplemental Fig. S9). Therefore, gene lineages containing genes of generalist ORs tend to have expanded more during evolution than those containing genes of specialist ORs. These observations may be explained by assuming that generalist ORs persist for long periods during evolution because they remain functional in changing environments, while specialist ORs may easily become useless as environments change and the respective genes get stuck in evolutionary “dead ends.” However, this analysis was based on a limited number of ORs, and we need more data on OR–ligand pairs to derive a clear conclusion.
In summary, we used the surprisingly large repertoire of African elephant OR genes to perform a successful phylogeny-based identification of OGGs among OR genes from 13 placental mammals. We then traced the differences in the evolutionary trajectories of OR gene lineages in terms of the extent of gene gains and gene losses. This study clearly illustrates that analyses of evolutionary dynamics of genes can provide insights into gene function. Nevertheless, more ORs should be deorphanized and more OR gene repertoires should be analyzed from many organisms to further elucidate the differences in the evolutionary fates of genes.
Methods
Data
Whole-genome sequences of placental mammals used in this study were downloaded from Ensembl (http://www.ensembl.org) (Flicek et al. 2014). The following data were used: African elephant (Loxodonta africana), loxAfr3; cow (Bos taurus), Btau_4.0; horse (Equus caballus), EquCab2; rabbit (Oryctolagus cuniculus), oryCun2; guinea pig (Cavia porcellus), cavPor3; mouse (Mus musculus), NCBIm36.
Identification of OR genes from whole-genome sequence
The method used to identify OR genes from the genome sequence was described in detail previously (Niimura 2013).
Construction of phylogenetic trees
Each neighbor-joining (NJ) tree (Saitou and Nei 1987) was constructed with Poisson correction (PC) distance using the program LINTREE (http://www.personal.psu.edu/nxm2/software.htm) (Takezaki et al. 1995). Multiple alignments of translated amino acid sequences were made by the program MAFFT (http://mafft.cbrc.jp/alignment/software/) (Katoh et al. 2005).
Assignment of intact OR genes to phylogenetic clades
Because the total number of OR genes in the 13 placental mammals was large (10,659), we classified each OR gene into phylogenetic clades defined in previous studies (Niimura and Nei 2003, 2005) and examined each of the clades separately for the identification of OGGs (see below). We used 33 monophyletic clades (one Class I clade and 32 Class II clades named A–S, AA–AJ, AT, BB, and BC), each of which is supported by a high (>90%) bootstrap value. Clade assignment has been performed for rat, dog (Niimura and Nei 2007), and five primate species (Matsui et al. 2010). For each of the other species (referred to here as species X), we constructed a phylogenetic tree using all intact genes in species X and those in either rat or dog. We then identified monophyletic clades with high bootstrap values that contained all member genes belonging to a given clade from either rat or dog; genes from species X that were included in a respective clade were assigned to the same clade as that for rat genes and then separately for dog genes. We confirmed that the results obtained using rat genes and those using dog genes were consistent with each other. Some Class II genes remained unclassified, and they were treated separately for OGG identification.
Identification of OGGs among 13 species
To identify OGGs, each phylogenetic gene clade identified above was treated separately. We first identified candidate orthologous gene pairs between two species. Because OGGs among the five primates were identified in a previous study (Matsui et al. 2010), we treated the five primates as one species in the following process. Therefore, there are 36 possible pairs of species among eight non-primate species and five primates treated as one species. For each species pair (e.g., elephant and horse), we constructed NJ trees using all intact genes from the two species belonging to a given clade. We therefore constructed 34 phylogenetic trees (one Class I clade, 32 Class II clades, and one clade of unclassified Class II genes) for a given species pair. Eight representative genes were chosen from clades A–H (one gene from each clade) and were used as the outgroup of each of the 34 trees. For constructing the clade A tree, for example, seven representative genes from clades B–H were used as the outgroup. Similarly, seven representative genes were used as the outgroup to construct the trees of clades B–H. For the other trees, all eight representative genes from clades A–H were used as the outgroup.
From each tree, we extracted candidate orthologous gene pairs between two species by taking monophyletic clades that contained genes from both species and were supported with >70% bootstrap values (Supplemental Fig. S3A). Note that pairwise orthologous relationships were often not a one-to-one relationship, but one-to-multiple or multiple-to-multiple. When gene losses occurred independently in each of the two lineages, paralogous genes may have been wrongly identified as candidate orthologs (Supplemental Fig. S3B). On average, the evolutionary distances for paralogous gene pairs between two species tend to be larger than those for orthologous gene pairs between the same two species. Therefore, to exclude paralogous gene pairs, we calculated the number of synonymous changes per synonymous site, dS, for all candidate orthologous gene pairs identified above. We used the modified Nei-Gojobori method (Nei and Gojobori 1986) to calculate dS values from pairwise alignments generated by ClustalW (Thompson et al. 1994). When an orthologous relationship was not one-to-one, dS values were calculated for all possible gene pairs between the two species, and their mean value was used for the following analysis. For a given pair of species, the mean (μ) and the standard deviation (σ) were calculated. By inspecting the distribution of dS values, we regarded any gene pairs showing dS > μ + 2.326σ (corresponding to the top 1% data for normally distributed samples) as false positives (paralogs) and discarded them (Supplemental Fig. S3C). This criterion was conservative and was designed to avoid the possibility that true orthologs were erroneously eliminated. The remaining orthologous gene pairs for the 36 combinations of two species were collected, and “a friend of a friend is a friend” strategy was used to merge these pairs into candidate OGGs among the 13 species (Supplemental Fig. S3D). After this procedure, we obtained 611 candidate OGGs that contained genes from at least two species.
By visually inspecting the phylogenetic trees of the 611 candidate OGGs identified above, we found that candidate OGGs often contained apparent paralogous genes. We separated such “entangled” candidate OGGs into true OGGs by using the following criteria. We first constructed a rooted NJ tree using all intact genes belonging to each of the 611 candidate OGGs. As the outgroup, we used eight genes, each of which was chosen from clades A–H. If the resultant NJ tree contained a monophyletic clade (denoted as clade X in Supplemental Fig. S3E) that met the conditions below, genes in clade X were considered to constitute an OGG. We used the three following conditions to make these determinations: (1) Clade X contains both elephant gene(s) and non-elephant gene(s), (2) clade X is supported with a >90% bootstrap value, and (3) clade X and clade Y (a sister clade of clade X) contain gene(s) from at least one common species; this third condition was necessary because sometimes a gene tree was inconsistent with the species tree. This procedure was repeated until no more clades could be separated.
The candidate OGGs obtained using the procedure described above were divided further. We next constructed an unrooted NJ tree using all genes in each of the candidate OGGs. If (1) a tree contained two clades, each of which contained both elephant and non-elephant genes, and (2) the separation of the two clades was supported with a >70% bootstrap value, then each clade was considered to form an independent OGG (Supplemental Fig. S3F). This procedure was also repeated until no more separation was possible. As a result, we obtained 731 OGGs that contained genes from two or more species.
There were 67 species-specific genes (“singletons”) that were not included in any of the 731 OGGs identified above. Some singletons from the same species may have originated from the same ancestral gene in the MRCA of placental mammals, and such singletons should be assigned to the same OGG. For such singletons, the evolutionary distances among them were assumed to be small. To identify combinations of such singletons, we calculated dS values between any pairs of singletons from the same species. If dS was smaller than a threshold value, the pair of singletons was considered to belong to the same OGG. The threshold values were determined in the following way. As described above, we examined the distribution of dS values for all candidate orthologous gene pairs between a given pair of species and calculated the mean value, μ. We examined all of the eight μ values for elephant vs. non-elephant comparisons, and took the largest μ among them. μ = 0.389, 0.386, 0.340, 0.447, 0.513, 0.566, 0.558, and 0.376 for elephant–cow, elephant–dog, elephant–horse, elephant–rabbit, elephant–guinea pig, elephant–rat, elephant–mouse, and elephant–primates comparisons, respectively; therefore, the threshold value for elephants was set to be 0.566. In the same manner, the threshold values for the other species were determined. This procedure generated 50 OGGs from 67 singletons. Eventually, 10,659 intact OR genes from 13 placental mammals were classified into 781 OGGs.
Finally, we assigned each of the non-intact genes (pseudogenes and truncated genes) to the 781 OGGs identified above. In this process, we did not use a phylogeny-based approach, because the presence of fragmented sequences lowers the accuracy of phylogenetic inference. Instead we conducted BLASTP searches using each non-intact gene as a query (Altschul et al. 1997) against all 10,659 intact OR genes. We assigned each non-intact gene to the OGG that contained its respective best-hit intact gene.
Estimation of the numbers of gene gains and losses
The reconciled-tree method was used to estimate the numbers of gene gains and gene losses in each branch of a species tree and the number of ancestral genes in the evolution of placental mammals (http://bioinfo.tmd.ac.jp/∼niimura/software.html) (Niimura and Nei 2007). A calculation was performed for each OGG separately using a 70% bootstrap value as a threshold for reconciliation. The reconciled-tree method requires a rooted tree for each given set of genes to make these estimates. To identify a root position of a tree, we did not use any outgroup sequences that were not included in a given OGG. Rather, we assumed the root position to be on a branch dividing all genes in a given OGG into two clades: One was the clade containing all genes from the first divergent species, and the other was the clade containing all genes from the other species. For example, if a given OGG contained at least one elephant gene, then the root was assumed to be located on the branch connecting the clade containing elephant gene(s) with that containing non-elephant gene(s). If an OGG did not contain elephant genes but contained cow, dog, and/or horse gene(s), the root was assumed to be on a branch between a Laurasiatheria clade and an Euarchontoglires clade. The results for all OGGs were compiled to generate Figure 6.
Extent of purifying selection
We used the maximum likelihood method implemented in PAML (http://abacus.gene.ucl.ac.uk/software/paml.html) (Yang and Nielsen 2000) to estimate the nonsynonymous/synonymous rate ratio, ω. In this analysis, 702 OGGs that contained three or more intact OR genes were used. An unrooted NJ tree was constructed separately using all intact genes contained in each of the 702 OGGs. The program codeml and a codon frequency model of F3×4 were then used to calculate the ω value from the phylogenetic tree.
Estimation of gene birth and death rates
The gene birth rate β, the number of gene gains per million years per gene, and the gene death rate δ, the number of gene losses per million years per gene, were calculated for each branch in the phylogeny of 13 placental mammals in the following way. For a given branch, β and δ were assumed to be constant with time. Suppose that at the initial time t = 0, there were A0 genes, and at time t = T, the number of genes became A0 + G – L due to G gene gains and L gene losses that occurred during time T. The number of gene gains that occurred until time t was denoted as g(t) and that of gene losses, l(t). Therefore, G = g(T) and L = l(T). We then obtained the following simultaneous differential equations:
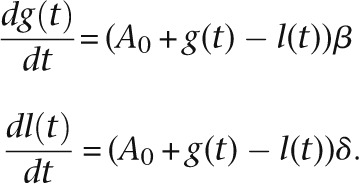 |
Solving these equations, we obtained
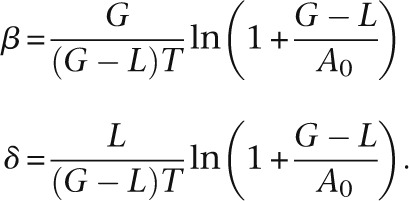 |
We used these formulas to calculate the results presented in Supplemental Figure S8.
Vieira and colleagues used the formulas β = G/TA0 and δ = L/TA0 to estimate the birth and death rates (Vieira et al. 2007). They assumed that the number of genes is constant along a branch; therefore, Vieira and colleagues’ estimates are inaccurate when β ≠ δ.
The mean birth and death rates, 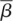 and
and 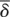 , are calculated by weighting the birth (βi) and death (δi) rates of the ith branch by the length of the branch, bi:
, are calculated by weighting the birth (βi) and death (δi) rates of the ith branch by the length of the branch, bi:
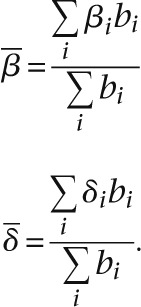 |
Data access
Nucleotide and predicted amino acid sequences for OR genes in African elephant, horse, cow, rabbit, guinea pig, and mouse are provided in Supplemental Data Sets S1 and S2, respectively.
Acknowledgments
This study was supported by a Grant-in-Aid for Young Scientists (B) (JSPS KAKENHI Grant Number 23770271) and the ERATO Touhara Chemosensory Signal Project from JST, Japan.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.169532.113.
References
- Adipietro KA, Mainland JD, Matsunami H. 2012. Functional evolution of mammalian odorant receptors. PLoS Genet 8: e1002821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloni R, Olender T, Lancet D. 2006. Ancient genomic architecture for mammalian olfactory receptor clusters. Genome Biol 7: R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates LA, Sayialel KN, Njiraini NW, Moss CJ, Poole JH, Byrne RW. 2007. Elephants classify human ethnic groups by odor and garment color. Curr Biol 17: 1938–1942 [DOI] [PubMed] [Google Scholar]
- Bates LA, Sayialel KN, Njiraini NW, Poole JH, Moss CJ, Byrne RW. 2008. African elephants have expectations about the locations of out-of-sight family members. Biol Lett 4: 34–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck L, Axel R. 1991. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell 65: 175–187 [DOI] [PubMed] [Google Scholar]
- Demuth JP, De Bie T, Stajich JE, Cristianini N, Hahn MW. 2006. The evolution of mammalian gene families. PLoS ONE 1: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J 1985. Phylogenies and the comparative method. Am Nat 125: 1–15 [Google Scholar]
- Flegel C, Manteniotis S, Osthold S, Hatt H, Gisselmann G. 2013. Expression profile of ectopic olfactory receptors determined by deep sequencing. PLoS ONE 8: e55368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2014. Ensembl 2014. Nucleic Acids Res 42: D749–D755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Takahashi T, Suzuki A, Kawashima K, Nara F, Koishi R. 2007. Deorphanization of Dresden G protein-coupled receptor for an odorant receptor. J Recept Signal Transduct Res 27: 323–334 [DOI] [PubMed] [Google Scholar]
- Gabaldon T 2008. Large-scale assignment of orthology: back to phylogenetics? Genome Biol 9: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldon T, Koonin EV. 2013. Functional and evolutionary implications of gene orthology. Nat Rev Genet 14: 360–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Przeworski M, Lancet D. 2004. Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates. PLoS Biol 2: E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y, Niimura Y. 2008. Similar numbers but different repertoires of olfactory receptor genes in humans and chimpanzees. Mol Biol Evol 25: 1897–1907 [DOI] [PubMed] [Google Scholar]
- Hayden S, Bekaert M, Crider TA, Mariani S, Murphy WJ, Teeling EC. 2010. Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res 20: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. 2006. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22: 2971–2972 [DOI] [PubMed] [Google Scholar]
- Jordan IK, Wolf YI, Koonin EV. 2004. Duplicated genes evolve slower than singletons despite the initial rate increase. BMC Evol Biol 4: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33: 511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida T, Hikida T. 2010. Degeneration patterns of the olfactory receptor genes in sea snakes. J Evol Biol 23: 302–310 [DOI] [PubMed] [Google Scholar]
- Kishida T, Kubota S, Shirayama Y, Fukami H. 2007. The olfactory receptor gene repertoires in secondary-adapted marine vertebrates: evidence for reduction of the functional proportions in cetaceans. Biol Lett 3: 428–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA, Rogozin IB, Wolf YI, Koonin EV. 2002. Selection in the evolution of gene duplications. Genome Biol 3: RESEARCH0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautwurst D, Yau KW, Reed RR. 1998. Identification of ligands for olfactory receptors by functional expression of a receptor library. Cell 95: 917–926 [DOI] [PubMed] [Google Scholar]
- Langbauer WR 2000. Elephant communication. Zoo Biol 19: 425–445 [Google Scholar]
- Leja J, Essaghir A, Essand M, Wester K, Oberg K, Totterman TH, Lloyd R, Vasmatzis G, Demoulin JB, Giandomenico V. 2009. Novel markers for enterochromaffin cells and gastrointestinal neuroendocrine carcinomas. Mod Pathol 22: 261–272 [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155 [DOI] [PubMed] [Google Scholar]
- Malnic B, Hirono J, Sato T, Buck LB. 1999. Combinatorial receptor codes for odors. Cell 96: 713–723 [DOI] [PubMed] [Google Scholar]
- Matsui A, Go Y, Niimura Y. 2010. Degeneration of olfactory receptor gene repertories in primates: no direct link to full trichromatic vision. Mol Biol Evol 27: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Meredith RW, Janecka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Eizirik E, Simao TL, Stadler T, et al. 2011. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 334: 521–524 [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426 [DOI] [PubMed] [Google Scholar]
- Nei M, Rooney AP. 2005. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39: 121–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Niimura Y, Nozawa M. 2008. The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat Rev Genet 9: 951–963 [DOI] [PubMed] [Google Scholar]
- Neuhaus EM, Zhang W, Gelis L, Deng Y, Noldus J, Hatt H. 2009. Activation of an olfactory receptor inhibits proliferation of prostate cancer cells. J Biol Chem 284: 16218–16225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman T, Trask BJ. 2003. Complex evolution of 7E olfactory receptor genes in segmental duplications. Genome Res 13: 781–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y 2012. Olfactory receptor multigene family in vertebrates: from the viewpoint of evolutionary genomics. Curr Genomics 13: 103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y 2013. Identification of olfactory receptor genes from mammalian genome sequences. Methods Mol Biol 1003: 39–49 [DOI] [PubMed] [Google Scholar]
- Niimura Y, Nei M. 2003. Evolution of olfactory receptor genes in the human genome. Proc Natl Acad Sci 100: 12235–12240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, Nei M. 2005. Comparative evolutionary analysis of olfactory receptor gene clusters between humans and mice. Gene 346: 13–21 [DOI] [PubMed] [Google Scholar]
- Niimura Y, Nei M. 2007. Extensive gains and losses of olfactory receptor genes in mammalian evolution. PLoS ONE 2: e708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Springer-Verlag, New York. [Google Scholar]
- O’Leary MA, Bloch JI, Flynn JJ, Gaudin TJ, Giallombardo A, Giannini NP, Goldberg SL, Kraatz BP, Luo ZX, Meng J, et al. 2013. The placental mammal ancestor and the post-K-Pg radiation of placentals. Science 339: 662–667 [DOI] [PubMed] [Google Scholar]
- Rajaram A, Krishnamurthy V. 2003. Elephant temporal gland ultrastructure and androgen secretion during musth. Curr Sci 85: 1467–1471 [Google Scholar]
- Rasmussen LEL, Krishnamurthy V. 2000. How chemical signals integrate Asian elephant society: the known and the unknown. Zoo Biol 19: 405–423 [Google Scholar]
- Rasmussen LE, Lee TD, Roelofs WL, Zhang A, Daves GD Jr. 1996. Insect pheromone in elephants. Nature 379: 684. [DOI] [PubMed] [Google Scholar]
- Rasmussen LE, Lee TD, Zhang A, Roelofs WL, Daves GD Jr. 1997. Purification, identification, concentration and bioactivity of (Z)-7-dodecen-1-yl acetate: sex pheromone of the female Asian elephant, Elephas maximus. Chem Senses 22: 417–437 [DOI] [PubMed] [Google Scholar]
- Rizvanovic A, Amundin M, Laska M. 2013. Olfactory discrimination ability of Asian elephants (Elephas maximus) for structurally related odorants. Chem Senses 38: 107–118 [DOI] [PubMed] [Google Scholar]
- Saito H, Chi Q, Zhuang H, Matsunami H, Mainland JD. 2009. Odor coding by a Mammalian receptor repertoire. Sci Signal 2: ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425 [DOI] [PubMed] [Google Scholar]
- Shirasu M, Yoshikawa K, Takai Y, Nakashima A, Takeuchi H, Sakano H, Touhara K. 2014. Olfactory receptor and neural pathway responsible for highly selective sensing of musk odors. Neuron 81: 165–178 [DOI] [PubMed] [Google Scholar]
- Shoshani J, Kupsky WJ, Marchant GH. 2006. Elephant brain. Part I: gross morphology, functions, comparative anatomy, and evolution. Brain Res Bull 70: 124–157 [DOI] [PubMed] [Google Scholar]
- Takezaki N, Rzhetsky A, Nei M. 1995. Phylogenetic test of the molecular clock and linearized trees. Mol Biol Evol 12: 823–833 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touhara K, Vosshall LB. 2009. Sensing odorants and pheromones with chemosensory receptors. Annu Rev Physiol 71: 307–332 [DOI] [PubMed] [Google Scholar]
- Touhara K, Sengoku S, Inaki K, Tsuboi A, Hirono J, Sato T, Sakano H, Haga T. 1999. Functional identification and reconstitution of an odorant receptor in single olfactory neurons. Proc Natl Acad Sci 96: 4040–4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira FG, Sanchez-Gracia A, Rozas J. 2007. Comparative genomic analysis of the odorant-binding protein family in 12 Drosophila genomes: purifying selection and birth-and-death evolution. Genome Biol 8: R235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Wang J, Hu X, Wang F, Ittmann M, Liu M. 2006. PSGR2, a novel G-protein coupled receptor, is overexpressed in human prostate cancer. Int J Cancer 118: 1471–1480 [DOI] [PubMed] [Google Scholar]
- Xu LL, Stackhouse BG, Florence K, Zhang W, Shanmugam N, Sesterhenn IA, Zou Z, Srikantan V, Augustus M, Roschke V, et al. 2000. PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res 60: 6568–6572 [PubMed] [Google Scholar]
- Yang Z, Nielsen R. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 17: 32–43 [DOI] [PubMed] [Google Scholar]
- Yoshikawa K, Nakagawa H, Mori N, Watanabe H, Touhara K. 2013. An unsaturated aliphatic alcohol as a natural ligand for a mouse odorant receptor. Nat Chem Biol 9: 160–162 [DOI] [PubMed] [Google Scholar]